

































Below is the uncorrected machine-read text of this chapter, intended to provide our own search engines and external engines with highly rich, chapter-representative searchable text of each book. Because it is UNCORRECTED material, please consider the following text as a useful but insufficient proxy for the authoritative book pages.
Atmospheric Transformations of Automotive Emissions ROGER ATKINSON University of California, Riverside Components of Atmospheric Pollution / 100 Physical and Chemical Transformations Under Atmospheric Conditions / 101 Physical Removal Processes / 101 Chemical Removal Processes / 102 Atmospheric Lifetimes, Fates, and Products of the Atmospheric Transformations of Automotive Emissions / 105 Atmospheric Lifetimes / 105 Atmospheric Transformations / 109 Analytical Techniques / 125 Summary / 126 Summary of Research Recommendations / 126 Air Pollution, the Automobile' and Public Health. (it) 1988 by the Health Effects Institute. National Academy Press, Washington, D.C. 99
100 Atmospheric Transformations of Automotive Emissions Components of Atmospheric Pollution A wide spectrum of inorganic and organic chemical compounds are emitted from au- tomotive use. These emissions arise from combustion as well as evaporative ~oro- cesses. They include the obvious water vapor and carbon dioxide (CO2), as well as carbon monoxide (CO), oxides of nitrogen (NOx), oxides and oxyacids of sulfur, re- duced sulfur compounds, a wide variety of volatile organic compounds comprising fuel components and partially oxidized products of combustion, and particulate matter. The identities of these emissions and a quantitative understanding of their emission rates are the focus of "Auto- motive Emissions" Johnson, this volume). In highly urbanized regions, automotive emissions contribute a significant, and of- ten major, fraction of the overall emission burden of NOx, volatile organic com- pounds, and elemental carbon and/or par- ticulate organic matter. For example, the 1979 mobile and stationary source contri- butions of NOx, volatile reactive organic gases (ROG), oxides of sulfur (SOx), total suspended particulate matter (TSP), CO, and lead (Pb) to the overall emissions in the Los Angeles South Coast Air Basin of California are given in table 1. In this particular urban air basin, mobile source emissions are predominantly automotive (since aircraft and ship emissions are rela- tively minor) and are major contributors to the overall emission inventory of NOx, ROG, CO, and Pb. Some of these emissions have a direct impact on the ecosystem, including human health. In addition, most of them can un- dergo chemical transformations in the at- mosphere (see, for example, Atkinson and Lloyd 1984; Atkinson 1986), sometimes leading to the production of more toxic products. The possible chemical transfor- mations and physical loss processes that occur in the atmosphere during transport of these primary automotive emissions from source to receptor are the main subjects of this chapter. The time scales of these atmo- spheric transformations and physical loss processes vary widely, with chemical life times ranging from '1 min for some highly reactive organic compounds to months or even years for other much more inert emissions (Atkinson 1986~. For exam- ple, the Los Angeles urban plume has been identified by ambient air monitoring mea- surements at Niwot Ridge, Colorado, and the transit time estimated at approximately four days (Roberts et al. 1984~. To understand effects on health and to assess risk, it is necessary to know the identities, the ambient concentrations, and the distributions between gaseous and con- densed phases of the chemical compounds impacting human receptors. Thus, it is necessary to determine the chemical and physical changes that primary automotive emissions undergo during their transport through the atmosphere, and the threats, if any, that the resulting products pose to human health. It must be borne in mind that automotive emissions cannot be considered in isolation. Synergistic chemical and physical interac- tions occur between automotive emissions and emissions arising from, for example, stationary sources and vegetation, giving rise to a further multitude of product spe- cies. Clearly, changes in emission rates or chemical characteristics of these nonauto- motive emissions can lead to changes in the photochemical reactivities of the overall atmospheric pollutant mixtures. The eluci- dation of the effect of automotive emissions on human health necessitates a complete knowledge of the emission inventories, the physical and chemical transformations, transport, and ambient atmospheric mea surements ot automotive, stationary source, and vegetative emissions, all com- bined within the framework of local, ur Table t. Average Emission Rates of NOX, ROG, SOx, TSP, CO, and Pb in the South Coast Air Basin of California During 1979 Emissions (tons/day) Source NOX ROG SO.r TSP CO Pb 91 7,060 9.1 522588 0.6 6137,650 9.7 Mobile Stationary Total 837853 406680 1724317533 73 201 274 SOURCE: Adapted with permission from South Coast Air Quality Management District 1982.
Roger Atkinson 101 ban, or regional photochemical computer models. This chapter assesses the atmospheric lifetimes of the various classes of automo- tive emissions, which, for compounds of low volatility, may vary markedly with their distribution between the gaseous and particulate phases. The state of knowledge about products formed by chemical reac- tions under atmospheric conditions, in- cluding indoor environments, is reviewed and discussed. In many cases, the products formed during the photodegradation of primary automotive emissions are not pres- ently known, and studies are needed to determine the general nature, and toxicity to humans, of these products. A list of research recommendations to obtain the necessary data base about these atmo- spheric transformations is presented. Physical and Chemical Transformations Under Atmospheric Conditions Two decades of laboratory, environmental chamber, and ambient atmospheric mea- surements have revealed the physical and chemical processes that transform and/or remove chemical compounds emitted into the atmosphere. These atmospheric emis- sions are partitioned between the gas and particulate phases, and the atmospheric loss processes for both phases must be evaluated separately. Physical Removal Processes The physical removal processes can be de- fined as accretion (or coagulation) of parti- cles, and dry and wet deposition of gases as well as particles. Removal of gases and particles at ground surfaces including snow-covered ground and other moist sur- faces is known as dry deposition, whereas removal of these species by raindrops is referred to as wet deposition. These pro- cesses are dynamic, and we do not yet have a quantitative understanding of them (see, for example, Eisenreich et al. 1981; Graedel et al. 1982; Slinn 1982; Colbeck and Harri son 1985; Dolske and Gatz 1985; Jonas and Heinemann 1985; Ligocki et al. 1985a,b; Sehmel et al. 1985; Terry Dana et al. 1985; van Noort and Wondergem 1985~. Dry Deposition of Gases and Particles. Gas-phase species and particles can be re- moved from the atmosphere by an overall process that involves downward transport from the atmospheric boundary layer to the ground surface. The complex atmospheric physical mechanisms that deliver gaseous and particulate species to the surface are generally combined with the chemical pro- cesses of mass transfer at the surface by use of a "deposition velocity" Vat. The dry deposition rate, F. is F = Vat [C] (1) where [C] is the concentration of the spe- cies at some reference height (generally defined as 1 m). The deposition velocity depends on the specific gaseous chemical and/or particle species, the surface to which the species is being deposited, and the reference height. It also depends on the atmospheric stability, being highest during unstable conditions (see, for example, Cadle et al. 1985; Colbeck and Harrison 1985~. The deposition velocity is often defined by three "resistance" terms all = (A + rb + rS) (2) where ra is the resistance between the ref- erence height and the laminar sublayer near the receiving surface; rb is the laminar sub- layer resistance; and rS is the surface resis- tance, specific to each pollutant and surface type. For certain species (for example, gas- eous nitric acid), the surface resistance rS is effectively zero, and transport to the surface becomes rate determining (Huebert and Robert 1985~. The deposition velocities of particles de- pend on the particle size, exhibiting a min- imum for particles of mean diameter of ~ O.1 ,um. It should also be noted that, for particles, a constant adsorption and desorp- tion of chemicals occurs, characterized by their Henry's law properties. Thus there is a dynamic equilibrium between the gaseous and adsorbed (or particulate) phases which,
102 in accordance with the Henry's law con- stants, depends on temperature, properties of the individual particles, vapor pressure, and liquid adsorption properties. Wet Deposition of Gases and Particles (Rainout). In addition to dry deposition, . . wet c .epos1tlon can remove gaseous com- pounds and particles from the atmosphere. This process occurs during precipitation. Slinn and coworkers (1978) showed that a falling raindrop attains equilibrium with a gaseous chemical over a distance of~ 10 m. As described by Eisenreich et al. (1981), the wet removal of gaseous chemicals arises from equilibrium partitioning, and a wash- out ratio, W. defining the scavenging eff~- ciency of a gas-phase species, is given by W = Crajs,/Cair = RT/H (3) where R is the gas constant, T is the temperature (°K), H is the Henry's law constant, and Grain and Cair are the concen- trations in rain and air, respectively. The deposition rate, F. is then given by F = wJcair (4) where ~ is the precipitation rate. Wet removal of gases is clearly most important for chemicals highly soluble in water, such as hydrogen peroxide, nitric acid, and phenols. Thus, following a pre- cipitation event, the atmospheric concen- trations of highly water soluble species may fall to near zero. For most gas-phase or- ganic chemicals, however, it is likely that wet removal is of minor importance. Atlas and Giam (1981) calculated atmospheric residence times ranging from ~ 60 days for phthalates and hexachlorohexanes up to ~ 6 years for the polychlorinated biphenyl mixture Aroclor 1242. Clearly, wet deposition is episodic. Thus, only average wet deposition veloci- ties can be ascribed, and these are strong functions of the climatological conditions at the particular geographic location in question. Chemical Removal Processes Many chemical processes contribute to the removal of compounds emitted into the Atmospheric Transformations of Automotive Emissions troposphere. For gas-phase chemicals, these removal processes involve · photolysis during daylight hours; · reaction with hydroxyl (OH) radicals during daylight hours; · reaction with ozone (O3) during daytime and nighttime; · reaction with hydroperoxyl (HO2) radi- cals during, typically, late daytime and early nighttime hours; · reaction with the gaseous nitrate (NO3) radical during nighttime hours; · reaction with dinitrogen pentoxide (N205) during nighttime hours; · reaction with NO2 during daytime and nighttime hours; and · reaction with gaseous nitric acid (HNO3) and other species such as nitrous (HNO2) and sulfuric (H2SO4) acids. Additionally, the following processes are likely to contribute to the degradation of chemical compounds present in the ad- sorbed phase: · photolysis; · reaction with O3; · reaction with N2O5 during nighttime hours; · reaction with NO2, typically present throughout a full 24-hr period; · reaction with H2O2; · reaction with HNO3, HNO2, and H2SO4. Synergism may be important in certain of these reactions involving adsorbed auto- motive emissions. For example, the pres- ence of HNO3 together with NO' may lead to enhanced nitration of adsorbed polycyclic aromatic hydrocarbons (PAHs) (see, for example, Pitts 1983~. In addition to photolysis and reactions of automotive emissions in the gaseous and adsorbed states with a variety of atmo- spherically important species, the chemical · . . . . . . reactions ot automotive emlsslons 1n rain, cloud, or fog water with other reactive components of these aqueous systems must be considered. This subject has recently received much attention because of an in- creasing emphasis on acid deposition (see, for example, Calvert 1984~. Reactions of chemicals in the aqueous phase with reac
Roger Atkinson tive intermediates such as H2O2 and vari- ous radical and ionic species have been dealt with in some detail in connection with these acidic deposition studies (Graedel and Weschler 1981; Chameides and Davis 1982; Graedel and Goldberg 1983; Jacob and Hoffmann 1983; Chameides 1986; Graedel et al. 1986~. Photolysis. Automobile emissions can be removed from the atmosphere by pho- tolysis. This process requires that a chem- ical compound absorb light in the actinic portion of the spectrum (that is, the wave- length region from ~ 290 to 1,000 nm) and, after absorption of a photon, undergo chemical change (Calvert and Pitts 1966~. For most compounds, breakage of a~chem- ical bond requires an energy in excess of ~ 40 kcal/mole (Benson 1976~. Therefore, photolytic wavelengths of ' 700 nm are necessary. One fundamental tenet is that absorption of a single photon (referred to hereafter as he cannot photodissociate more than one molecule (Calvert and Pitts 1966~. Formation of Ozone. Ozone is formed in the troposphere from the Photolysis of NO2 NO2 + hv~ NO + 0 (3P) (5) followed by reaction of the ground-state oxygen atom, CROP), with O2 M 0(3P) + O2 ' O3 (6) where M denotes a third body, air in this case. Tropospheric O3 is also transported downward from the stratosphere (Logan 1985~. In the clean troposphere, O3 mixing ratios are typically 30 + 10 parts per billion (ppb) at3ground level (~ 7 x 10~ mole- cules/cm ), and increase with altitude (Logan 1985~. The relative contributions to tropospheric O3 of photochemical forma- tion and downward transport from the stratosphere are discussed by Logan (1985~. Formation of Hydroxyl Radicals. The OH radical is the major reactive species in the troposphere (Logan et al. 1981), and is formed by Photolysis of 03, Photolysis of HNO2, and reaction of the HO2 radical 103 with nitric oxide (NO) (DeMore et al. 1985~. Photolysis of Ozone. Ozone photodisso- ciates at wavelengths of < 319 nm to yield, in part, electronically excited oxygen at- oms, O(~D), O3+hu ~O(~D)+O2 (7) which react with water vapor (eq. 8) or N2 and O2 (eq. 9) O(iD) + H2O ~ 2 OH (8) O('D) + N2, Of ~ 0(3P) + No, O2 (9) For a relative humidity of~ 50 percent at 298°K, ~ 0.2 OH radicals are formed for each O(~D) atom formed. Photolysis of Nitrous Acid. Nitrous acid, which is present during nighttime hours in urban atmospheres (Platt et al. 1980a; Harris et al. 1982; Pitts et al. 1984a), is rapidly photolyzed at wavelengths of < 400 nm (eq. 10) during daylight hours to yield OH radicals (DeMore et al. 1985), HNO2 + hv~ OH + NO (10) with a lifetime due to Photolysis of~ 1~15 min under noontime conditions. Reaction of Hydroperoxyl Radicals with Ni- tric Oxide. Hydroperoxyl radicals, formed from the photodissociation of aldehydes and other photochemical processes (see be- low, and Atkinson and Lloyd 1984), react with NO to yield the OH radical HO2 + NO ~ OH + NO2 (1l) In the troposphere, under conditions where NO concentrations are less than 5 x 108 molecules/cm3, HO2 radicals are expected to react with HO2 and alkyl peroxy (RO2) radicals in competition with reaction with NO. Hence a knowledge of tropospheric NO concentrations is important for assess- ing the conversion of HO2 to the more reactive OH radical (Logan et al. 1981; Crutzen 1982; Logan 1983~. Formation of Hydroperoxyl Radicals. Hydroperoxyl radicals are produced under tropospheric conditions from the pho- tolysis of aldehydes (Atkinson and Lloyd 1984), for example from formaldehyde (HCHO),
104 Atmospheric Transformations of Automotive Emissions HCHO + ho {:H + HCO lengths between 400 and 650 nm (Graham H2 + co (12) and Johnston 1978; Magnotta and Johnston 1980), followed by the rapid reactions of hydro gen atoms and HCO radicals with O2 M H + O2 ~ HO2 (13) HCO + O2 ~ HOT + CO (14) The higher aldehydes also photodissociate to ultimately yield HO2 radicals RCHO + ho ~ R + HCO ~ O2 HO2 + CO (15) (Here and below, R. R', R" . . ., represent unspecified groups and the dot () repre- sents an incomplete chemical bond or un- paired electron.) Additionally, HO2 radicals are formed from reactions of alkoxy and a-hydroxy radicals, which are reactive intermediates produced during the photooxidation pro- cesses of most organic compounds (At- kinson and Lloyd 1984). For example, R: CHO + Of ~ RCOR' + HOT (16) R'' RCHOH + Of ~ RCHO + HOT (17) Daytime tropospheric HO2 radical concen- trations are calculated to range from ~ 108 to 109 molecules/cm3 (Hov and Isaksen 1979; Stockwell and Calvert 1983a). Formation of Nitrate Radicals. The gas- eous NO3 radical has been shown to be an important constituent of nighttime atmo- spheres (see, for example, Winer et al. 1984; Atkinson et al. 1986b). Nitrate radi- cals are formed by the reactions NO2 + O3 ~ NO3 + O2 (18) NO2 + NO3 ~ N2Os ( 1 9) N2Os ~ NO2 + NO3 (20) NO2 + 0 (3P) NO3 + by tNo + O2 (21) with a lifetime of~ 5 sec at noontime (Magnotta and Johnston 1980), and react rapidly with NO, NO3 + NO ~ 2 NOT (22) NO3 radical concentrations are essentially negligible during daylight hours. After sunset, they can rise rapidly to levels of ups to ~ 400 parts per trillion (ppt) (~ 1 x 10 molecules/cm3) over continental areas (see, for example, Platt et al. 1984; Atkinson et al. 1986b). For example, at several semiarid and desert sites in southern California, Platt and coworkers (1984) consistently observed nighttime NO3 radical concentrations of~ 2 x 108 to 2 x 109 molecules/cm3. Formation of Dinitrogen Pentoxide. As shown above (eq. 19 and 20), N2O5 is in equilibrium with NO2 and the NO3 radi cal. Maximum nighttime N2O5 concentra tions of~ (2-3) x 10~ molecules/cm3 can be inferred from the equilibrium constant for these reactions, which is uncertain by a factor of + ~ 1.2-1.5 (Graham and John ston 1978; Tuazon et al. 1984b; Burrows et al. 1985a; Perner et al. 1985); and concen trations greater than ~ 2 x 10~° mol ecules/cm3 were calculated to be exceeded ~ 30 percent of the nights for which data are available (Atkinson et al. 1986b). Formation of Gas-Phase Acids. Chemi cals that are basic can react with gas-phase acids to form their salts. As presently un derstood, the major gas-phase acidic spe cies are HNO3 and HNO2. Nitric acid is formed in the gas phase from the reaction of OH radicals with NOB OH + NO2 ~ HNO3 (23) and can be formed, probably in the ad with N2O5 being in relatively rapid (< 1 sorbed phase, from the heterogeneous hy min at 298°K and 760 torr total pressure) drolysis of N2O5 equilibrium with NO2 and the NO3 radical (DeMore et al. 1985). Since NO3 radicals photolyze at wave heeerogeneous N2Os + H2O > 2 HNO3 (24)
Roger Atkinson 105 though this initially adsorbed HNO3 may be desorbed back into the gas phase (Tua- zon et al. 1983; Atkinson et al. 1986a). Nitrous acid is formed from the reaction of OH radicals with NO OH + NO ~ HNO2 (25) although its rapid photolysis during day- light hours (eq. 10) leads to a low ambient daytime concentration. However, HNO2 has been identified and measured in night- time Los Angeles atmospheres at up to 8 ppb (~ 2 x 10i ~ molecules/cm3) (Harris et al. 1982~. Indeed, nighttime HNO2 levels of~ 2 x 10~° to 2 x 10~ molecules/cm3 are probably typical of many, if not most, urban environments. In environmental chambers and indoor environments, HNO2 has been shown to be formed from the heterogeneous hydrolysis of NO2 (Sakamaki et al. 1983; Pitts et al. 1984c, 1985d). For automotive emissions associated with the particulate phase, reactions with NO2, HNO3, HNO2, N2O5, and O3 must be considered (see, for example, Pitts 1983~. Many, if not most, of these reactions probably proceed by reaction of the ad- sorbed automotive emissions with ad- sorbed, rather than gas-phase, reactive atmospheric species. Additionally, photol- ysis of adsorbed automotive emissions also occurs and may be highly important (Behymer and Hites 1985~. Recommendation 1. Study is required on the physical removal processes leading to wet and dry deposition of gases and particles. Investigations of the processes occurring on and in particulate and aerosol matter should focus on gas-to-particle con- version processes and the chemical pro- cesses that occur within aerosols (including fogs and clouds). Atmospheric Lifetimes, Fates, and Products of the Atmospheric Transformations of Automotive Omissions This section reviews and summarizes the present status of knowledge concerning the atmospheric loss processes and atmospheric lifetimes of automotive emissions and the products formed from them under atmo- spheric conditions. Reference is made whenever possible to existing reviews and/or evaluations, from which further details can be obtained. Atmospheric Lifetimes Data obtained during the past two decades have provided a comprehensive view of the chemical and physical removal processes that occur in the troposphere, and of the reaction rate constants for many of these processes. Table 2 lists the rate constants at room temperature (298°K, 77°F) for the known tropospheric chemical removal re- actions for selected automotive emissions. The corresponding calculated lifetimes in the lower troposphere of these chemicals due to reaction with each of the atmospher- ically important reactive species listed in table 2 are given in table 3. Although the individual rate constants are known to a reasonable degree of accu- racy (sometimes to within + 25 percent, and in most cases to within a factor of two), the calculated atmospheric lifetimes are much more uncertain because of the larger uncertainties about the ambient atmo- spheric concentrations of several of these key tropospheric species. For example,-the ambient atmospheric OH radical concen- trations at any given time and/or location are uncertain to a factor of at least five, and more likely ten (Hewitt and Harrison 1985~. The tropospheric diurnally and an- nually averaged OH radical concentrations are better known, to within possibly a factor of two (Crutzen 1982), being ~ 5 x 105 and ~ 6 x 105 molecules/cm3 in the northern and southern hemispheres, re- spectively. Similar arguments apply for the ambient nighttime tropospheric concentra- tions of the NO3 radical and of N2O5 (Atkinson et al. 1986b). In addition to these chemical loss pro- cesses of automotive emissions in the tro- posphere, physical loss processes must also be taken into account. Tables 4 and 5 give selected examples from the literature of dry deposition velocities and of washout ratios for several inorganic and organic species.
106 Atmospheric Transformations of Automotive Emissions Table 2. Room Temperature Rate Constants at Atmospheric Pressure of Air for the Gas-Phase Reactions of Selected Automotive Emissions with Atmospherically Important Intermediate Species Rate Constant [cm3/(molecule see)] O3 NO3 HO2 N2Os Emlsslon OH 1.1 x 10-11 6.6 x 10-12 6.6 x 10-'2 1.3 x 10-13 9 x 10-13 1.6 x 10-13 2.2 x 10-" 3 x 10-14 4.7 x 10-12 3.3 x 10-'l 1.7 x 10-12 1.2 x 10-12 2.5 x 10-'2 8.7 x 10-'2 2.2 x 10-13 2.5 x 10-13 8.5 x 10-12 2.6 x 10-11 3.1 x 10-" 6.4 x 10-" 7.8 x 10-13 6.1 x 10-'2 2 x 10-11 9.0 x 10-12 1.6 x 10 1.3 x 10-11 2.0 x 10-" 3.6 x 10 1.9 x jo-" 2.3 x 10-13 1.0 x 10-'2 3.0 x 10-'2 9 x 10-13 2.9 x 10-12 4.6 x 10-13 ~ 1.8 x 10-13 1.3 x 10-'2 6.2 x 10-12 2.5 x 10-1' 4.0 x 10-' 2.8 x 10-'1 2.2 x 10-" 5.2 x 10-" 7.7 x 10-" 3.2 x 10-1' 1.3 x 10-'° NO2a NO HNO2a HNO3b SO2 NH3C CH3NH2C HCN H2S CH3SH H2O2a Propane e-Butane e-Octane 1,2-Dichloroethane 1,2-Dibromoethane Ethene Propene 1-Butene trans-2-Butene Acetylene Propyne Butadiyne Formaldehydea Acetaldehydea Benzaldehydea Acrolein Crotonaldehyde Methyl vinyl ketone Acetonea 2-Butanone`' Dimethyl ether Methanol Ethanol Formic acid Methyl nitritea Benzene Toluene m-Xylene 1 ,2,4-Trimethyl- benzene Phenol Naphthalene 2-Methylnaphthalene 2,3-Dimethylnaph- thalene Phenanthrene Anthracene 3.2 x 10-17 1.8 x 10-'4 < 5 x 10-19 <2 x 10-22 2.1 x 10-2° < 2 x 10-2° < 6 x 10-24 < 1 X 10-23 1.8 x 10-'8 1.1 X 10-17 1.1 X 10-~7 2.0 x 10-16 8 x 10-21 1.4 x 10-2° 6 x 10-2° 2 x 10-24 6 x 10-2~ 2.8 x 10-~9 9.0 x 10-~9 4.8 x 10-'8 1.3 x 10-2° 7 x 10-23 1.5 x 10-22 6 x 10-22 1.3 x 10-21 2 x 10-19 4 x 10-~9 4 x 10- i9 1.2 x 10-'2 3.0 x 10-" < 7 x 10-2' <3 x 10-'4 1 X 10-12 < 2 x 10- 15 3.6 x 10-17 9.9 x 10-~7 1.1 X 10-'6 7.5 x 10-~5 9.7 x 10-~5 3.8 x 10-'3 s 2.3 x 10-~7 9.4 x 10-~7 5.8 x 10-'6 2.4 x 10-~5 2.0 x 10-~5 < 3 x 10- ~s < 6 x 10- 16 <9 x 10-'6 <2 x 10-~7 3.6 x 10-~7 1.3 x 10-'6 9.7 x 10-'6 3.8 x 10-'2 1.4 x 1()-~2 8.3 x 1()-~2 < 1 x 10-'8 < 4.2 x 10-23 < 4 x 10-'8 ~ 1 x 10-'4 1.4 x 10-~7 4.2 x 10-~7 5.7 x 10-~7 a Photolysis also occurs at a significant rate (see table 3). b Also reacts with NH3 to form NH4NO3. c Also reacts with gaseous HNO3 to form nitrate salts.
Roger Atkinson 107 Table 3. Calculated Atmospheric Lifetimesa for the Gas-Phase Reactions of the Selected Automotive Emissions with Atmospherically Important Intermediate Speciesb Atmospheric Lifetime Due to Reaction with EmissionOHO3NO3HO2 ho' NO22 days12 hr1 hr2 hr 2 min NO4 days1 min3 min20 min HNO24 days> 33 days ~ 10 min ANON180 days SO2e26 days> 200 yr> 4.5 x 104 yr> 600 yr NH3f140 days CH3NH2f12 hr2 yr HCN2 yr HIS5 days> 2 yr> 4 days CH3SH8 hr 1 hr H20214 days > 60 days 36 hr Propane19 days> 7,000 yr e-Butane9 days> 4,500 yr9 yr e-Octane3 days 3 yr 1,2-Dichloroethane100 days 1,2-Dibromoethane90 days Ethene3 days9 days3 yr Propene11 hr1.5 days15 days 1-Butene9 hr1.5 days12 days trans-2-Butene4 hr2 hr4 hr> 150 yr Acetylene30 days6 yr-14 yr Propyne4 days3 yr3.4 yr Butadiyne1 day~ 270 days Formaldehyde3 days> 2 x 104 yr210 days23 days 4 hr Acetaldehyde1 day> 7 yr50 days 60 hr Benzaldehyde2 days 60 days Acrolein1 day60 days Crotonaldehyde8 hr18 days Methyl vinyl ketone1 day3 days Acetone100 days 15 days 2-Butanone23 days Dimethyl ether7 days > 40 days Methanol26 days > 190 days Ethanol8 days > 130 days Formic acid50 days Methyl nitrite~ 120 days3 yr 8 min Benzene18 days600 yr> 16 yr Toluene4 days300 yr9 yr m-Xylene11 hr75 yr2 yr 1,2,~Trimethylbenzene7 hr35 yr120 days Phenol10 hr 20 min Naphthalenee1 day> 80 days 2-Methylnaphthalenee5 hr> 40 days 2,3-Dimethylnaphthalenee4 hr> 40 days Phenanthrene9 hr Anthracene2 hr a The time for the compound to decay to 37 percent of its original concentration. b For concentrations of OH, 12-hr average of 1 x 106 molecules/cm3 (Crutzen 1982); 03, 2thr average of 7 x 10~' molecules/cm3 (Singh et al. 1978); NO3, 12-hr average of 2 x 108 molecules/cm3 (Platt et al. 1984); HO2, 12- hr average of 108 molecules/cm3 (Hov and Isaksen 1979). c For solar zenith angle of 0°. Also reacts with NH3 to form NH4NOs. e Lifetimes due to gas-phase reaction with a 12-hr average concentration of N~Os of 2 x 10'° molecules/cm3 (Atkinson et al. 1986b) are SO2, > 7.5 x 104 yr; naphthalene, ~80 days; 2-methylnaphthalene, ~ 30 days; 2,3 dimethylnaphthalene, ~ 20 days. f Also reacts with gaseous HNO3 to form nitrate salts.
108 Atmospheric Transformations of Automotive Emissions Table 4. Dry Deposition Velocities for Several Inorganic and Organic Chemicals Depositing Species Mean Deposition Velocity (cm/see) o3 Particulate sulfur Particles 0.18-,um median diameter 0.25-m median diameter Calcium sulfate (CaSO4) particles 1-,um diameter 2-,um diameter 5-,um diameter 10-,um diameter SO2 HNO3 Tetrachloroethene Nitrobenzene Polychlorinated biphenyls (PCBs) 0.3_0.5a 0.08 - 0.91b 0.17a 0.16a 0.35a o.olc 0.o3c 0.44c 4.6c 2.la 2.5a ~ 10-4 ~ 10 ~ 0.1-0.5 a From Dolske and Gatz 1985, with grass as the surface. b From Colbeck and Harrison 1985, with grass as the surface. c From Jonas and Heinemann 1985, with grass as the surface. ~ From Sehmel et al. 1985. e From Eisenreich et al. 1981. Table 5. Washout Ratios for Selected Organic Chemicals For the particle-associated chemical species, the washout ratios W given in table 5 reflect the loss of the particles. Thus, as discussed by Eisenreich et al. (1981), the washout ratios W for aerosols are typically ~ 105 to 106, in comparison to values of~ 10° to 104 for gaseous chemicals. For particles, the atmospheric lifetimes due to dry deposition are of the order of several days for 0.1-1 ,um diameter parti- cles, and table 6 gives the average lifetimes of atmospheric particles as a function of their diameter. The dry deposition lifetimes of many organic compounds are also weeks or months (see, for example, Atlas and Giam 1981~. However, for certain chemi- cals that have relatively slow gas-phase chemical loss rates, such as HNO3 and SO2, dry deposition can be the major loss process under typical atmospheric condi- tions. Because of the potential importance of the dry deposition atmospheric removal process, measurements of the deposition velocities of gaseous and particulate species need to be carried out for a variety of terrains. This is a difficult and time-con- suming task, and emphasis must be given to extending the presently available exper- imental techniques and to developing and testing new experimental, and possibly the- oretical, approaches (see, for example, Dolske and Gatz 1985~. . , . Phase Chemical Washout Ratio (W) Gas Ethene oxide 4-6a Phenol (0.7-25) x 1o4b Nitrobenzene (2-4) x 103a Naphthalene 100-300C Phenanthrene 2, 000-4, 000C Pyrene 3 000-9 000C Benz ~a j anthracene 7,000-22,000C Hexachlorobenzene 1,500 Particle PCBs ~ (1-10) x 104 Particles 0.1-1.0-,um diameter ' 10S~ 10-,um diameter ~ 106~ Tricosane through hexacosane ~ 2 x 104e a From Terry Dana et al. 1985. b From Leuenberger et al. 1985. c From Ligocki et al. 1985a. From Eisenreich et al. 1981. e From Ligocki et al. 1985b.
Roger Atkinson 109 - J For nonpolar organic compounds, wet deposition appears to be of minor impor- tance as an atmospheric loss process. How- ever, for highly water-soluble gases such as HNO~ and H2O2, wet deposition can be important (Jacob and HofEmann 1983: Chang 1984), and in fog and cloud systems this process leads to removal of these and other compounds from the gas phase into the aqueous phase where reactions can oc- cur that lead ultimately to the formation of acids and other oxygenated products. Wet deposition rates are somewhat better understood, with the experimental results for gas-phase chemicals agreeing to within a factor of~ 10 or better with theoretical expectations (see, for example, Ligocki et al. 1985a,b; Terry Dana et al. 1985~. Fur- ther research is needed to provide a wider data base concerning the washout ratios of chemical compounds present in the has phase and of aerosols and particles. These data will then allow the importance of this wet deposition process to be better evalu- ated, both as a loss process for primary automotive emissions as well as for the formation and deposition of acid species resulting from aqueous-phase reactions. The major atmospheric loss process for most of the automotive emissions present in the gas phase is by daytime reaction with the OH radical. However, for certain classes of automotive emissions, pho- tolysis, reaction with NO3 radicals during nighttime hours, and reaction with O3 can be important removal routes. Furthermore, reactions that are relatively minor removal processes may need to be considered if they generate products with potential health risks to humans. For example, the reactions of gas-phase N2O5 with PAHs appear to be of minor significance as a PAH loss proc- ess, but they form toxic nitropolycyclic aromatic hydrocarbons (nitro-PAHs) (Pitts et al. 1985b; Sweetman et al. 1986~. Clearly, a knowledge of the atmospheric loss processes and lifetimes for automotive . . . . . , . ~ . emissions Is important, since t nese lifetimes determine the geographic extent of the influ- ence of the parent automotive emission. Thus, a short lifetime leads to local exposure, whereas a long lifetime leads to regional or global exposure at lower concentrations. Table 6. Average Atmospheric Lifetimes for Particles Due to Dry Deposition Diameter (,um) Lifetime (days) 0.002 0.02 0.2 2 20 200 0.01 10 10 1 0.01 SOURCE: Adapted with permission from Graedel and Weschler 1981. The atmospheric lifetimes of automotive emissions present in the particulate phase are less well known. Dry and wet deposi- tion constitute the physical loss processes, and photolysis and/or reaction with gas- phase and coadsorbed reactive intermedi- ates constitute the possible chemical loss processes. These chemical processes are substrate dependent, with photolysis, reac- tion with O3, reaction with NO2 and/or HNO3, and reaction with N205 occurring (see, for example, Nielsen et al. 1983; Pitts 1983~. However, due to the dependence of these loss processes on the nature of the substrate, it is presently impossible to cite any meaningful atmospheric lifetimes for adsorbed automotive emissions, except to remark that the reaction of PAHs with O3 may lead to lifetimes on the order of hours, photolysis is probably slow, and reaction with N2O5 though slow as a loss proc- ess leads to the formation of direct-acting mutagenic and possibly carcinogenic nitro- PAH products. _ ~1 Atmospheric Transformations Oxides of Nitrogen. Oxides of nitogen emitted into the atmosphere as a result of automotive use comprise NO, NO2, N2O, HNO2, and possibly HNO3. N2O has been shown to be chemically inert in the troposphere, being transported into the stratosphere where it photodissociates at wavelengths of ~ 220 nm (Liu et al. 1977), NATO + he ~ No + OILED) (26) and reacts with electronically excited oxy- gen atoms, O(iD), leading to formation of NO.
110 Atmospheric Transformations of Automotive Emissions O('D) + N2O ~ 2 NO (27) Hence, tropospheric N2O emissions are a major source of stratospheric NO, and the effect of increasing tropospheric N2O emis- sions on stratospheric O3 has been well documented (Liu et al. 1977~. In the troposphere, the other NOX are in- terrelated by means of their reactions (At- kinson and Lloyd 1984; DeMore et al. 1985~. At high mixing ratios ~-1 part per million (ppm)], NO is oxidized by O2 to NO2. 2 NO + O2 ~ 2 NO2 (28) Additionally, NO reacts rapidly with O3 to yield NO2 (29) Nitrogen dioxide photolyzes rapidly at wavelengths of < 430 nm (with a lifetime of~ 2 min at solar noon, 40°N latitude, sum- mertime) to yield NO and an oxygen atom, NO + O3 ~ NO2 + O2 NO2 + by ~ NO + 0 (3P) (30) with the 0~3P) atom reacting with O2 to yield O3 (eq. 6~. With this series of reac- tions, NO, NO2, and O3 are in a photosta- tionary state, NO + O3 ~ NO2 + O2 , ' Lit 0(3P) with (31) [o ] k30 [NO2] k29 [NO] where k29 and k30 are the rate constants for the reactions given in equations 29 and 30, respectively, and brackets ~ l signify con- centrations. As shown below, this pho- tostationary state is strongly affected by NO-to-NO2 conversions caused by reac- tions involving organic compounds. Further reactions of NO and NO2 under atmospheric conditions involve the forma- tion of NO3 radicals and N205 (eq. 1~20~. Removal of N2O5 by dry and wet deposi- tion (eq. 24) may be an important night- time NOX sink in the lower troposphere (Heikes and Thompson 1983; Atkinson et al. 1986b). However, measurements of the sticking coeff~cient of N2O5 to particles and of its dry deposition velocity are necessary before the importance of this nighttime NOX re- moval process can be quantified. Thus, nighttime reactions involving NO3 radicals and N2O5 need to be investigated, together with the health impacts of N2O5. Dinitro- gen pentoxide is known from laboratory experiments to be in equilibrium with NO3 radicals and NO2, but it has not been observed in the ambient troposphere, al- though it is calculated to be present at mixing ratios of up to ~ 1~15 ppb (At- kinson et al. 1986b). Furthermore, it is well recognized that NO3 and/or N2O5 are re- moved from the troposphere by presently unknown processes (Noxon et al. 1980; Platt et al. 1980b; Atkinson et al. 1986b). These NO3 and/or N2O5 removal pro- cesses may be due to loss of NO3 radicals or to loss of N205 (the latter by dry or wet deposition), and are important as nighttime loss processes for NOX as well as for acid deposition. If N2O5 is hydrolyzed in the gas phase, presently viewed as unlikely (Atkinson et al. 1986a), significant gas- phase HNO3 concentrations could result in the presunrise hours. Conversely, hetero- geneous processes may be, at least in part, an explanation of the highly acidic fogs and rains observed in and around Los Angeles (Waldman et al. 1982~. Thus these hetero- geneous loss processes of N2O5 and/or NO3 require investigation, since they directly in- fluence the gas-phase concentrations of NO3 radicals and N2O5. During daylight hours, NO3 radicals photolyze rapidly (eq. 21) with a photolysis lifetime at solar noon of~ 5 sec (Magnotta and Johnston 1980~. In addition, N(93 radicals react with NO (e4. 22) suff~ciently rapidly that NO and NO3 cannot coexist at concentrations of a few ppt or higher. Nitric oxide and NO2 also react with HO2 radicals (eq. 11, 32) _ NO + HO2~ NO2 + OH (11) NO2 + HO2 ~ HO2 NO2 . . . . . . . .. . . (32) w~th the latter react~on be~ng revers~b~e aue to the rapid (~ 10 sec lifetime at 298°K and
Roger Atkinson 111 760 torr total pressure) back-decomposi- tion of HO2NO2 (DeMore et al. 1985~. Additionally, NO and NO2 react with OH radicals (eq. 25, 23~. The formation of HNO2 is balanced during daytime hours (when OH radicals are present at apprecia- ble concentrations) by its photolysis (eq. 10~. In addition to these homogeneous reac- tions of NOX, heterogeneous reactions may also be atmospherically important. Thus, the formation of HNO2 from the hetero- geneous hydrolysis of NO2, 2 NO2 + HERO ~ HNO2 + (HNO3) (33) has been observed to occur in environmen- tal chambers and in an indoor environment (Sakamaki et al. 1983; Pitts et al. 1984c, 1985d). This reaction may be a formation pathway for the HNO2 observed during nighttime hours in a number of urban airsheds, at levels ~5 percent of the NO2 present (Perner 1980; Platt et al. 1980a; Harris et al. 1982; Pitts et al. 1984a), and in automobile exhaust (Pitts et al. 1984b). In environmental chambers, the formation rate of HNO2 is first order with respect to the NO2 concentration, and the potential coproduct HNO3 has not been observed in the gas phase (Sakamaki et al. 1983; Pitts et al. 1984c). The HNO2 formation rate in large-volume enclosures at ~ 50 percent relative humidity is ~ 0.1 ppb/min at an NO2 concentration of 1 ppm (Sakamaki et al. 1983; Pitts et al. 1984c, 1985d). Surpris- ingly, if the observed nighttime HNO2 concentrations and formation rates in the Los Angeles air basin are ascribed to this heterogeneous reaction, the calculated HNO2 formation rates are similar to those observed in environmental chambers, de- spite the greatly different surface-to- volume ratios. Thus, nighttime HNO2 can arise from direct automotive emissions, heteroge- neous hydrolysis of NO2, and/or other processes. However, the relative contribu- tions of direct emissions and heterogeneous formation of HNO2 to observed nighttime HNO2 levels need to be quantified. Fur- thermore, if HNO2 formation from the heterogeneous hydrolysis of NO2 is re- sponsible for a major portion of the night- time HNO2 levels observed, the impor tance of this process during daylight hours needs to be investigated, especially since the photolysis of HNO2 directly yields the key chain-carrying OH radical. In particu- lar, research is needed to investigate whether this heterogeneous hydrolysis of NO2 to HNO2 is accelerated by light. If so, this process could explain the "chamber-dependent" radical sources ob- served in environmental chambers (Carter et al. 1982), and would have a profound effect on the validity of chemical models developed for urban and regional airshed modeling. Present chemical mechanisms take into account the presence of cham- ber-dependent radical sources during their testing against environmental chamber ex- periments, but these radical sources are removed from the chemical mechanism for ambient atmospheric simulations. If HNO2 (and hence, by photolysis, OH radicals) is produced at significant rates from NO2, then the chemical computer models are being incorrectly applied under atmo- spheric conditions. The occurrence of this reaction would lead to significant under- prediction of the reactivity of many present ambient atmospheric pollutant mixtures by computer modeling studies. The observation of HNO2 formation from NO2 by heterogeneous hydrolysis is also of importance from an indoor air pol- lution viewpoint. For ppm mixing ratios of NO2, HNO2 formation rates of ~ 0.1 ppb/min may be readily attained (Pitts et al. 1984c, 1985d), leading to indoor HNO2 lev- els of up to ~ 10 ppb, especially in the presence of mercury strip-lighting, which photolyzes HNO2 slowly, if at all (Pitts et al. 1985d). Since HNO2 can attain ppb mixing ratios in ambient atmospheres as well as indoor environments, it is imperative that its health effects be investigated. In summary, these various nitrogen ox- ide species are readily interconverted in the lower atmosphere, but the major tropo- spheric loss process of NOX occurs by the formation of gas-phase HNO3 from the daytime reaction of OH radicals with NO2 (the gas-phase HNO3 being removed from the troposphere mainly by dry and wet deposition) and by wet and/or dry deposi- tion of N2O5 during nighttime hours.
112 Recommendation 2. Further investi- gations of the transformations of NOX un- der atmospheric conditions are needed. The kinetics and mechanism of gas-phase HNO2 formation in indoor environments as well as ambient nighttime atmospheres should be investigated. Further research is also required on the nighttime reactions of the NO3 radical, especially with respect to the products formed from its reactions with organic compounds, and on the heteroge- neous and/or homogeneous reactions of N2O5. Reduced Nitrogen Compounds. Reduced nitrogen compound emissions include am- monia (NH3), hydrogen cyanide (HCN), and possibly their higher homologues such as the aliphatic and aromatic amines RNH2, RR'NH and RR'R"N, and the nitrites RCN, where R. R', R" are alkyl or aryl groups. The atmospheric reactions of these auto- motive emissions are not totally under- stood. These reactions are best considered separately as the amine and nitrite classes. Amines. Reactions of amines with O3 have been shown to be slow, and are of minor significance under atmospheric con- ditions (Atkinson and Carter 1984~. The major atmospheric removal processes in- volve gas-phase reactions with the OH radical OH + NH3 ~ H2O + NH2 (34' OH + CH3NH2 ~ H2O + CH3NH `35' Atmospheric Transformations of Automotive Emissions NRR'R" + HNO3 ~ [NRR'R''Hl4-NO3- (39) to form the corresponding nitrate salts. As shown in tables 2 and 3, the OH radical reactions are rapid, leading to lifetimes on the order of hours during daylight. In polluted atmospheres, however, reactions with gaseous HNO3 could well be impor- tant, if not dominant, especially since this process continues during nighttime hours. Indeed, under certain conditions the reac- tion of ammonia with gaseous nitric acid is an important atmospheric loss process for NH3 and HNO3 (see, for example, ~acob et al. 1986~. The resulting nitrate salts un- dergo dry and/or wet deposition. The radical species (NRR') resulting from these OH radical reactions can react with 02, NO, NO2, or O3. NRR' + O2 ~ products ¢40y NRR' + NO ~ RR'NNO (nitrosamine) <41' RR'NNO2 (nitramine) NRR' + NO2- ~ RN CHR" + HNO2 (42) NRR' + O3 ~ products (43) To date, few data are available concern ing the absolute or relative rate constants for these reactions. For the NH2 radical, the reaction rate constants are (Lesclaux 1984) NH2 + O2 ~ products k40 < 3 x 10-18 cm3/(molecule~sec), OH + (CH3)2NH ~ H2O + NH2 + NO ~ products (including N2 + H2O) t(CH3)2N 1 lCH2NHCH3 | (36) OH + (CH3)3N ~ H2O + CH2N(CH3)2 (37) NH NH~ + NO2 ~ products NH2 OH + :} and with gaseous HNO3, k4, ~ 2 X 10- l l cm3/(molecule~sec) - - - - f~ +H2O NH2 OH ~ ~ 1 k42~ 2 X lo-ll cm3/(molecule~sec). The only other data available for the sub- sequent reactions of NR2 radicals concern the dimethylamino ((CH3~2N) radical, for which Lindley and coworkers (1979) determined the rate constant ratios k4O:k4l:k42a:k42b = 3.9 x 10 7:0.26:1.0:0.22. `38' For these aliphatic amines, the formation of . carclnogenlc nltrosamlnes, w :llC. :1 p. :10 tolyze rapidly (Tuazon et al. 1984a)
Roger Atkinson 113 RR'NNO + he ~ RR'N + NO (44) and nitramines, which are longer-lived and react mainly with OH radicals (Tuazon et al. 1984a), can be important. Nitrites. The available experimental data suggest that these reduced nitrogen compounds react mainly with OH radicals under tropospheric conditions. HCN reacts only slowly with OH radicals, with a room temperature rate constant at atmospheric pressure of 3 x 10-~4 cm3/(molecule see) (Fritz et al. 1984), by an addition process: gas-phase reaction with the OH radical is dominant, M OH + SO2 ~ HSO3 (48) followed by the formation of the chain- carrying HO2 radical and H2SO4 (Stock- well and Calvert 1983b). HSO3 + O2 ~ HO2 + SO3 ~ H2O H2SO4 (49) OH + HCN ~ [HCN.OH] ~ products (45) Reactions of SO2 with the Cringed b~rad~- cals (RR'COO) formed from alkene-O3 reactions The products of this reaction, as well as the subsequent reactions that occur under at- mospheric conditions, are not known (Ci- cerone and Zellner 1983~. For the organic nitrites, the available data show that the OH radical reactions are more rapid than that for HCN, and that they probably occur by hydrogen-atom abstraction from the alkyl groups, OH + CH3CN ~ H2O + CH2CN (46) which ultimately leads to the formation of CN radicals (Atkinson 1986~. . ~ ~ . ~ ~ ~ ~ r Nitrites. Methyl nitrite is the most im- portant compound of this class of organics, and photolysis is the only significant atmo- spheric loss process, CH3ONO + he ~ CH3O + NO i02 HCHO + HO2 resulting in a lifetime of~ 1~15 min at solar noon (Taylor et al. 1980~. Sulfur Oxides and Oxyacids. Sulfuric acid (H2SO4) exists almost entirely in the particulate phase under atmospheric condi- tions, and generally is neutralized by reac- tion with metal cations or ammonia to form salts such as Na2SO4, CaSO4, (NH4~2SO4, and NH4HSO4. Wet and dry deposition of particulate matter lead to its removal from the troposphere. Sulfur dioxide is removed from the tro- posphere by gas- and aqueous-phase chem- ical reactions and by dry and wet deposi- tion. With respect to chemical removal, H2O RR'COO + SO2 ~ RR'CO + H2SO4 ¢50) are of minor importance under most tro- pospheric conditions (Calvert and Stock- well 1983~. Since SO2 is moderately soluble in the aqueous phase, it undergoes scavenging by fog, cloud water, and raindrops, leading to its inclusion in aqueous systems. Under these conditions, SO2 is readily oxidized to sulfate by a variety of reactions, as dis- cussed in detail by Graedel and Weschler (1981), Chameides and Davis (1982), Grae- del and Goldberg (1983), Jacob and Hoff- mann (1983), Chameides (1986), and Grae- del et al. (1986~. Reduced Sulfur Compounds. Laboratory data show that the reduced sulfur-con- taining species (RSH) react with OH radi- cals during daytime hours and with NO3 radicals during nighttime hours. For H2S, the NO3 radical reaction is slow (Wal- lington et al. 1986~; therefore the dominant tropospheric removal process involves re- action with the OH radical (DeMore et al. 1985~. OH + H2S ~ H2O + SH (51) Although the SH radical apparently un- dergoes a series of reactions resulting in the formation of SO2 (Thiemans and Schwartz 1978), the details of the reaction sequence are still not totally understood. The data of
114 Friedl et al. (1985) suggest that, under atmospheric conditions, SH radicals react with NO2 SH + NO2 ~ HSO + NO (52) and/or with O3 SH + O3 ~ HSO + O2 (53) with, typically, a lifetime of ' 1 sec. The resulting HSO radicals react with O2 to yield SO2, by the intermediate formation of SO radicals. Reaction of HSO radicals with O3 can apparently re-form SH radicals (Friedl et al. 1985~. For the aliphatic thiols, both NO3 radical as well as OH radical reactions are impor- tant. For example, for CH3SH ~ OH OH + CH3SH ~ ~ CH3S + H2O (54) CH3SH ~ HCHO, SO2, and CH3SO3H ~ ONO2 NO3 + CH3SH ~l ~ ( ,H3S + HNO3 CH3SH (55) The reaction mechanisms and products formed under atmospheric conditions are, however, incompletely understood (At- kinson 1986; Mac Lead et al. 1986~. Oxides of Carbon. CO2 is chemically sta- ble under tropospheric conditions, and its removal from the troposphere involves transport to the stratosphere and absorp . . tlon into t be oceans. CO is relatively long lived (~ 3 months), with its tropospheric lifetime determined by reaction with the OH radical (DeMore et al. 1985) OH + CO ~ HOCO ~ CO2 + H CHOW + CO2 O2 (56) which generates the chain-carrying HO2 radical. Hydrogen Peroxide (H202) and Organic Peroxides. These compounds are readily absorbed into aqueous phases, including Atmospheric Transformations of Automotive Emissions cloud and fog water Jacob and Hoffmann 1983~. In the gas phase, photolysis, ROOH + ha ~ OH + RO where R represents either hydrogen or an alkyl radical, and reaction with OH radicals OH + H2O2 ~ H2O + HO2 (58) ~ H2O + CH3OO OH + CH3OOH tH2O + CH2OOH HCHO + OH (59) are the important chemical loss processes (DeMore et al. 1985; Atkinson 1986~. The nighttime reaction of H2O2 with the NO3 radical has been shown to be slow (Bur- rows et al. 1985b). Alkanes. Alkanes, together with the aro- matic hydrocarbons, comprise a major cat- egory of automotive emissions. Gasoline contains alkanes ranging in size from 2 C4 through C~2-C~ 5, and diesel fuels from ~ Cal through ' C20 (Carter et al. 1981~. The atmospheric chemistry of the alkanes has been discussed by Carter and Atkinson (1985~. Under tropospheric conditions, the alkanes react with OH radicals during day- light hours and with the NO3 radical dur- ing nighttime hours, with the NO3 reac- tion being of minor (< 10 percent) importance as an atmospheric loss process. Both reactions proceed by hydrogen- atom abstraction from C H bonds TOH |+RH )TH2O |+R- (60) ~ NO31 (HNO3) For the OH radical reaction, the contribu- tions of hydrogen-atom abstraction at the various nonequivalent C-H bonds, and hence the distribution of the alkyl (R ~ radicals formed, can be calculated using the estimation method of Atkinson (1986~. Under atmospheric conditions, alkyl radi- cals react rapidly and exclusively with O2 to yield alkyl peroxy (ROT) radicals. M R + O2 ~ RO2 (61) Under tropospheric conditions, these RO2 radicals react with NO by two pathways,
Roger Atkinson 115 , ~ RO- + NO2 RO2-+NO~ M ~ ~ RONO2 (62) with HO2 radicals, RO2- + HOT ~ RO2H + O2 (63) and with other RO2 radicals (including the same RO2 species). RO2- + R'O2- ~ products (typically alkoxy radicals, alcohols, and carbonyls) (64) This competition between the reactions with NO and HO2 and other RO2 radicals typically occurs for NO concentrations of the order of (2~) X 108 molecules/cm3. Under polluted urban atmospheric condi- tions, and possibly for much of the lower troposphere in the eastern United States, reaction with NO is the dominant reaction pathway for RO2- radicals. In addition, the reaction with NO (eq. 62) forms the corresponding alkoxy (RO ~ radical and NO2 or the corresponding alkyl nitrate. The yield of the alkyl nitrate in- creases with increasing pressure and with decreasing temperature. For secondary RO2 radicals at 298°K and 760 torr total pressure, the yields of the alkyl nitrates increase monotonically from ~ 0.04 for a C3 alkane up to ~ 0.33 for a C8 alkane (Carter and Atkinson 1985~. The resulting alkoxy radicals react under tropospheric conditions by unimolecular decomposition, unimolecular isomeriza- tion or with O2, as shown, for example, for the 2-pentoxy radical: o , _ decomposition~ CH3CHO + CH3CH2CH2 CH`CHCH2CH2CH3 \ isomerization CH3CHOHCH2CH2CH2 O2 HO2 + cH3cOcH2cH2CH3 (65) The isomerization reactions are calculated to occur by 1,5-H-atom shifts, as shown. These isomerization reactions thus require a tcarbon chain, and where this is avail- able, isomerization is calculated to domi- nate over decomposition and/or reaction with O2. For a complete discussion of these alkoxy radical reactions, the review of Carter and Atkinson (1985) should be con- sulted. The reactions that occur after alkoxy radical isomerization have not been dem- onstrated experimentally. However, it is expected that, for example, the reaction sequence for the 2-pentoxy radical will proceed by o CH3CHCH2CH2CH3 CH3CHOHCH2CH2CH2 NC) O2 ~ NO2 CH3CHOHCH2CH2CH2O 1 . . . . secona lsomenzatlon CH3COHCH2CH2CH2OH i02 HO2 + cH3cOCH2cH2cH2OH (66) . . . . . . . . Wlt ~ t 11S lsomerlzatlon process glvlng rise to the bifunctional product pentan-4-one- 1-ol. Thus, for the alkanes the observed first- generation products are aldehydes, ke- tones, and alkyl nitrates, all of which react further under atmospheric conditions. The health effects of these first-generation prod- ucts need to be assessed. For the alkanes containing 2 C4 chains, the formation of ~bifunctional compounds, of the general formula RCOCH2CH2CHOHR', is also predicted. At the present time, however, no definitive evidence for the formation of these compounds has been reported. Whether this class of organic compounds is formed, and if so, what their health effects are, needs to be investigated. Our understanding of alkane chemistry also suggests the possibility that ~hy- droxynitratoalkanes (RCHOHCH2CH2- CH(ONO2)R') could be formed. Investi- gations to determine whether or not these compounds are formed and, if so, their health effects, are also necessary. These studies apply mainly to the longer chain alkanes ~-C5) that are the major constitu- ents of gasoline and, especially, diesel fuel.
116 For diesel fuel in which alkanes of ' C20 are present, many of the products are ex- pected to be partitioned almost entirely into the particulate phase. Thus, the atmo- spheric concentrations and subsequent at- mospheric transformations and health ef- fects of these products should be studied. · Recommendation 3. The products arising from the OH radical-initiated reac- tions of alkanes-the major component of automobile emissions require study. These products are likely to be distributed between the gas and particulate phases, and data are especially needed for the alkanes with eight or more carbon atoms. Haloalkanes. The two major haloalkanes emitted from automotive use are 1,2-di- chloroethane and 1,2-dibromoethane. These two haloalkanes react with the OH radical under atmospheric conditions with a room temperature rate constant of ~ 2.5 x 10- i3 cm3/(molecule see) (Atkinson 1986), corre- sponding to a lower tropospheric lifetime of~ 100 days (tables 2 and 3~. The products of these reactions have not been experimentally determined, but a likely reaction sequence is, OH + CH2XCH2X ~ H2O + CHXCH2X Atmospheric Transformations of Automotive Emissions with HO2 and/or other RO2 radicals may occur. The products arising from these radi- cal-radical reactions should be investigated. Alkenes. The major alkenes present in gasoline and diesel fuels as well as in auto- motive exhaust are the smaller ones such as ethene (CH2-CH2), propene (CH3CH CH2), 1-butene (CH3CH2CH CH2) and cis- and trans-2-butene (CH3CH CHCH3~. Laboratory research has shown that the alkenes are removed from the troposphere by reaction with OH rad- icals, NO3 radicals, and O3 (Atkinson and Lloyd 1984~. These reaction pathways are discussed briefly below. H:ydroxyl Radical Reaction. These reac- tions are rapid, with the corresponding lifetimes for the smaller alkenes being given in table 3. All of these reactions proceed by OH radical addition to the >C-C< dou- ble bond (Atkinson 1986), R R" OH + C=C / \ R' R"' R OH R" R OH 2 \1 ./ \. 1/ C ~and C~ / \ / \ R' R"' R' R'', (68) followed by rapid addition of O2 to yield the corresponding hydroxyperoxy radi oocHxcH2x cars, for example, I N(~ OH \ I . / / -C\ + O2 O2 1-' HO2 + CH 2 XCXO ~ OCHXCH2X + NO2 / R' R"' H2X + HCXO 1 ~ O2 CH2XCHO + X HO2 + HCXO (67) where X represents bromine (Br) or chlo- rine (Cl) and the first-generation products are underlined. Further work is needed to elucidate the reaction routes of the 1,2-diha- loethanes under atmospheric conditions. Since their tropospheric lifetimes are long (~ 100 days), their degradation will take place under low NO mixing ratio conditions where reaction of CH2XCHXOO radicals M Reaction with NO then yields the ,B hy- droxyalkoxy radical or the ,~hydroxynitrate. R OH OO R" \~t ~ / + NO / \ R' R"' M
Roger Atkinson R OH O n \1 1/ C C R ' R"' R OH ONO2 R M \1 1 / R' R"' (70) The ,~hydroxynitrate formation pathway accounts for ~ 1-1.5 percent of the overall NO reaction at ~ 298°K and atmospheric pressure for propene (Shepson et al. 1985a). Thus, for the alkenes emitted from auto- motive use, formation of ,~hydroxyni- trates appears to be minor, accounting for only a few percent of the overall reaction pathways. The ,~hydroxyalkoxy radicals react with 02, unimolecularly decompose, or unimo- lecularly isomerize (Atkinson and Lloyd 1984~. This is shown, for example, for the OCH2CHOHCH2CH3 radical formed from internal OH radical addition to 1- butene. on HO2 + CH3CH2CHOHCHO _ 1 CH3CH2CHCH2O / \, isomerization l CH2CH2CHOHCH2OH . . . aecomposltlon CH3CH2CHOH + HCHO IO2 CH3CH2CHO + HO2 (71) The available data show that at room tem- perature, decomposition of the ,l3 hydrox- yalkoxy radicals dominates over their isomerization (Atkinson et al. 1985~. Fur- thermore, only for the ,(3 hydroxyalkoxy radical formed from ethene does the reac- tion with O2 (eq. 72) compete with decom- position (eq. 73), HOCH2CH2O + O2 ~ HOCH2CHO + HO2 (72) . . HOCH2CH2O ~ HOCH2 + HCHO 117 with reaction 72 accounting for ~ 22 per- cent of the overall reaction for ethene at 298°K and 760 torr total air pressure (Niki et al. 1981a). Thus, for the alkenes with more than two carbons, these OH radical reactions lead, by ultimate cleavage of the >C C< double bonds, to the formation of aldehydes and/or ketones. Nitrate Radical Reaction. Only for those alkenes more reactive than the 2-butenes does the NO3 radical reaction become im- portant under atmospheric conditions (At- kinson 1986~. As presently understood, the initial reaction involves NO3 radical addi- tion to the >C-C< double bond, R R" \ ~ NO3 + /C=C\ R' R"' R ONO2 jR ~C-C + other isomer R' R"' (74) followed by reaction with O2 R ONO2 R" R ONO2 OO R" \C-C/ + O2 ~ \ I I / / \ / 1 R' R"' R' | R"' NO or RO2 radicals R~1 NO2 I R" C C / \ R' R"' R. ~ , _ . RCOR' + NO2 ~ ~CONO2 + R"COR"' R' (75) The limited data available suggest that de composition of the ,~nitratoalkoxy radicals predominates, with minor amounts of di nitrates being formed by reaction of the ,l3 nitratoalkoxy radicals with NO2 (Band ow et al. 1980; Shepson et al. 1985a). However, because of the reported high toxicity of these dinitrates, further work is necessary to better define their yields from `1 O2 a variety of alkenes as a function of tem HCHO + HO2 (73) perature and pressure. Furthermore, the
118 reaction sequences that occur in the absence of NO, typically during nighttime hours when these NO3 radical reactions are im- portant, are not known (see Recommenda- tion 2~. Reaction with Ozone. These reactions compete with the daytime OH radical re- actions and the nighttime NO3 radical re- actions as a tropospheric loss process for the alkenes (table 3~. These reactions pro- ceed by initial O3 addition to the >C Cat double bond, followed by rapid decompo- sition of the resulting energy-rich (denoted as ~ ]~) "molozonide," R .;El." y +O3' R' ,,, /C~\ , a,/ \~b L ~[ ~ , ~, R\ R" 'C=0 R' + \C=0 R"' (76) with the relative importance of pathways a and b assumed to be equal (see Atkinson and Carter 1984~. The major uncertainty concerns the fate under atmospheric conditions of the ini- tially energy-rich biradicals [RR'COO]t, which can be collisionally stabilized or un- dergo unimolecular decomposition. M RR'COO [RR C00] tdeComposirion (77) Recent data of Hatakeyama and coworkers (1984) show that, at atmospheric pressure, the fraction of initially energy-rich Criegee biradicals which is stabilized decreases from ~ 0.39 for ethene (in agreement with the earlier data of Su et al. 1980, Kan et al. 1981, and Niki et al. 1981b) to ~ 0.18 for trans-2-butene, and to ~ 0.04 for cyclohex Atmospheric Transformations of Automotive Emissions ene. The decomposition products, how- ever, are not well known, and further work . . . concerning t~ aese reactions 1S necessary. The thermalized biradicals have been shown or postulated to react with a number of species, including H2O (eq. 78), NO (eq. 79), NO2 (eq. 80), SO2 (eq. 81), CO (eq. 82), and aldehydes (eq. 83) (Calvert et al. 1978; Hatakeyama et al. 1981; Martinez and Herron 1981; Herron et al. 1982; At- kinson and Lloyd 1984~. . . RCHOO + H2O ~ RCOOH + H2O (78) RCHOO + NO ~ RCHO + NO2 (79) . . RCHOO + NO2 ~ RCHO + NO3 (80) RCHOO + SO2 ~ RCHO + H2SO4 (81) . . RCHOO + CO ~ products (82) R 0 ~p.' RCHOO + R'CHO ~ C C H \O H (83) In addition, biradicals such as (CH3)2COO may undergo unimolecular isomerization (Carter et al. 1986). CH3 CH3 / OO ~ ~COOH CH3 CH2 (84) Again, further research is needed to eluci- date the mechanisms and products of these reactions. Oxygen-Containing Organics. The non . . . . aromatic, oxygen-contalnlng organic com- pounds that are either directly emitted as a result of automotive use or are formed as atmospheric transformation products in- clude aldehydes, ketones, c~,,l3 unsaturated carbonyls, carboxylic acids, esters, and ethers. Our current understanding of the atmospheric chemistry of these organic compounds is discussed below (see Atkin- son 1986 for further details). Aldehydes. These include aliphatic and aromatic aldehydes (unsaturated aldehydes such as acrolein and crotonaldehyde are dealt with below). These aldehydes are removed from the troposphere by pho- tolysis and reaction with OH and NO3
Roger Atkinson 119 radicals (and, for formaldehyde, with HO2 radicals). The reaction with NO3 radicals is a relatively minor tropospheric loss process (table 3), but contributes to HO2 radical and peroxyacetyl nitrate (PAN) formation during nighttime hours (Calvert and Stock- well 1983; Stockwell and Calvert 1983a; Cantrell et al. 1985, 1986~. Thus the major loss processes involve photolysis and reac- tion with OH radicals. Recent studies (De- More et al. 1985) show that the photodis- sociation quantum yields for the aliphatic aldehydes are lower than previously as- sumed, and for essentially all aldehydes, except HCHO, reaction with the OH rad- ical represents the dominant daytime re- moval process (table 3~. For HCHO the OH radical reaction yields the HCO radical, which reacts rap- idly with O2 to form HO2 and CO. OH + HCHO ~ H2O + HCO lo2 HO2 + co (85) For the higher aldehydes, the RCO radical initially formed, OH + RCHO ~ HERO + RCO (86) rapidly adds O2 to yield an acylperoxy radical: RCO + O2 ~ RC(0)00 (87) Under polluted atmospheric conditions, these acylperoxy radicals then react with NO (eq. 88) or NO2 (eq. 89), RC(0)00 + NO ~ RC(0)0 + NO2 R + CO2 (88) RC(0)00 + NO~ ~ RC(0)00NO. Peroxyacyl nitrates (89) with the peroxyacyl nitrates being in ther- mal equilibrium with NO2 and the acylper- oxy radicals. For PAN, the lifetime with respect to thermal decomposition (as ob- served in the presence of excess NO) is ~ 45 min at 298°K (Atkinson and Lloyd 1984~. PAN has been shown to be a direct- acting mutagen toward Ames Salmonella typhimurium strain TA100 (Kleindienst et al. 1985) and is phytotoxic; peroxypro pionyl nitrate (PPN) is even more phyto- toxic. Thus, further studies concerning the health effects of these peroxyacyl nitrates need to be conducted. Benzaldehyde is the simplest aromatic aldehyde, and is a primary automotive emission as well as being a product (see below) of the atmospheric transformations of toluene. Data concerning the atmo- spheric reactions of aromatic aldehydes are available only for benzaldehyde. For benz- aldehyde, as for the aliphatic aldehydes, photolysis and OH radical reaction are the major loss processes (Atkinson and Lloyd 1984), with the nighttime NO3 radical re- action being of minor importance (table 3~. Reaction with the OH radical initiates a series of reactions leading to the formation of peroxybenzoyl nitrate (PBzN) and nitro- phenols (Niki et al. 1979; Atkinson and Lloyd 1984~. OH + C6H5CHO ~ H2O + C6H5CO :02 o ,, C6H5COO (90) o 11 11 C6H5COO + NO2 ~ C6H5COONO2 (PBzN) (91) o o C6H5COO- + NO ~ C6H5CO + NO2 c6H5 + CO2 (92) C6H5 ~ O2 ~ C6H5OO NO ~l NO2 HO2 / C6H5OH + O2 C6H5O (phenoxy radical) / \ ~NO2 o- and p-nitrophenol (93) Ketones. This class of organic emissions is exemplified by acetone and its higher homologues. As with the aldehydes, pho- tolysis and reaction with the OH radical are the major atmospheric loss processes (At- kinson 1986~. For acetone, the calculated lifetime of~ 15 days due to photolysis (Gardner et al. 1984) is significantly shorter
120 Atmospheric Transformations of Automotive Emissions than that of~ 100 days due to OH radical reaction. For the other ketones, reaction with OH radicals appears to be the major tropospheric loss process, proceeding by, for example, for 2-butanone, OH + CH3CH2COCH3- O2 H2O 00 + CH3CHCOCH3 2 H2O 2, H2O + CH3CH2COCH2OO + ·00CH2CH2COCH3 (94) with the top reaction being the major pathway (Atkinson 1986~. Subsequent re- action of the radical formed in this reaction pathway with NO leads to the formation of acetaldehyde and the acetyl radical: OH + CH3CH=CHCHO~ OH OH CH3CHCHCHO and CH3CH(tHCHO H2O + CH3CH=CHCO (96) Analogous to the OH radical-initiated re- actions with the alkenes and aliphatic al- dehydes, respectively, these initial reactions are expected to be followed by the sequence of reactions shown below, where the f~rst- generation products are underlined: CH3CHOHCHCHO + O 00 CH3CHOHCHCHO NO ~ NO2 I . CH3CHOHCHCHO I 00 0 CH3C! IOH + (CHOP [glyoxal] CH3CHCOCH3 + NO ~ NO2 + CH3CHCOCH3 CH3CHO + CH3CO (95) Thus the major products from the atmo- spheric reactions of the ketones appear to be carbonyls and PAN precursors, al- though bifunctional oxygen-containing compounds may be formed in small amounts. The atmospheric degradation pathways need further investigation, and the health impacts of the products formed should be studied. a,/3-Unsaturated Carbonyls. These com- pounds, exemplified by acrolein, crotonal- dehyde, and methyl vinyl ketone, are known to react with 03, OH radicals, NO3 radicals as well as undergo photolysis. Un- der atmospheric conditions the OH radical reaction is the major loss process (Atkinson and Carter 1984~. For the aldehydes, OH radical reaction can proceed by two reac- tion pathways: OH radical addition to the double bond and hydrogen-atom abstrac- tion from the CHO group (Atkinson 1986): O2 CH`CHO + HO~ CH3CH=CHCO + O o ll NO CH3CH CHCOO ~ CH3CH=CH + CO2 NO2 NO2 o 11 CH3CH=CHCOONO2 CH3CH CH + O2 ~ (97) (98) [CH3CH CH00] ~ products (99) When the reaction of the vinyl radical (CH2 CH) with O2 is used as an analogy (Slagle et al. 1984), the products from the reaction of O2 with CH3CH- CH may be CH3CHO and HCO. Hence these c~,,~ unsaturated aldehydes are expected to ulti- mately give rise to a-dicarbonyls and car- bonyls. For the c~,,l3 unsaturated ketones, such as methyl vinyl ketone, the major atmospheric reaction with the OH radical occurs only by OH radical addition to the double bond:
Roger Atkinson O2 OH + CH2=CHCOCH3 ~ OO OH HOCH2CHCOCH3 and ·0OCH2CHCOCH3 NO ~ NO2 NO _ NO2 O OH 1 . 1 HOCH2CHCOCH3 OCH2CHCOCH3 I 1 (100) HOCH:CHO HCHO + CH3CO + CH3COCHOH 1 02 _3COCHO + HO2 where the first-generation intermediate products are again underlined. a-Dicar- bonyls, together with aldehydes and hy- droxyaldehydes, are expected to be formed. Ethers. Under atmospheric conditions the aliphatic ethers, such as dimethyl and diethyl ether, react primarily with the OH radical. These reactions proceed by hydro- gen-atom abstraction from the C-H bonds (Atkinson 1986), as shown below for diethyl ether (first-generation products are underlined). OH + CH3CH2OCH2CH3 ~ H2O + CH3CHOCH2CH3 O2 NO- ~ NO2 f CH3CHOCH2CH3 / \ ~\ (101) 11 CH3COCH2CH3 + HO2 CH3CHO + C2HsO lo2 CH3CHO + HO2 Few experimental data are available, but E. C. Tuazon (unpublished data) has shown that dimethyl ether yields mainly methyl 121 formate (HC(O)OCH3), presumably by the O2 reaction route. Alcohols. The reaction sequences for the simple aliphatic alcohols under atmospheric conditions are known (Atkinson 1986~. These involve hydrogen-atom abstraction, mainly from the a-C H bonds. For ex- ample, ~ H2O + CH2OH OH + CH3OH ~ H2O + CH3O (102) with the top reaction pathway accounting for ~ 85 percent of the overall reaction at 298°K. Since the CH2OH and CH3O rad- icals react with O2 to yield formaldehyde, the overall reaction can be written as OH + CH3OH ~H2O + HCHO + HO2 (103) The reaction pathways for ethanol are anal- ogous, with acetaldehyde being the major product formed. For the higher alcohols, other, more complex, products are ex- pected to be formed. Carboxylic Acids. The carboxylic acids most likely to be emitted from automotive use are formic and acetic acids. The avail- able data suggest that these carboxylic acids react with the OH radical under atmo- spheric conditions (Atkinson 1986~. For formic acid, hydrogen-atoms are pro- duced, o 11 OH + HCOH ~ ~ H2O + CO2 + H (104) with the reaction probably proceeding through the initial formation of an addition complex. The reaction products formed from the reactions of OH radicals with the higher carboxylic acids are presently not known. Esters . The es ters, RC (O ~ O R ' als 0 re- act primarily with the OH radical under atmospheric conditions (Atkinson 1986~. The limited data available show that these reactions proceed by reaction with the alkoxy moiety.
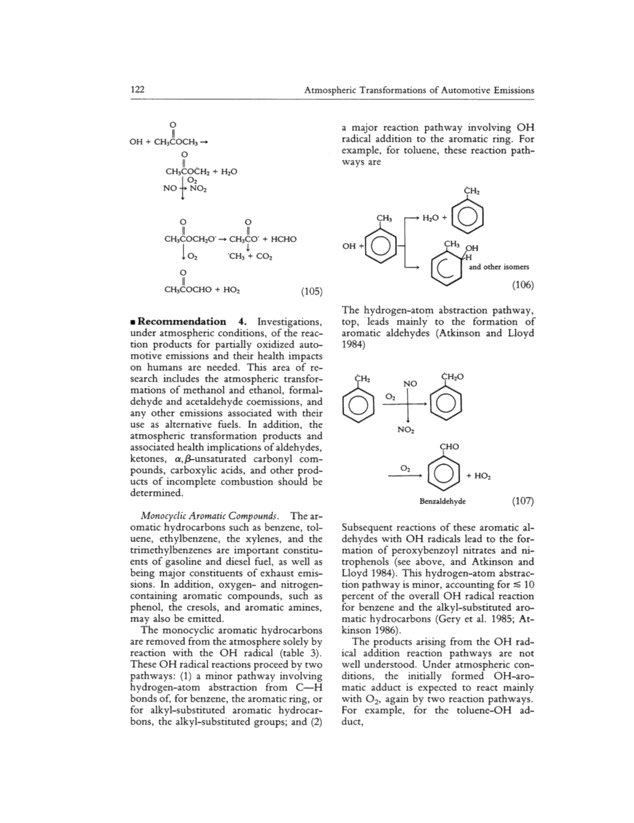
122 Atmospheric Transformations of Automotive Emissions o 11 OH + CH3COCH3 o 11 CH3COCH2 + H2O O2 NO- ~ NO2 O O 11 11 CH3COCH2O ~ CH3CO- + HCHO 1 O2 CH3 + CO2 o 11 CH3COCHO + HO2 (105) Recommendation 4. Investigations, under atmospheric conditions, of the reac- tion products for partially oxidized auto- motive emissions and their health impacts on humans are needed. This area of re- search includes the atmospheric transfor- mations of methanol and ethanol, formal- dehyde and acetaldehyde coemissions, and any other emissions associated with their use as alternative fuels. In addition, the atmospheric transformation products and associated health implications of aldehydes, ketones, cr,,l3 unsaturated carbonyl com- pounds, carboxylic acids, and other prod- ucts of incomplete combustion should be determined. Monocyclic Aromatic Compounds. The ar- omatic hydrocarbons such as benzene, tol- uene, ethylbenzene, the xylenes, and the trimethylbenzenes are important constitu- ents of gasoline and diesel fuel, as well as being major constituents of exhaust emis- sions. In addition, oxygen- and nitrogen- containing aromatic compounds, such as phenol, the cresols, and aromatic amines, may also be emitted. The monocyclic aromatic hydrocarbons are removed from the atmosphere solely by reaction with the OH radical (table 3). These OH radical reactions proceed by two pathways: (1) a minor pathway involving hydrogen-atom abstraction from C H bonds of, for benzene, the aromatic ring, or for alkyl-substituted aromatic hydrocar- bons, the alkyl-substituted groups; and (2) c~3 OH +~- a major reaction pathway involving OH radical addition to the aromatic ring. For example, for toluene, these reaction path- ways are CH2 - ~ H2O + CH3 OH ~H and other isomers ~(106) The hydrogen-atom abstraction pathway, top, leads mainly to the formation of aromatic aldehydes (Atkinson and Lloyd 1984) CH7 NO CH2O +~ O2 NO2 CHO ~ + HO2 Benzaldehyde (107) Subsequent reactions of these aromatic al- dehydes with OH radicals lead to the for- mation of peroxybenzoyl nitrates and ni- trophenols (see above, and Atkinson and Lloyd 1984). This hydrogen-atom abstrac- tion pathway is minor, accounting for c 10 percent of the overall OH radical reaction for benzene and the alkyl-substituted aro- matic hydrocarbons (Gery et al. 1985; At- kinson 1986). The products arising from the OH rad- ical addition reaction pathways are not well understood. Under atmospheric con- ditions, the initially formed OH-aro- matic adduct is expected to react mainly with O2, again by two reaction pathways. For example, for the toluene-OH ad- duct,
Roger Atkinson 123 CH3 + O2 CH3 ~1' + HO2 CH3 OH 7 ~ ~. {-)- The hydrogen-atom abstraction reaction of the OH-aromatic adduct to yield phenolic compounds has been shown to be relatively minor, accounting for ~ 20 percent of the overall OH radical reaction mechanism for toluene (Atkinson and Lloyd 1984; Gery et al. 1985; Leone et al. 1985~. The major reaction pathway involves other reactions of the OH-aromatic-O2 adducts, and these have been shown to involve ring cleavage. Thus, the cY-dicarbonyls glyoxal, methyl- glyoxal, and biacetyl have been identified and quantified from benzene and the methyl-substituted benzenes (Bandow et al. 1985; Bandow and Washida 1985a,b; Tuazon et al. 1986), and reaction pathways that lead to these products have been pro- posed (Atkinson et al. 1980; Killus and Whitten 1982~. The reaction pathways that form a-dicarbonyls, phenolic products, and hydrogen-atom abstraction products, however, fail to account for ~ 3~50 per- cent of the overall reaction products. The recent semiquantitative or qualitative, but highly important, product studies of Dum- dei and O'Brien (1984) and Shepson et al. (1984) have identified a variety of other bifunctional ring cleavage products from toluene and o-xylene, which include, from toluene, CH3COCOCH-CH2, CHOCOCH CH2, CH3COCH-CH2, CH3COCH-CHCH CH2, CHOC (OH) CHCHO, and CH3COCH-CHCH- CHCHO. Much less information is available for the other aromatic compounds either directly emitted from automobiles, or formed as products from the primary aromatic emis- sions during their atmospheric transport. Indeed, most of the information has been derived from kinetic rather than direct product studies. For example, the phenolic compounds, which are known to be re- moved from the atmosphere primarily by chemical reaction with OH and NO3 radi- cals (tables 2 and 3; Atkinson and Lloyd 1984) and by wet deposition from the gas phase (Leuenberger et al. 1985), react with OH radicals mainly by initial OH radical addition to the ring. However, the NO3 radical reaction appears to proceed by hy (108) drogen-atom abstraction from the substitu ent-OH group (Atkinson et al. 1984~. OH NO3 + ~ HNO3 + [~ (109) 1 NO2 o- and p-nitrophenol For other classes of monocyclic aromatic compounds, product data are not available. Product yields under atmospheric condi- tions are reliably known for only a few of the many aromatic hydrocarbons emitted from automotive use. Moreover, the health effects of most of these compounds are not known, although methylglyoxal has re- cently been reported to be mutagenic toward Salmonella typhimurium strain TA100 (Shepson et al. 1985b). Since the observed product yields typically account for ' 50 percent of the overall reaction products, an understanding of the remain- ing products and their health effects is necessary. This will include products formed in the particulate and gas phases. Recommendation 5. The products arising from the OH radical-initiated reac- tions of the aromatic hydrocarbons, a ma . . . . Jor emlsslon category trom automotive use, need to be identified. These studies are important because of the relatively high reactivity of aromatic hydrocarbons, and will involve the identification and quantifi- cation of a plethora of bifunctional organic compounds, many of which will probably be present in low yield. Polycyclic Aromatic Hydrocarbons (PAHsJ and Their Derivatives and Analogues. A large number of these chemical com
124 Atmospheric Transformations of Automotive Emissions Table 7. Vapor Pressures at 298°K for a Series of PAHs PAH Vapor Pressure at 298°K (torr) Naphthalene Phenanthrene Anthracene Fluoranthrene Pyrene Bench janthracene Benzota~pyrene Chrysene 8.0 x 10 a 1.2 x 10 - 4 a 6.0 x 10 9.2 x 106 a 4.5 x 106 a 2.1 x 10a 5.6 x 10 6.4 x 10 a From Sonnefeld et al. 1983. b From Yamasaki et al. 1984. pounds, including PAHs (such as naphtha- lene, phenanthrene, anthracene, fluoran- thene, pyrene, perylene, and benzo~a] pyrene), and their alkyl-substituted or ox- ygen-, sulfur-, and nitrogen-containing de- rivatives, as well as oxygen-, nitrogen- and sulfur-containing heterocyclic analogues, are, or may be, emitted from combustion sources. Although these compounds are relatively minor components of automo- tive emissions, they have assumed a "spot- light" position in automotive-related health risk assessments because of their potential toxicity. The PAHs and their analogues and de- rivatives have relatively low vapor pres- sures (table 7), and are distributed between the gas and particulate phases, with this distribution being highly temperature de- pendent. As presently understood, the atmo- spheric transformations of these PAHs and their derivatives are highly dependent upon the phase with which they are associated. The available data show that for the PAHs present in the gas phase, reaction with the OH radical predominates, leading to atmo- spheric lifetimes of a few hours or less (table 3~. The nighttime reaction with N205 is of minor significance as a PAH loss process (table 3) (Pitts et al. 1985a), but may be important for the formation of nitro-PAHs (see below, and Arey et al. 1986~. Recent ambient atmospheric data from Norway, Denmark, and the United States show that 2-nitrofluoranthene and 2-ni tropyren~ nitro-PAHs not observed from combustion sources are the major nitro- PAH components of particulate organic matter (POM) (Nielsen et al. 1984; Pitts et al. 1985c; Ramdahl et al. 1986~. Since these two nitro-PAHs are not formed during the collection of POM, they must be formed in the atmosphere from the parent PAH dur- ing transport from source to receptor (Nielsen et al. 1984; Pitts et al. 1985c; Arey et al. 1986; Ramdahl et al. 1986; Sweetman et al. 1986; Zielinska et al. 1986~. Indeed, it now appears that the majority of the nitro- PAHs present in ambient POM are formed via atmospheric transformations during transport from source to receptor. xecent env~ronmenta~ chamber studies have shown that 2-nitrofluoranthene as well as 2-nitropyrene are formed from the gas-phase reactions of fluoranthene and py- rene with OH radicals in the presence of NOx (Arey et al. 1986~. 2-Nitrofluoran- thene is also formed from the gas-phase reaction of N2O5 with fluoranthene (Sweetman et al. 1986; Zielinska et al. 1986~. Since many PAHs and their analogues and derivatives are partitioned primarily into the adsorbed phase under atmospheric conditions, a large number of experimental studies have been performed to delineate the reaction processes occurring for the adsorbed-phase compounds. However, most of these studies have been done using nonatmospherically realistic adsorbents such as glass fiber and Teflon-impregnated glass fiber and silica surfaces. The data obtained from these and from more realis- tic surfaces, such as carbon black and fly- ash, show that the reactions (including photolysis) are strongly dependent on the nature of the adsorbent species (see, for example, Pitts 1983; Behymer and Hites 1985~. Photolysis, reaction with 03, NO2, and/ or HNO3, and N2O5 have all been shown to lead to loss of PAHs on several sub- strates (see, for example, Pitts 1983; Pitts et al. 1985b, 1986~. Certain of these reac- tions, in particular, those with NO2 and HNO3 and with N2O5, appear to be rela- tively slow under atmospheric conditions (Pitts et al. 1985b). However, because of
Roger Atkinson 125 the substrate dependence of these adsorbed- phase reactions (see, for example, Behymer and Hites 1985), no firm conclusions can be drawn about the importance of these reac- tions under atmospheric conditions. It does appear that photolysis and reaction with O3 may be important for certain PAHs and their derivatives. Clearly, a comprehensive and systematic investigation of the gas- and adsorbed- phase reactions of this class of automotive emissions is necessary before further risk assessment studies concerning these com- pounds can be carried out. This is a difficult research area because of the partitioning of many, if not most, of these emissions and their products between the gas and partic- ulate phases, and because of the high potential for analytical artifacts during sam- pling with the currently available tech- niques. · Recommendation 6. The atmospheric transformation products of PAHs and their oxygen-, nitrogen-, and sulfur-containing analogues and homologues require study, in the gaseous and the adsorbed phases. In particular, the reaction pathways that lead to nitro-PAHs need to be quantitatively established. In addition, the atmospheric removal processes and resulting products of these nitro-PAHs should be studied fur- ther. Particulate Matter. A variety of other chemical compounds, including metals such as Pb, are emitted from automotive use into the atmosphere in particulate form. As shown in table 6, particles are removed from the atmosphere at rates that depend markedly on the particle size. For particles of diameter 0.1-1 Em (the size that corre- sponds to most particles present in the atmosphere), dry and wet deposition pro- cesses lead to lifetimes of several days or more. Since the metals emitted are expected to be present mainly in their oxidized form for example, PbBrC1-chemical reactions are unlikely and their removal will occur principally by these physical processes. For organic chemicals emitted from automo tive use and present on ambient POM, reactions may occur during atmospheric transport. This topic has assumed impor- tance because of the recent interest in acidic deposition and the role of aerosols in the formation of nitric, sulfuric, and organic acids. The reactions that can occur are complex and involve aqueous chemistry, gas-to-particle conversion, and heteroge- neous reactions (see, for example, Graedel and Weschler 1981; Chameides and Davis 1982; Graedel and Goldberg 1983; Jacob and Hoffmann 1983; Chameides 1986; Graedel et al. 1986~. However, this field is in a state of rapid change, and further research is necessary before a full under- standing can be reached (see Recommenda- tion 1~. Analytical Techniques It has become apparent during the past several years that a major experimental . . . . . lnltlatlVe IS necessary to c .eve op new ana- lytical techniques to allow the products of these complex atmospheric reactions to be identified and quantified. Fourier transform infrared absorption spectroscopy as well as gas chromatography/mass spectrometry (now often used in gas-phase studies) are subject to significant limitations when the organic products are complex and because of the possibility of the formation of arti- facts. Similarly, for particle-associated chemicals, studies of their atmospheric degradation reactions and associated rates are often complicated by artifacts. Clearly, there is a need for new in situ analytical techniques allowing real-time analyses de- void of artifact product formation prob- lems. This area is a major research topic in its own right and should be recognized as such. If this research area is not ag- gressively pursued, any advances in our knowledge about the atmospheric mecha- nisms and reaction products of automotive emissions may well become limited by the available analytical procedures. As re- search progresses in this area, it is also apparent that further techniques for study- ing low-volatility organics in the gas and the adsorbed phases must be devel- oped.
126 Atmospheric Transformations of Automotive Emissions Recommendation 7. A major research effort is needed to develop the necessary analytical techniques for identifying and quantifying the products of complex atmo- spheric reactions. Of prime importance is the development of nondestructive, nonin- trusive, in situ analytical techniques that will allow the atmospheric transformations of gaseous and particulate-associated chem- ical species to be studied. Summary As a result of the last two decades of laboratory, computer modeling, and ambi- ent atmospheric experiments, a large body of data now exists concerning the atmo- spheric loss processes and transformations of automotive emissions. However, signif- icant gaps in our knowledge still remain, mainly about the products formed under atmospheric conditions. As discussed in the sections above, the physical and chemical processes leading to the removal of automotive emissions from the atmosphere include · wet and dry deposition of gases and particles; · chemical reactions of gaseous automotive emissions with OH, NO3, and HO2 radi- cals, 03, N2O5, and gaseous HNO3; · photolysis; · reaction of particulate-associated organic compounds with a variety of gas- and adsorbed-phase species; and · reactions in the aqueous phase with a vari- ety of reactive species that are of importance in clouds, raindrops, and fog droplets, and lead to the formation of acidic precipita- tion. Chemical reactions dominate the removal of most organic chemicals, with atmospheric lifetimes ranging from less than 1 min for highly reactive organic compounds reacting with the NO3 radical during nighttime hours, to months or even years for the less reactive alkalies, haloalkanes, and substituted benzenes. Inorganic compounds emitted as a result of automotive use also exhibit a wide range of atmospheric lifetimes, with NO2, HNO2, and alkyl nitrites having photolysis lifetimes measured in minutes. In contrast, H2O2 and HNO3 are readily removed at surfaces, and are predominantly removed from the gas phase by wet and/or dry depo- sition processes that can take several days or more. The chemical transformations of auto- motive emissions lead to the formation of a wide variety of products. Many of these transformation products are unknown, and the health impacts on humans of those that are known have not been investigated. Future research programs must first re- quire studies to determine the general chemical classes of products formed from the atmospheric transformations of auto- motive emissions. For those products sus- pected to threaten human health, additional work will then be necessary to better de- fine their amounts and formation mecha- nisms. Summary of Research Recommendations Significant gaps still exist in our understanding of the physi- cal and chemical transformations of automotive emissions that occur in the atmosphere during transport from source to recep- tor. The areas requiring further study are ranked in order of . . prlorlty. HIGH PRIORITY Recommendlation2 Further investigations of the transformations of NOX under atmospheric conditions are needed. This topic is important for
Roger Atkinson 127 indoor environments as well, since certain NOx undergo important heterogeneous transformations. In particular, laboratory research has shown that, under conditions representative of certain indoor environments, NO2 hydrolyzes on surfaces to yield gas-phase HNO2 at significant rates. The kinetics and mechanism of this heterogeneous reaction should also be investigated in ambient nighttime atmospheres as should the reactions of the NO3 radical (especially with respect to the products formed from its reactions with organic compounds) and the heterogeneous and/or homoge- neous reactions of N2O5. Recommendlation 5 The products arising from the OH radical-initiated reactions of the aromatic hydrocarbons, a major emission category from auto motive use, need to be identified. These studies are important because of the relatively high reactivity of aromatic hydrocarbons, and will involve the identification and quantification of a plethora of bifunctional organic compounds, many of which will probably be present in low yield. Recommendlation6 The atmospheric transformation products of PAHs and their oxygen-, nitrogen-, and sulfur-containing analogues and homolo gues require study in the gaseous and the adsorbed phases. In particular, the reaction pathways that lead to nitro-PAHs need to be quantitatively established. In addition, the atmospheric removal processes and resulting products of these nitro-PAHs should be studied further. These studies will be difficult to perform because of the high potential for artifact formation. MEDIUM PRIORITY Recommendlation 1 Study is required on the physical removal processes leading to wet and dry deposition of gases and particles. Investigations of the processes occurring on and in particulate and aerosol matter should focus on As-to-particle conversion processes and the chem ical processes that occur within aerosols (including fogs and clouds). Recommendlation 3 The products arising from the OH radical-initiated reactions of alkanes the major component of automobile emissions require study. These products are likely to be distributed between the gas and particulate phases, and data are especially needed for the alkanes with eight or more carbon atoms. - Recommendation4 Investigations, under atmospheric conditions, of the reaction products for partially oxidized automotive emissions and their health impacts on humans are needed. This area of research includes the atmospheric transformations of methanol and ethanol, formaldehyde and acetaldehyde co-emissions, and any other emis sions associated with their use as alternative fuels. In addition, the atmospheric transformation products and associated health impli cations of aldehydes, ketones, a,,~unsaturated carbonyl com pounds, carboxylic acids, and other products of incomplete com bustion should be determined.
128 Atmospheric Transformations of Automotive Emissions Recommendation 7 A major research effort is needed to develop the necessary analytical techniques for identifying and quantifying the products of complex atmospheric reactions. Of prime importance is the development of nondestructive, nonintrusive, in situ analytical techniques that will allow the atmospheric transformations of r gaseous and particulate-associated chemical species to be studied. This is clearly a long-term ideal, but utterly crucial in order to advance our current knowledge of the atmospheric transformations ~ . . . 01 automotive emlsslons. References Arey, J., Zielinska, B., Atkinson, R., Winer, A. M., Ramdahl, T., and Pitts, J. N., Jr. 1986. The forma- tion of nitro-PAH from the gas-phase reactions of fluoranthene and pyrene with the OH radical in the presence of NOX, Atmos. Environ. 20:2339-2345. Atkinson, R. 1986. Kinetics and mechanisms of the gas phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions, Chem. Rev. 86:69-201. Atkinson, R., and Carter, W. P. L. 1984. Kinetics and . ~ mechanisms ot the gas-phase reactions of ozone with organic compounds under atmospheric condi- tions, Chem. Rev. 84:437070. Atkinson, R., and Lloyd, A. C. 1984. Evaluation of kinetic and mechanistic data for modeling of pho- tochemical smog, J. Phys. Chem. Ref: Data 13:315- 444. Atkinson. R.. Carter W. P. L. Darnall K R. A. _ , 7 ~ 7 Winer, A. M., and Pitts, J. N., Jr. 1980. A smog chamber and modeling study of the gas phase NOx-air Photooxidation of toluene and the cresols, Int.J. Chem. Kinet. 12:779-836. Atkinson, R., Carter, W. P. L., Plum, C. N., Winer, A. M., and Pitts, J. N., Jr. 1984. Kinetics of the gas-phase reactions of NO3 radicals with a series of aromatics of 296 + 2 K, Int. J. Chem. Kinet. 16:887-898. Atkinson, R., Tuazon, E. C., Carter, W. P. L. 1985. The extent of H-atom abstraction from the reaction of the OH radical with 1-butene under atmospheric conditions, Int.J. Chem. Kinet. 17:725-734. Atkinson, R., Tuazon, E. C., Mac Lead, H., Asch- mann, S. M., and Winer, A. M. 1986a. The gas- phase reaction of chlorine nitrate with water vapor, Geophys. Res. Lett. 13:117-120. Atkinson, R., Winer, A. M., and Pitts, J. N., Jr. 1986b. Estimation of nighttime N205 concentra- tions from ambient NO2 and NO3 radical concen- trations and the role of N2O5 in nighttime chemis- try, Atmos. Environ. 20:331-339. Atlas, E., and Giam, C. S. 1981. Global transport of Correspondence should be addressed to Roger Atkin- son, Statewide Air Pollution Research Center, Uni versity of California, Riverside, CA 92521. organic pollutants: ambient concentrations in the remote marine atmosphere, Science 211:16~165. Bandow, H., and Washida, N. 1985a. Ring-cleavage reactions of aromatic hydrocarbons studied by FT-IR spectroscopy. II. Photooxidation of o-, m-, and p-xylenes in the NOx-air system, Bull. Chem. Soc.Jpn. 58:2541-2548. Bandow, H., and Washida, N. 1985b. Ring-cleavage reactions of aromatic hydrocarbons studied by FT-IR spectroscopy. III. Photooxidation of 1,2,3-, 1,2,4- and 1,3,5-trimethylbenzenes in the NOx-air system, Bull. Chem. Soc.Jpn. 58:2549-2555. Bandow, H., Okuda, M., and Akimoto, H. 1980. Mechanism of the gas-phase reactions of C3H6 and NO3 radicals, J. Phys. Chem. 84:3604-3608. Bandow, H., Washida, N., and Akimoto, H. 1985. Ring-cleavage reactions of aromatic hydrocarbons studied by FT-IR spectroscopy. I. Photooxidation of toluene and benzene in the NOx-air system, Bull. Chem. Soc.Jpn. 58:2531-2540. Behymer, T. D., and Hites, R. A. 1985. Photolysis of polycyclic aromatic hydrocarbons adsorbed on sim- ulated atmospheric particulates, Environ. Sci. Tech- nol. 19:100~1006. Benson, S. W. 1976. Thermochemical Kinetics, 2nd ea., Wiley, New York. Burrows, J. P., Tyndall, G. S., and Moortgat, G. K. 1985a. A study of the N2O5 equilibrium between 275 and 315 K and determination of the heat of formation of NO3, Chem. Phys. Lett. 119:193-198. Burrows, J. P., Tyndall, G. S., and Moortgat, G. K. 1985b. Absorption spectrum of NO3 and kinetics of the reactions of NO3 with NO2, Cl and several stable atmospheric species at 298 K, J. Phys. Chem. 89:48480856. Cadle, S. H., Dasch, J. M., and Mulawa, P. A. 1985. Atmospheric concentrations and the deposition ve- locity to snow of nitric acid, sulfur dioxide and various particulate species, Atmos. Environ. 19: 1819-1827. Calvert, J. G. (ed.) 1984. S02, NO and NO2. Oxida- tion Mechanisms: Atmospheric Considerations, Butter- worth, Boston. Calvert, J. G., and Pitts, J. N., Jr. 1966. Photochemis- try, Wiley, New York. Calvert, J. G., and Stockwell, W. R. 1983. Acid generation in the troposphere by gas-phase chemis- try, Environ. Sci. Technol. 17:428A - 43A. Calvert, J. G., Su, F., Bottenheim, J. W., and Strausz,
Roger Atkinson O. P. 1978. Mechanism of the homogeneous oxi- dation of sulfur dioxide in the troposphere, Atmos. Environ. 12:197-226. Cantrell, C. A., Stockwell, W. R., Anderson, L. G., Busarow, K. L., Perner, D., Schmeltekopf, A., Calvert, J. G., and Johnston, H. S. 1985. Kinetic study of the NO3-CH2O reaction and its possible role in nighttime tropospheric chemistry, J. Phys. Chem. 89:139-146. Cantrell, C. A., Davidson, J. A., Busarow, K. L., and Calvert, J. G. 1986. The CH3CHO-NO3 reaction and possible nighttime PAN formation, J. Geophys. Res. 91:5347-5353. Carter, W. P. L., and Atkinson, R. 1985. Atmo- spheric chemistry of alkalies, J. Atmos. Chem. 3: 337005. Carter, W. P. L., Ripley, P. S., Smith, C. G., and Pitts, J. N., Jr. 1981. Atmospheric chemistry of hydrocarbon fuels. Vol. 1: Experiments, results and discussion, Final Report ESL-TR-81-53, Engineer- ing and Services Laboratory, Air Force Engineering and Services Center, Tyndall AFB, Fla. (November 1981). Carter, W. P. L., Atkinson, R., Winer, A. M., and Pitts, J. N., Jr. 1982. An experimental investigation of chamber-dependent radical sources, Int.J. Chem. Kinet. 14:1071-1103. Carter, W. P. L., Lurmann, F. W., Atkinson, R., and Lloyd, A. C. 1986. Development and testing of a surrogate species chemical reaction mechanism, EPA-600/3-86-031 (August 1986). Chameides, W. L. 1986. Possible role of NO3 in the nighttime chemistry of a cloud, J. Geophys. Res. 91 :5331-5337. Chameides, W. L., and Davis, D. D. 1982. The free radical chemistry of cloud droplets and its impact upon the composition of air, J. Geophys. Res. 87:4863 4877. Chang, T. Y. 1984. Rain and snow scavenging of HNO3 vapor in the atmosphere, Atmos. Environ. 18:191-197. Cicerone, R. J., and Zellner, R. 1983. The atmo- spheric chemistry of hydrogen cyanide (HCN), J. Geophys. Res. 88:10689-10696. Colbeck, I., and Harrison, R. M. 1985. Dry deposi- tion of ozone: some measurements of deposition velocity and of vertical profiles to 100 metres, Atmos. Environ. 19:1807-1818. Crutzen, P. J. 1982. The global distribution of hy- droxyl, In: Atmospheric Chemistry (E. D. Goldberg, ed.), pp. 31~328, Springer-Verlag, New York. DeMore, W. B., Margitan, J. J., Molina, M. J., Watson, R. T., Golden, D. M., Hampson, R. F., Kurylo, M. J., Howard, C. J., and Ravishankara, A. R. 1985. Chemical kinetics and photochemical data for use in stratospheric modeling, NASA Eval- uation No. 7, Jet Propulsion Laboratory Publication 85-37 Quly 1, 1985). Dolske, D. A., and Gatz, D. F. 1985. A field inter- comparison of methods for the measurement of particle and gas dry deposition, J. Geophys. Res. 90:2076-2084. Dumdei, B. E., and O'Brien, R. J. 1984. Toluene 129 degradation products in simulated atmospheric con- ditions, Nature 311 :248-250. Eisenreich, S. J., Looney, B. B., and Thornton, J. D. 1981. Airborne organic contaminants in the Great Lakes ecosystem, Environ. Sci. Technol. 15:30-38. Friedl, R. R., Brune, W. H., and Anderson, J. G. 1985. Kinetics of SH with NO2, 03, O2 and H202, J. Phys. Chem. 89:5505-5510. Fritz, B., Lorenz, K., Steinert, W., and Zellner, R. 1984. Rate of oxidation of HCN by OH radicals at lower temperatures, Oxid. Commun. 6:36~370. Gardner, E. P., Wijayaratne, R. D., and Calvert, J. G. 1984. Primary quantum yields of photodecomposi- tion of acetone in air under tropospheric conditions, J. Phys. Chem. 88:5069-5076. Gery, M. W., Fox, D. L., Jeffries, H. E., Stockbur- ger, L., and Weathers, W. S. 1985. A continuous stirred tank reactor investigation of the gas-phase reaction of hydroxyl radicals and toluene, Int. J. Chem. Kinet. 17:931-955. Graedel, T. E., and Goldberg, K. I. 1983. Kinetic studies of raindrop chemistry. 1. Inorganic and organic processes, J. Geophys. Res. 88:10865-10882. Graedel, T. E., and Weschler, C. J. 1981. Chemistry within aqueous atmospheric aerosols and raindrops, J. Geophys. Res. 19:50~539. Graedel, T. E., Ayers, G. P., Duce, R. A., Georgii, H. W., Klockow, D. G. A., Morgan, J. J., Rodhe, H., Schneider, B., Slinn, W. G. N., and Zafiriou, O. C. 1982. Aqueous chemistry in the atmosphere, group report, In: Atmospheric Chemistry (E. D. Goldberg, ed.), pp. 9~118, Springer-Verlag, New York. Graedel, T .E., Mandich, M. L., and Weschler, C. J. 1986. Kinetic model studies of atmospheric droplet chemistry. 2. Homogeneous transition metal chem- istry in raindrops, J. Geophys. Res. 91 :5205-5221. Graham, R. A., and Johnston, H. S. 1978. The photochemistry of NO3 and the kinetics of the N2O5-O3 system, J. Phys. Chem. 82:254-268. Harris, G. W., Carter, W. P. L., Winer, A. M., Pitts, J. N., Jr., Platt, U., and Perner, D. 1982. Obser- vations of nitrous acid in the Los Angeles atmo- sphere and implications for predictions of ozone- precursor relationships, Environ. Sci. Technol. 16:411 419. Hatakeyama, S., Bandow, H., Okuda, M., and Aki- moto, H. 1981. Reactions of CH2OO and CH2(~A~) with H2O in the gas phase, J. Phys. Chem. 2249-2254. Hatakeyama, S., Kobayashi, H., and Akimoto, H. 1984. Gas-phase oxidation of SO2 in the ozone- olefin reactions, J. Phys. Chem. 88:473~4739. Heikes, B. G., and Thompson, A. M. 1983. Effects of heterogeneous processes on NO3, HONO and HNO3 chemistry in the troposphere, J. Geophys. Res. 88:1088~10895. Herron, J. T., Martinez, R. I., and Huie, R. E. 1982. Kinetics and energetics of the Criegee intermediate in the gas phase. I. The Criegee intermediate in ozone-alkene reactions, Int. J. Chem. Kinet. 14: 201-224. Hewitt, C. N., and Harrison, R. M. 1985. Tropo 85:
130 spheric concentrations of the hydroxyl radical a review, Atmos. Environ. 19:545-554. Hov, 0., and Isaksen, I. S. A. 1979. Hydroxyl and peroxy radicals in polluted tropospheric air, Geo- phys. Res. Lett. 6:219-222. Huebert, B. J., and Robert, C. H. 1985. The dry deposition of nitric acid to grass, J. Geophys. Res. 90:2085-2090. Jacob, D. J., and Hoffmann, M. R. 1983. A dynamic model for the production of H+, NO3- and SO42- in urban fog, J. Geophys. Res. 88:6611-6621. Jacob, D. J., Munger, J. W., Waldman, J. M., and Hoffmann, M. R. 1986. The H2SO4-HNO3-NH3 system at high humidities and in fogs. 1. Spatial and temporal patterns in the San Joaquin Valley of California,J. Geophys. Res. 91:107~1088. Jonas, R., and Heinemann, K. 1985. Studies on the dry deposition of aerosol particles on vegetation and plane surfaces, J. Aerosol Sci. 16:463-471. Kan, C. S., Calvert, J. G., and Shaw, J. H. 1981. Mechanism of the ozone-ethene reaction in dilute N2/O2 mixtures near 1-atm pressure, J. Phys. Chem. 85:2359-2363. Killus, J. P., and Whitten, G. Z. 1982. A mechanism describing the photochemical oxidation of toluene in smog, Atmos. Environ . 16: 1973-1988. Kleindienst, T. E., Shepson, P. B., Edney, E. O., and Claxton, L. D. 1985. Peroxyacetyl nitrate: measure- ment of its mutagenic activity using the Salmonella/ mammalian microsome reversion assay, Mutat. Res. 157:123-128. Leone, J. A., Flagan, R. C., Grosjean, D., and Sein- feld, J. H. 1985. An outdoor smog chamber and modeling study of toluene-NOx photooxidation, Int. J. Chem . Kinet. 17: 177-216. Lesclaux, R. 1984. Reactivity and kinetic properties of the NH2 radical in the gas phase, Rev. Chem. Intermed. 5:347-392. Leuenberger, C., Ligocki, M. P., and Pankow, J. F. 1985. Trace organic compounds in rain. 4. Identi- ties, concentrations and scavenging mechanisms for phenols in urban air and rain, Environ. Sci. Technol. 19:1053-1058. Ligocki, M. P., Leuenberger, C., and Pankow, J. F. 1985a. Trace organic compounds in rain. II. Gas scavenging of neutral organic compounds, Atmos. Environ. 19:160~1617. Ligocki, M. P., Leuenberger, C., and Pankow, J. F. 1985b. Trace organic compounds in rain. III. Par- ticle scavenging of neutral organic compounds, Atmos. Environ. 19:1619-1626. Lindley, C. R. C., Calvert, J. G., and Shaw, J. H. 1979. Rate studies of the reactions of the (CH3)2N radical with 02, NO and NO2, Chem. Phys. Lett. 67:57~2. Liu, S. C., Cicerone, R. J., Donahue, T. M., and Chameides, W. L. 1977. Sources and sinks of atmospheric N2O and the possible ozone reduction due to industrial fixed nitrogen fertilizers, Tellus 29:251-263. Logan, J. A. 1983. Nitrogen oxides in the tropo- sphere: global and regional budgets, J. Geophys. Res. 88:10785-10807. Logan, J. A. 1985. Tropospheric ozone: seasonal Atmospheric Transformations of Automotive Emissions behavior, trends and anthropogenic influence, J. Geophys. Res. 90:10463-10482. Logan, J. A., Prather, M. J., Wofsy, S. C., and McElroy, M. B. 1981. Tropospheric chemistry: a global perspective, J. Geophys. Res. 86:7210-7254. Mac Leod, H., Aschmann, S. M., Atkinson, R., Tuazon, E. C., Sweetman, J. A., Winer, A. M., and Pitts, J. N., Jr. 1986. Kinetics and mechanisms of the gas phase reactions of the NO3 radical with a series of reduced sulfur compounds, J. Geophys. Res. 91:5338-5346. Magnotta, F., end Johnston, H. S. 1980. Photodisso- ciation yields for the NO3 free radical, Geophys. Res. Lett. 7:769-772. Martinez, R. I., and Herron, J. T. 1981. Gas-phase reaction of SO2 with a Criegee intermediate in the presence of water vapor, J. Environ. Sci. Health A16:623-636. Nielsen, T., Ramdahl, T., and Bjorseth, A. 1983. The fate of airborne polycyclic organic matter, Environ. Health Perspect. 47:103-114. Nielsen, T., Seitz, B., and Ramdahl, T. 1984. Occur- rence of nitro-PAH in the atmosphere of a rural area, Atmos. Environ. 18:215942165. Niki, H., Maker, P. D., Savage, C. M., and Brei- tenbach, L. P. 1979. Fourier transform infrared (FT-IR) studies of gaseous and particulate nitroge- nous compounds, In: Nitrogenous Air Pollutants (D. Grosjean, ed.), pp. 1-16, Ann Arbor Press, Ann Arbor, Mich. Niki, H., Maker, P. D., Savage, C. M., and Brei- tenbach, L. P. 1981a. An FT-IR study of mecha- nisms for the HO radical initiated oxidation of C2H4 in the presence of NO: detection of glycolal- dehyde, Chem. Phys. Lett. 80:499-503. Niki, H., Maker, P. D., Savage, C. M., and Brei- tenbach, L. P. 1981 b. A FT-IR study of a transitory product in the gas-phase ozone-ethylene reaction, J. Phys. Chem. 85:1024-1027. Noxon, J. F., Norton, R. B., and Marovich, E. 1980. NO3 in the troposphere, Geophys. Res. Lett. 7:125-128. Perner, D. 1980. HNO2 in urban atmospheres and its photochemical significance, In: Proceedings of the International Workshop on Test Methods and Assessment Procedures for the Determination of the Photochemical Degradation Behavior of Chemical Substances, Decem- ber 2-4, Berlin, pp. 159-173. Perner, D., Schmeltekopf, A., Winkler, R. H., John- ston, H. S., Calvert, J. G., Cantrell, C. A., and Stockwell, W. R. 1985. A laboratory and field study of the equilibrium N2O5 ~ NO3 + NO2, J. Geophys. Res. 90:3807-3812. Pitts, J. N., Jr. 1983. Formation and fate of gaseous and particulate mutagens and carcinogens in real and simulated atmospheres, Environ. Health Per- spect. 47:115-140. Pitts, J. N., Jr., Biermann, H. W., Atkinson, R., and Winer, A. M. 1984a. Atmospheric implications of simultaneous nighttime measurements of NO3 rad- icals and MONO, Geophys. Res. Lett. 11 :557-560. Pitts, J. N., Jr., Biermann, H. W., Winer, A. M., and Tuazon, E. C. 1984b. Spectroscopic identification
Roger Atkinson and measurement of gaseous nitrous acid in dilute auto exhaust, Atmos. Environ. 18:847-854. Pitts, J. N., Jr., Sanhueza, E., Atkinson, R., Carter, W. P. L., Winer, A. M., Harris, G. W., and Plum, C. N. 1984c. An investigation of the dark forma- tion of nitrous acid in environmental chambers, Int. J. Chem. Kinet. 16:919-939. Pitts, J. N., Jr., Atkinson, R., Sweetman, J. A., and Zielinska, B. 1985a. The gas-phase reaction of naphthalene with N205 to form nitronaphthalenes, Atmos. Environ. 19:701-705. Pitts, J. N., Jr., Sweetman, J. A., Zielinska, B., Atkinson, R., Winer, A. M., and Harger, W. P. 1985b. Formation of nitroarenes from the reaction of polycyclic aromatic hydrocarbons with dinitro- gen pentaoxide, Environ. Sci. Technol. 19:1115- 1121. Pitts, J. N., Jr., Sweetman, J. A., Zielinska, B., Winer, A. M., and Atkinson, R. 1985c. Determi- nation of 2-nitrofluoranthene and 2-nitropyrene in ambient particulate organic matter: evidence for atmospheric reactions, Atmos. Environ. 19:1601- 1608. Pitts, J. N., Jr., Wallington, T. J., Biermann, H. W., and Winer, A. M. 1985d. Identification and mea- surement of nitrous acid in an indoor environment, Atmos. Environ. 19:76~767. Pitts, J. N., Jr., Paur, H.-R., Zielinska, B., Arey, J., Winer, A. M., Ramdahl, T., and Media, V. 1986. Factors influencing the reactivity of polycyclic aro- matic hydrocarbons adsorbed on filters and ambient POM with ozone, Chemosphere 15:675085. Platt, U., Perner, D., Harris, G. W., Winer, A. M., and Pitts, J. N., Jr. 1980a. Observations of nitrous acid in an urban atmosphere by differential optical absorption, Nature 285:312-314. Platt, U., Perner, D., Winer, A. M., Harris, G. W., and Pitts, J. N., Jr. 1980b. Detection of NO3 in the polluted troposphere by differential optical absorp- tion, Geophys. Res. Lett. 7:89-92. Platt, U. F., Winer, A. M., Biermann, H. W., Atkinson, R., and Pitts, J. N., Jr. 1984. Measure- ment of nitrate radical concentrations in continental air, Environ. Sci. Technol. 18:365-369. Ramdahl, T., Zielinska, B., Arey, J., Atkinson, R., Winer, A. M., end Pitts,J. N.,Jr. 1986. Ubiquitous occurrence of 2-nitrofluoranthene and 2-nitropy- rene in air, Nature 321:425027. Roberts, J. M., Fehsenfeld, F. C., Liu, S. C., Bollin- ger, M. J., Hahn, C., Albritton, D. L., and Sievers, R. E. 1984. Measurements of aromatic hydrocarbon ratios and NOX concentrations in the rural tropo- sphere: observation of air mass photochemical ag- ing and NOX removal, Atmos. Environ. 18:2421- 2432. Sakamaki, F., Hatakeyama, S., and Akimoto, H. 1983. Formation of nitrous acid and nitric oxide in the heterogeneous dark reaction of nitrogen dioxide and water vapor in a smog chamber, Int. J. Chem. Kinet. 15:101~1029. Sehmel, G. A., Lee, R. N., and Horst, T. W. 1985. Hazardous air pollutants: dry-deposition phenom- ena, EPA-600/3-8~114 January 1985). Shepson, P. B., Edney, E. O., and Corse, E. W. 1984. 131 Ring fragmentation reactions in the photooxida- tions of toluene and o-xylene, J. Phys. Chem. 88:41220126. Shepson, P. B., Edney, E. O., Kleindienst, T. E., Pittman, J. H., Namie, G. R., and Cupitt, L. T. 1985a. The production of organic nitrates from hydroxyl and nitrate radical reaction with propy- lene, Environ. Sci. Technol. 19:849-854. Shepson, P. B., Kleindienst, T. E., Edney, E. O., Namie, G. R., Pittman, J. H., Cupitt, L. T., and Claxton, L. D. 1985b. The mutagenic activity of irradiated toluene/NOx/H2O/air mixtures, Environ. Sci. Technol. 19:249-255. Singh, H. B., Ludwig, F. L., and Johnson, W. B. 1978. Tropospheric ozone: concentrations and vari- abilities in clean remote atmospheres, Atmos. Envi- ron. 12:2185-2196. Slagle, I. R., Park, J.-Y., Heaven, M. C., and Gut- man, D. 1984. Kinetics of polyatomic free radicals produced by laser photolysis. 3. Reaction of vinyl radicals with molecular oxygen, J. Am. Chem. Soc. 106:4356 4361. Slinn, W. G. N. 1982. Some influences of the atmo- spheric water cycle on the removal of atmospheric trace constituents. In: Atmospheric Chemistry (E. D. Goldberg, ed.), pp. 57-90, Springer-Verlag, New York. Slinn, W. G. N., Hasse, L., Hicks, B. B., Hogan, A. W., Lal, D., Liss, P. S., Munnich, K. O., Sehmel, G. A., and Vittori, O. 1978. Some aspects of the transfer of atmospheric trace constituents past the air-sea interface, Atmos. Environ. 12:2055-2087. Sonnefeld, W. J., Zoller, W. H., and May, W. E. 1983. Dynamic coupled-column liquid chromato- graphic determination of ambient temperature va- por pressures of polynuclear aromatic hydrocar- bons, Anal. Chem. 55:275-280. South Coast Air Quality Management District. 1982. Draft Air Quality Management Plan, Revision, Appendix No. IV-A, Final 1979 Emissions Inven- tory for the South Coast Air Basin. Stockwell, W. R., and Calvert, J. G. 1983a. The mechanism of NO3 and HONO formation in the nighttime chemistry of the urban troposphere, J. Geophys. Res. 88:6673-6682. Stockwell, W. R., and Calvert, J. G. 1983b. The mechanism of the HO-SO2 reaction, Atmos. Envi- ron. 17:2231-2235. Su, F., Calvert, J. G., and Shaw, J. H. 1980. An FT-IR spectroscopic study of the ozone-ethene reaction mechanism in O2-rich mixtures, J. Phys. Chem. 84:239-246. Sweetman, J. A., Zielinska, B., Atkinson, R., Ram- dahl, T., Winer, A. M., and Pitts, J. N., Jr. 1986. A possible formation pathway for the 2-nitrofluoran- thene observed in ambient particulate organic mat- ter, Atmos. Environ. 20:235-238. Taylor, W. D., Allston, T. D., Moscato, M. J., Fazekas, G. B., Kozlowski, R., and Takacs, G. A. 1980. Atmospheric photodissociation lifetimes for nitromethane, methyl nitrite and methyl nitrate, Int. J. Chem. Kinet. 12:231-240. Terry Dana, M., Lee, R. N., and Hales, J. M. 1985.
132 Atmospheric Transformations of Automotive Emissions Hazardous air pollutants: wet removal rates and mechanisms, EPA-600/3-84-113 January 1985). Thiemans, M. W., and Schwartz, S. E. 1978. The fate of HS radical under atmospheric conditions, paper presented at the 13th International Conference on Photochemistry, Clearwater Beach, Fla. Tuazon, E. C., Atkinson, R., Plum, C. N., Winer, A. M., and Pitts, J. N., Jr. 1983. The reaction of gas phase N2O5 with water vapor, Geophys. Res. Lett. 10:953-956. Tuazon, E. C., Carter, W. P. L., Atkinson, R., Winer, A. M., and Pitts, J. N., Jr. 1984a. Atmo- spheric reactions of N-nitrosodimethylamine and dimethylnitramine, Environ. Sci. Technol. 18:49- 54. Tuazon, E. C., Sanhueza, E., Atkinson, R., Carter, W. P. L., Winer, A. M., Pitts, J. N., Jr. 1984b. Direct determination of the equilibrium constant at 298 K for the NO2 + NO3 ~ N2O5 reactions, J. Phys. Chem. 88:309~3098. Tuazon, E. C., Mac Leod, H., Atkinson, R., and Carter, W. P. L. 1986. c'-Dicarbonyl yields from the NOx-air photooxidation of a series of aromatic hydrocarbons in air, Environ. Sci. Technol. 20: 383-387. van Noort, P. C. M., and Wondergem, E. 1985. Scavenging of airborne polycyclic aromatic hydro- carbons by rain, Environ. Sci. Technol. 19: 1011 1048. Waldman, J. M., Munger, J. W., Jacob, D. J., Flagan, F. C., Morgan, J. J., and Hoffmann, M. R. 1982. Chemical composition of acid fog, Science 218:677-680. Wallington, T. J., Atkinson, R., Winer, A. M., and Pitts, J. N., Jr. 1986. Absolute rate constants for the gas-phase reactions of the NO3 radical with CH3SH, CH3SCH3, CH3SSCH3, H2S, SO2, and CH3OCH3 over the temperature range 280-350 K, J. Phys. Chem. 90:5393-5396. Winer, A. M., Atkinson, R., and Pitts, J. N., Jr. 1984. Gaseous nitrate radical: possible nighttime atmo- spheric sink for biogenic organic compounds, Sci- ence 224:156-159. Yamasaki, H., Kuwata, K., and Kuge, Y. 1984. Determination of vapor pressure of polycyclic aro- matic hydrocarbons in the supercooled liquid phase and their adsorption on airborne particulate matter, Nippon Kagaku Kaishi 13201329. Zielinska, B., Arey, J., Atkinson, R., Ramdahl, T., Winer, A. M., and Pitts,J. N., Jr. 1986. Reaction of dinitrogen pentoxide with fluoranthene, J. Am. Chem. Soc. 108:4126-4132.
