
Page 47
The Human Genome Project: Elucidating Our Genetic Blueprint
ERIC D. GREEN
National Human Genome Research Institute National Institutes of Health Bethesda, Maryland
The human genome consists of approximately 3 billion base pairs (bp) of DNA contained within 24 chromosomes that range in size from approximately 50 million to 260 million bp. Encoded within this DNA are an estimated 25,000 to 75,000 genes and the necessary elements that control the regulation of their expression. A listing of the sequence of the human genome's bases in single-letter symbols (G, A, T, C) would fill about 13 sets of the Encyclopaedia Britannica,approximately 750 megabytes of computer disk space, or roughly 1 CD-ROM.
THE HUMAN GENOME PROJECT
The Human Genome Project is a coordinated, international effort to map and sequence the human genome and, in parallel, that of several well-studied model organisms. In the United States, the National Institutes of Health ( www.nhgri.nih.gov) and the Department of Energy ( jgi.doe.gov) orchestrate the relevant programs, with the project officially beginning on October 1, 1990. Since its inception, the Human Genome Project has been associated with carefully crafted, milestone-oriented goals, the most recent set being established in 1998 (Collins et al., 1998).
The first phase of the project involved constructing relatively low-resolution maps of the human genome and refining the approaches for large-scale DNA sequencing. The second phase has focused on establishing the complete sequence of the human and other vertebrate genomes, as well as beginning to decipher the encoded information in a systematic fashion.
Page 48
MAPPING THE HUMAN GENOME
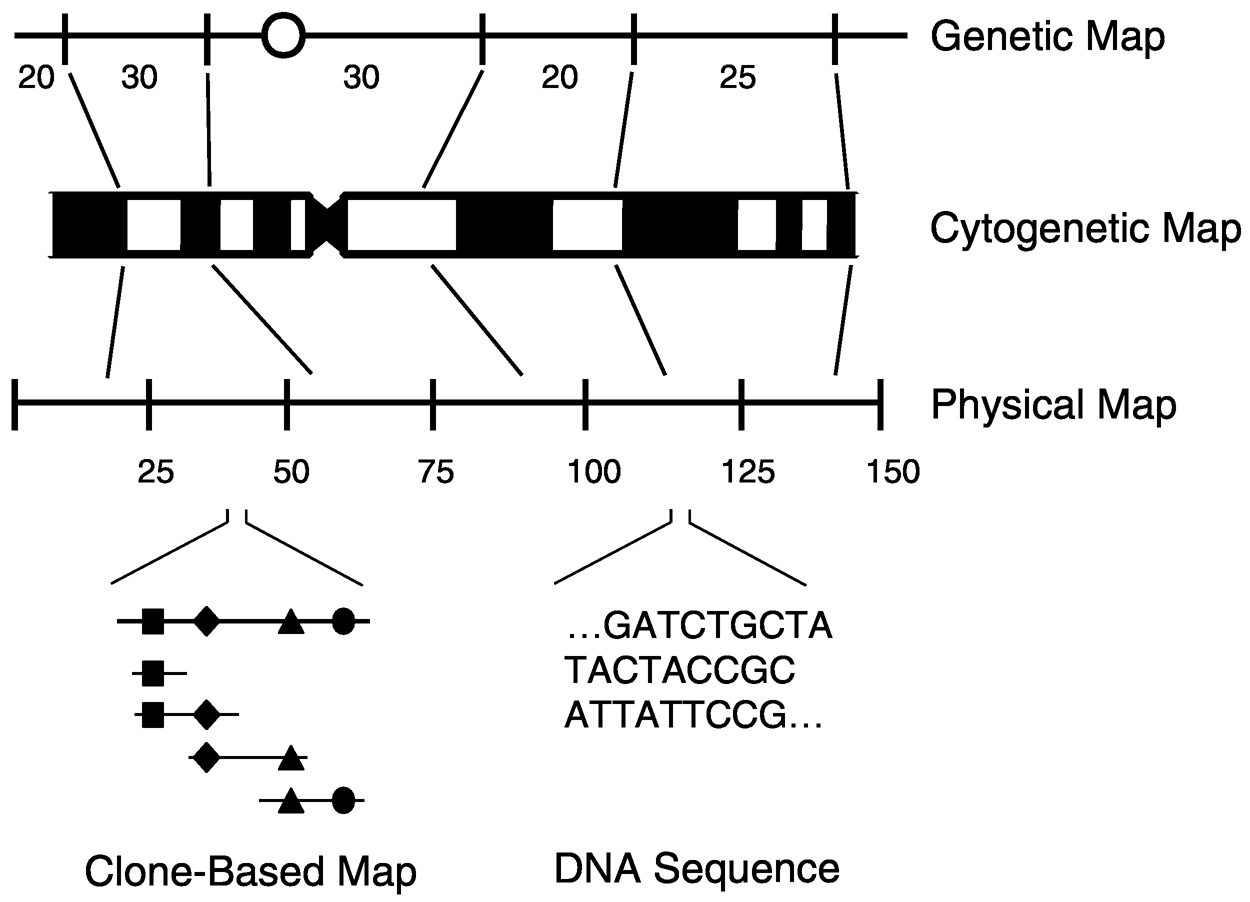
Genomic maps are organizational representations that reflect specific features of the corresponding DNA. There are three major classes of genomic maps: cytogenetic, genetic, and physical ( Figure 1). In each case, the coordinates on which the maps are based reflect the experimental method(s) used to establish the relative positions of landmarks.
-
Cytogenetic maps. Cytogenetic maps represent the appearance of chromosomes when properly stained and examined microscopically. Particularly important is the appearance of differentially staining regions (called bands) that render each chromosome uniquely identifiable.
~ enlarge ~
FIGURE 1 Schematic representations of the genetic, cytogenetic, and physical maps of a human chromosome. For the genetic map, the positions of several hypothetical genetic markers are indicated, along with the relative genetic distances between them. The circle indicates the position of the centromere. For the cytogenetic map, the classic Giemsabanding pattern of a chromosome is shown. For the physical map, the approximate physical locations of the above genetic markers are indicated, along with the relative distances between them in millions of bp. Two types of physical maps—an overlapping clone-based map and the highest-resolution physical map, the actual DNA sequence—are depicted along the bottom. SOURCE: Reprinted with permission from The McGrawHill Companies (Green, 2001).
Page 49
-
Genetic maps. Also known as linkage maps or meiotic maps, genetic maps depict the relative locations of genetic markers across a stretch of DNA. Genetic markers reflect sequences that vary among different individuals (i.e., they are polymorphic). Such sequence variation allows genetic markers to be followed during passage from one generation to the next.
-
Physical Maps. Physical maps depict the relative locations of physical landmarks across a stretch of DNA. The conventional approach for physical mapping involves fragmenting and cloning the DNA, and then reassembling the pieces in an organized fashion. A collection of ordered, overlapping clones is called a contig, since the clones together contain a contiguous segment of DNA.
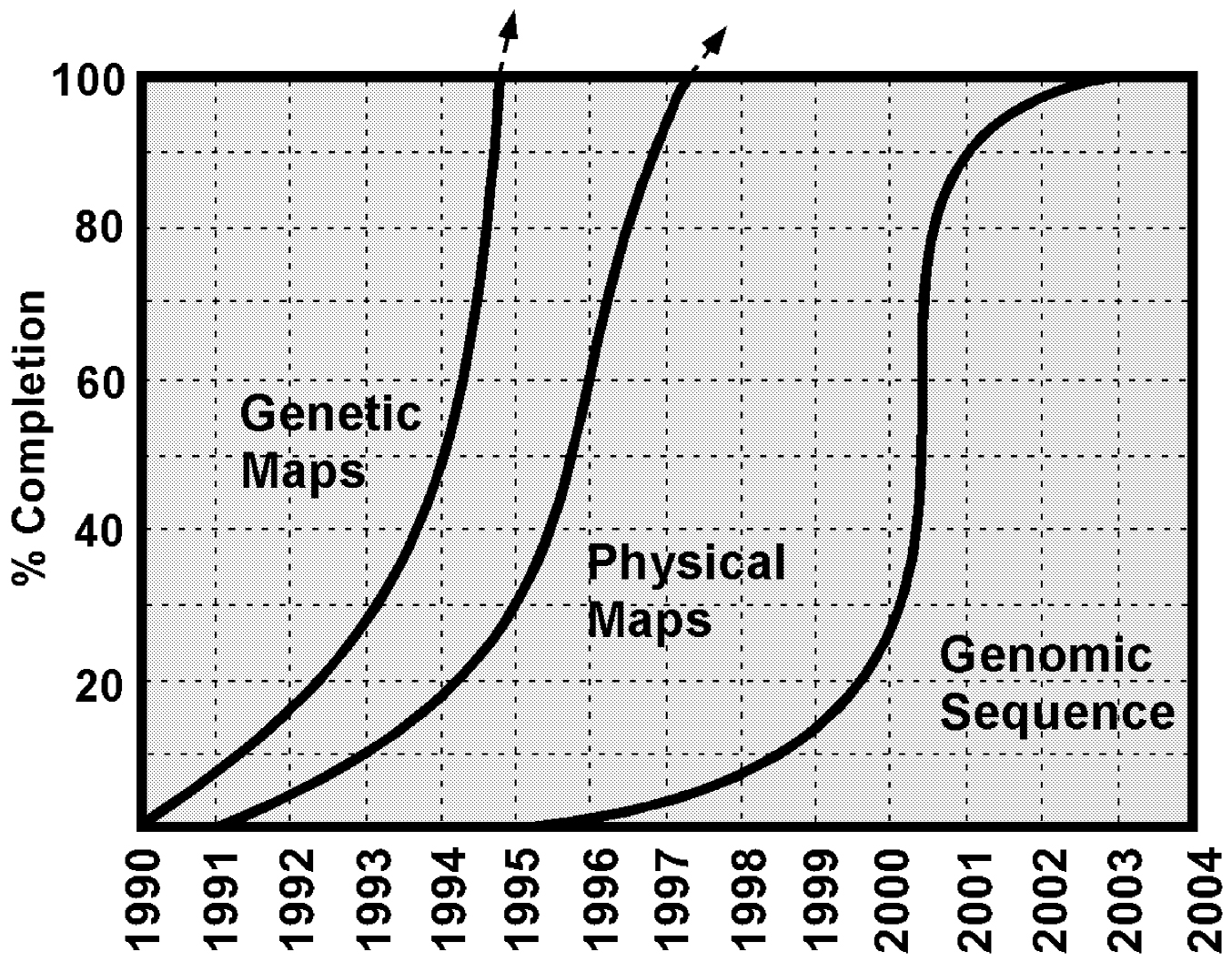
The development of high-quality genetic and physical maps of the human genome represented key early activities of the Human Genome Project. These goals were largely reached by the mid-1990s ( Figure 2). Attention then turned to sequencing the human genome.
~ enlarge ~
FIGURE 2 Timetable for human genome analysis in the Human Genome Project. The approximate schedule for completing the human genetic map, physical map, and complete genomic sequence is depicted. SOURCE: Reprinted with permission from The McGraw-Hill Companies (Green, 2001).
Page 50
SEQUENCING THE HUMAN GENOME
The highest-resolution physical map is the actual DNA sequence, which depicts the precise order of bases (adenine [A], guanine [G], cytosine [C], and thymine [T]). A handful of techniques have been developed for sequencing DNA. The approach described by Sanger and coworkers in 1977 (Sanger et al., 1977)—dideoxy chain-termination sequencing—is the most widely used sequencing method. This technique involves the in vitrosynthesis of DNA molecules in the presence of artificial (dideoxy) bases, which prevent chain extension when incorporated into a growing DNA strand. The resulting population of DNA molecules, which terminate at different base positions, is then analyzed by gel electrophoresis and the relative migration of the various DNA fragments is used to deduce the sequence of the starting DNA. The most contemporary methods for large-scale DNA sequencing involve the incorporation of fluorescent tags into the DNA, followed by their real-time detection using laser-based instrumentation (Hunkapiller et al., 1991; Smith et al., 1986).
In the past decade, there have been numerous advances in fluorescence-based DNA sequencing. In parallel, researchers have gained tremendous experience through the Human Genome Project's efforts to sequence several model organisms, including the yeast S. cerevisiae (genome www.stanford.edu/Saccharomyces), the bacterium E. coli (www.tigr.org/tdb/mdb/mdbcomplete.html), the nematode C. elegans (www.sanger.ac.ukand genome.wustl.edu/gsc/gschmpg.html), and the fruit fly D. melanogaster (www.fruitfly.org). As a result, the project has focused most recently on sequencing the human genome, with the aim of generating a complete human genome sequence by 2003 (Figure 2). This endeavor has involved two major steps (Waterston and Sulston, 1998). First, suitable overlapping clones are being selected for sequencing—specifically, well-ordered sets of bacterial artificial chromosome (BAC) clones, each containing approximately 100,000 to 200,000 bp of DNA. Second, individual clones are being subjected to a process known as shotgun sequencing (Wilson and Mardis, 1997).
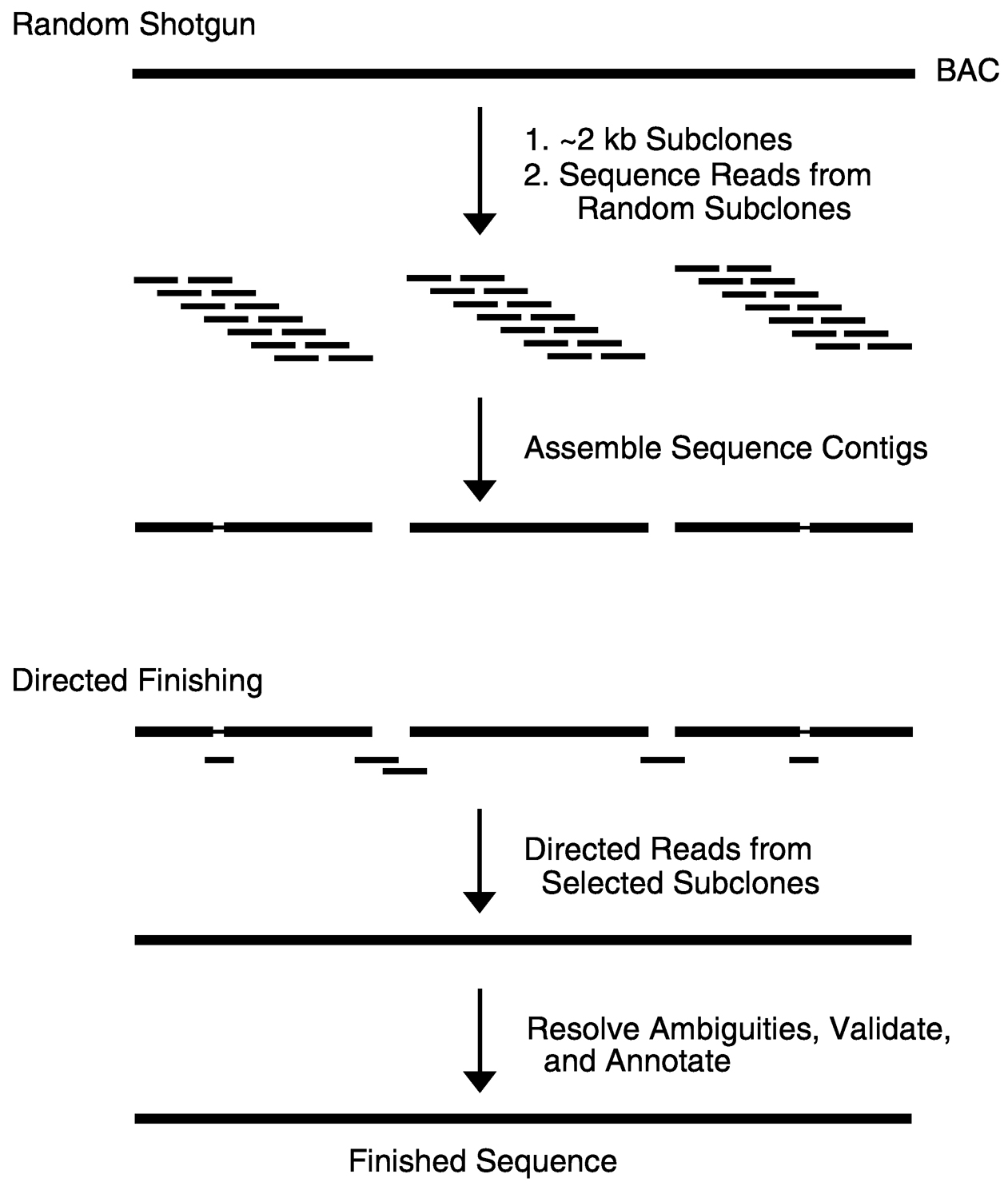
Shotgun sequencing ( Figure 3) begins with the generation of subclones that each contain a small (e.g., ~2000 bp), random piece of the starting clone (e.g., BAC). Sequence reads are then obtained from one or both ends of a large number of subclones. Sufficient sequence data are generated such that each base of the starting DNA is read, on average, 6 to 10 times. Computational tools are then used to analyze the resulting sequences so as to identify those that overlap to form sequence contigs (each associated with an assembled consensus sequence). The initial shotgun sequencing data typically result in the assembly of a small number of sequence contigs. The next phase involves generating sequence data in a highly directed fashion so as to fill in the remaining gaps, merge the sequence contigs together, and refine any low-quality regions. This “finishing” process involves a number of specialized computational and experimental
Page 51
~ enlarge ~
FIGURE 3 Shotgun DNA sequencing. The dominant genomic sequencing strategy being used in the Human Genome Project is shotgun sequencing, which consists of two major phases. In the first random shotgun phase a genomic clone (e.g., BAC) is subcloned into approximately 2 kb fragments. Sequence reads are then obtained from one or both ends of a large number of randomly selected subclones. Sufficient sequence data are generated such that each base of the starting clone is read, on average, about 6 to 10 times. These redundant sequence data are then analyzed with various computational tools, allowing the assembly of sequence contigs. Typically, only a handful of gaps between sequence contigs and other problems (e.g., low-quality regions) remain at this stage. In the second directed finishing phase, additional data are generated to complete the sequence, most often by obtaining directed reads from strategically selected subclones. This usually allows the merger of the remaining sequence contigs and the refinement of any low-quality regions to yield a final contiguous, high-accuracy (i.e., finished) sequence. SOURCE: Reprinted with permission from The McGraw-Hill Companies (Green, 2001).
Page 52
tools, requires highly trained technical personnel, and often involves generating data from particularly difficult sequences (Wilson and Mardis, 1997). The last step of shotgun sequencing involves careful review of the entire assembled sequence, which includes both checking for any ambiguities or problem areas and analyzing the sequence for features of interest.
Several additional points about sequencing the human genome deserve mention. First, this activity is being carefully coordinated among five major and about twelve smaller sequencing centers around the world so that the sequence can be completed as efficiently as possible. Second, all new sequence data being generated by the project are made freely available over the Internet every night (e.g., via the public sequence database GenBank [ www.ncbi.nlm.nih.gov]). Note that this includes both the final, finished sequence of individual clones and the evolving, preliminary sequence data of clones whose analyses are still in progress. Finally, the public Human Genome Project's effort to sequence the human genome is occurring in parallel with the much-publicized private effort being performed at the company Celera Genomics (see www.celera.com). The latter group is using a different (albeit complementary) sequencing strategy, whereby the entire human genome is shotgun sequenced en masse (called a “whole-genome shotgun”; Venter et al., 1998).
In the summer of 2000, a major milestone was reached in the Human Genome Project, with completion of a “working draft” sequence for approximately 90 percent of the human genome. In essence, this reflects the acquisition of preliminary sequence data for virtually all of the readily clonable human DNA. This sequence will be refined (i.e., finished to high accuracy) over the next 2 to 3 years. Indeed, the complete, finished sequence for two human chromosomes has already been achieved.
SUMMARY
The Human Genome Project is one of the most important projects—if not themost important project—ever undertaken in biomedical research. It is fundamentally an endeavor to develop tools for the study of biology and medicine. These tools reflect both an information resource in the form of genetic maps, physical maps, and the underlying sequence of the human genome and that of several model organisms, as well as an ever-increasing number of experimental technologies that are becoming standard techniques in the armamentarium of biomedical researchers. In this regard, an exciting new “genomic revolution” has started and will forever change the way research is performed. The new and powerful foundation of genetic information is empowering investigators to tackle complex problems relating to disease, development, and evolution that were previously unapproachable. Ultimately, the central legacy of the Human Genome Project will be the provision to future generations of scientists and clinicians of an unprecedented resource—the human “genetic blueprint”—that will
Page 53
allow them to understand fundamental features of human biology (Lander, 1996) and to elucidate the genetic bases of disease (Green, 2001).
REFERENCES
, , , , , , and . 1998. New goals for the U.S. Human Genome Project:1998–2003. Science 282: 682–689.
2001. The Human Genome Project and its impact on the study of human disease. Pp.259–298 in The Metabolic and Molecular Bases of Inherited Disease, C. R. Scriver, A. L.Beaudet, W. S. Sly, and D. Valle, eds. New York: McGraw–Hill.
, , , and . 1991. Large-scale and automated DNA sequence determination. Science 254: 59–67.
1996. The new genomics: Global views of biology. Science 274: 536–539.
, , and . 1977. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the USA 74: 5463–5467.
, , , , , , , , and . 1986. Fluorescence detection in automated DNA sequence analysis. Nature 321: 674–679.
, , , , , and . 1998. Shotgun sequencing of the human genome. Science 280: 1540–1542.
, and . 1998. The Human Genome Project: Reaching the finish line. Science 282: 53–54.
Wilson, R. K., and . 1997. Shotgun sequencing. Pp. 397–454 in Genome Analysis: A Laboratory Manual. Vol. 1: Analyzing DNA, B. Birren, E. D. Green, S. Klapholz, R. M. Myers, and J. Roskams, eds. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.









