
Executive Summary
Consumer interest in health and self-care has expanded the market for a wide range of products including dietary supplements. Consumer use of dietary supplements has grown exponentially in the past decade, signifying increases for both traditional as well as new uses. As new formulations and products have come on the market, estimates of total sales have grown to $15.7 billion per year (Blendon et al., 2001; Nutrition Business Journal, 2000). As with conventional foods, many dietary supplements are probably safe when used as recommended. However, increased use of supplements, the broad spectrum of products that qualify as dietary supplements under the Dietary Supplement Health and Education Act of 1994 (DSHEA), and its requirement that the Food and Drug Administration (FDA) determine what is unsafe without requiring specific information on safety be presented by manufacturers prior to marketing, make regulation of dietary supplements a sizeable challenge.
THE COMMITTEE’S TASK
To monitor the continually evolving patterns of dietary supplement use and potential interactions with other consumed substances, FDA needs a cost-effective and scientifically based approach to considering the safety of dietary supplements. For these reasons, FDA turned to the Institute of Medicine (IOM) of The National Academies to provide a framework for evaluating the safety of dietary supplement ingredients. FDA requested that a committee of experts (1) develop a proposed framework for categorizing and prioritizing dietary supplement ingredients sold in the United States based on safety issues, (2) describe a process for developing a system of scientific reviews with specifications for evaluating the safety of dietary supplement ingredients, and (3) utilize the proposed framework to develop at least six scientific reviews or monographs as prototypes for the system after release of the proposed framework. The proposed framework is to include a methodology to review data with regard to the safety of dietary supplement ingredients, taking into consideration methods other expert bodies have used to categorize and review supplement safety and efficacy issues.
The proposed framework described in this report is now being released for comment and discussion to interested organizations and individuals; it is intended that at least one open forum will be held specifically to solicit input about the framework and its process for setting priorities and categorizing dietary supplement ingredients, as well as about the process for review and
TABLE ES-1 Current Status of Foods, Drugs, and Dietary Supplements under Food and Drug Administration (FDA) Regulation
|
Status |
Dietary Supplements |
Foodsa |
Food Additives |
New Drugsb |
|
Premarket approval required |
Noc |
Nod |
Yes |
Yes |
|
Risk-benefit analysis conducted by FDA prior to marketing |
No |
No |
No |
Yes |
|
Postmarket reporting or surveillance by industry required |
No |
No |
Rarely |
Yes |
|
Burden of proof for demonstrating safety or lack thereof |
FDA |
FDA |
Manufacturer |
Manufacturer |
|
a Foods (including conventional foods and dietary supplements), unlike drugs, are considered to be safe (reasonable certainty of no harm), and thus risk-benefit analysis is not applicable. b This description applies to “new” drugs. Many over-the-counter drugs that are not “new drugs” are regulated under FDA’s Over-The-Counter Drug Review procedures, which do not provide for postmarketing surveillance. c A 75-day premarket notification, but not premarket approval, is required for dietary supplements containing ingredients not marketed before 1994. d In 2001 FDA proposed a rule requiring marketers of food developed through biotechnology to notify the agency at least 120 days before commercial distribution and to provide information to demonstrate that the product is as safe as its conventional counterpart (FDA, 2001). |
||||
evaluation of information in the development of the prototype monographs. Based on a review of comments received and experience gained from completion of the prototype monographs, the proposed framework will be modified as appropriate. The revised framework will be released in a final report of the committee. This final report will also include the six prototype monograph reviews as examples of how the framework as revised can be implemented.
BACKGROUND
Current regulatory approaches to safety evaluation of dietary supplements in the United States are a product of several key pieces of legislation that span from the beginning to the end of the 20th century, culminating in the passage of DSHEA in 1994. The major controversy in considering the safety of dietary supplements has been whether supplements should be regulated as if they were conventional foods, food additives, or as drugs; foods are considered to be safe unless demonstrated otherwise, thus the government bears the burden to prove conventional foods are unsafe (see Table ES-1).
Since 1938 the drug industry has borne the burden of proof in establishing the safety of new drugs before they can be marketed, while the burden of establishing a food as unsafe has continued to remain with FDA. The 1938 Federal Food, Drug, and Cosmetic Act, which established the different burdens of proof, did not address when vitamins, minerals, and botanical products should be regulated as drugs as opposed to foods.
In 1958 the Food Additives Amendment defined food additives and provided that they must undergo a premarket approval process unless they were considered as generally recognized as safe (GRAS). FDA attempted to regulate the botanical industry by alleging that individual
|
BOX ES-1 The term dietary supplement:
Dietary supplements are further defined as products that are labeled as dietary supplements and are not represented for use as a conventional food or as a sole item of a meal or the diet. Supplements can be marketed for ingestion in a variety of dosage forms including capsule, powder, softgel, gelcap, tablet, liquid, or, indeed, any other form so long as they are not represented as conventional foods or as sole items of a meal or of the diet (FDCA, as amended, § 402). |
botanical products were unapproved food additives, an effort eventually struck down by the courts.
In the 1970s FDA tried to implement tighter regulations on vitamin and mineral supplements, but its actions were restricted by Congress via the 1976 Vitamins and Minerals Amendments. After FDA made another attempt to enforce stricter adherence to regulations in 1993, Congress acted further to contain FDA’s authority by passing DSHEA in 1994.
DSHEA established the first comprehensive definition of dietary supplements as foods (Box ES-1), along with legislative language defining procedures and regulations governing their marketing. Specifically, substances and products on the market in the United States prior to October 15, 1994 could continue to be marketed, but introduction of new products would require notification by the manufacturer to FDA 75 days prior to marketing. Most importantly, DSHEA established a regulatory framework for dietary supplements that defines FDA’s authority over these products. It establishes that dietary supplements are to be considered equivalent to foods in that they are assumed safe unless FDA has evidence that proves otherwise.
It is this postmarket burden of proof that makes FDA’s consideration of dietary supplement ingredients profoundly different from its consideration of substances such as food additives or drugs. Before marketing, food additives and drugs are required to undergo extensive safety evaluations by manufacturers that must prove them to be safe under conditions of use (see Table ES-1).
FINDINGS
In preparation for developing a framework and then prototype monographs of six selected dietary supplement ingredients, the committee was also charged with reviewing methods used by other expert bodies to categorize and review safety issues related to dietary supplements. The
committee reviewed published information about the approaches several organizations have taken to learn more about the limitations in the approaches, as well as their attributes. In reviewing these frameworks, the committee noted the following:
-
The purpose of the efforts varied substantially from organization to organization, focusing on quality, efficacy, safety, or a combination of these.
-
Most of the approaches were focused exclusively on botanical ingredients, others focused on medicinal substances. The reviewed approaches did not focus on the safety of dietary supplements of all types.
-
The approaches did not develop a systematic method to provide a categorized list of ingredients based on their need for more immediate attention, although several placed ingredients after review in general categories such as unsafe, safe, or unsafe for particular populations.
-
Often the approaches were not sufficiently detailed or transparent to give a complete picture of the data considered, the rationale behind the conclusions, and remaining unanswered questions regarding safety.
After reviewing approaches of other groups, the committee’s first objective was to develop a clear understanding of the purpose of the study and the expectations of FDA and those of industry. The second primary objective was to develop a collective understanding of what was meant by a “framework” and to identify common characteristics of effective frameworks already in place. To this end, the Committee defined a “framework” for safety evaluation of dietary supplement ingredients as “the processes by which FDA can screen, set priorities, and evaluate available information to make regulatory decisions regarding dietary supplement ingredients.”
In reviewing the methods used by other expert bodies to consider the safety of substances, and in reviewing the discussions with the sponsors and other interested representatives, the following attributes of an ideal framework were identified:
-
it must be workable and able to be integrated into FDA’s program of work;
-
it should provide guidance to organizing diverse information already available;
-
it should categorize the diverse substances classified as dietary supplements based on a scientifically valid metric;
-
it should establish a database for collection of information regarding potential safety concerns that can be updated as new information is available; and
-
it should provide a method to integrate diverse information into a prioritization scheme so that efforts and resources can be maximally directed toward those dietary supplement ingredients with the greatest safety concerns.
Once the definition and attributes of a safety framework were understood, the committee then identified key factors that could be used in such a framework. Following this, the committee developed a methodology to screen, set priorities, and then conduct critical evaluations of safety, with the results being collated into a monograph format. As a result of the review of types of information thought to be available for some dietary supplement ingredients, “guiding principles” for consideration during all of the steps were established.
Finally, the initial steps of the framework were applied to a variety of dietary supplement ingredients. Six diverse dietary supplement ingredients that would be expected to be flagged in the screening step were identified to serve as prototypes to test the proposed framework during the second phase of the study. During this phase, monograph reviews will be developed and put through the final critical safety evaluation step of the proposed framework.
PROPOSED FRAMEWORK FOR EVALUATING THE SAFETY OF DIETARY SUPPLEMENT INGREDIENTS
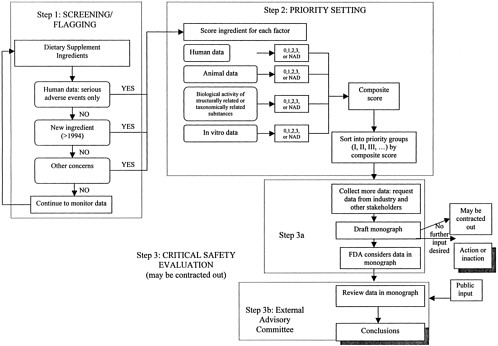
The Proposed Framework for Evaluatiing the Safety Dietary Supplement Ingredients consists of three steps: Step One, screening/flagging; Step Two, priority setting; and Step Three, critical safety evaluation (See Table ES-2 and Figure ES-1). Ideally, a critical safety evaluation for each dietary supplement ingredient could eventually be completed, but to best leverage available resources, it is necessary to determine which supplement ingredients warrant attention first. The first two steps in the process, screening/flagging and priority setting, are designed to set priorities for reviewing dietary supplement ingredients based on concern.
Key Factors Used in the Framework
In any scientific evaluation there are different types of data that are useful, or “factors” to consider, when collecting and sorting information (see Chapter 4). Different factors contribute to each step of the framework to a different degree, with different sources of information necessary to examine and evaluate the factors in the various steps of the processes proposed.
One key factor that should contribute to decision making at all three steps of the process is human data. Additional factors that should be considered are animal data, followed by information about the biological activity of structurally related and taxonomically related substances, and in vitro evidence of adverse effects. The potential for interactions among dietary supplement ingredients and other ingested substances or medical treatments are considered within each of these categories. An additional question considered only in the initial screening/flag step is whether the ingredient is new to the United States, as defined by DSHEA.
Other important factors integral to the framework are referred to as modifying factors. Whether particular subpopulations are especially vulnerable to the adverse effects of particular dietary supplement ingredients is considered with the data for each of the above factors. Second, the overall prevalence of use of the dietary ingredient in the United States is considered during the sorting process to increase the priority for review within a priority group.
Step One: Screening/Flagging
The screening process was developed on the premise that it is not feasible for FDA to extensively search for information about each and every dietary supplement ingredient immediately. Readily available information can be used to flag substances that warrant further attention, while maintaining enough sensitivity to minimize false negatives and not omit any items with potential safety concerns.
To flag substances warranting some level of attention, “yes or no” questions were developed to identify ingredients to undergo Step Two, priority setting:
TABLE ES-2 Overall Framework
|
Step in the Process |
Step One: Screening/Flagging |
Step Two: Priority Setting |
Step Three, Part A: Draft Monograph Preparation and Monograph Review by the Food and Drug Administration (FDA) |
Step Three, Part B: Critical Safety Evaluation |
|
Which ingredients |
All ingredients are considered “New” ingredients are automatically flagged |
Ingredients flagged in screening step |
Ingredients with highest priority based on Step Two ranking |
Monographed ingredients for which a decision is not clear cut or for which further input is desired |
|
Completed by |
FDA |
FDA |
FDA or contractor |
External advisory committee |
|
Factors and modifiers used |
Human data: serious adverse events only Other concerns,a as they come to FDA’s attention |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use (modifies other factors) Prevalence of use (modifies priority ranking) |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use considered with other factors |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use (considered with other factors) |
|
Level of information search |
Easily obtainable information (see Table 4–1) |
Literature search is more comprehensive |
Comprehensive Request industry data and data from other stakeholders |
Comprehensive Public input |
|
Depth of evaluation |
Low level evaluation: is there evidence suggesting a concern may exist? |
Weighting based on evidence of possible risk, potential seriousness of harm, and relative importance of factor |
Comprehensive: totality of evidence is considered, including data requested from industry and other stakeholders |
Totality of evidence; monograph reviewed and revised |
|
Goal |
Ingredients warranting further investigation are flagged |
Table of ingredients sorted into priority groups for further evaluation |
Monograph AND FDA decision for action/inaction OR Referral to external advisory committee |
Monograph with conclusions of external advisory committee |
|
a The term “other concerns,” as described in Chapter 3, encompasses concerns FDA becomes aware of without extensive information searching. These may include concerns expressed by other regulatory agencies, concerns expressed in secondary literature, or concerns expressed by other organizations. |
||||
-
Has a 75-day new ingredient notification been filed with FDA for the ingredient in question?
-
For the ingredient in question, are there potentially serious1 adverse events in humans reported through MedWatch, poison control centers, or clinical studies that illustrate a pattern (in terms of the type of incident reported) that are well-documented in the medical literature, or that may be plausibly linked to the dietary supplement ingredient? Does the number of serious adverse events reported in humans appear high compared to the ingredient’s prevalence of use? Does it seem plausible that particular subpopulations are particularly susceptible to serious adverse events?
-
Has the ingredient been brought to FDA’s attention because of concerns other than new ingredient status or human adverse event data described above? A preliminary evaluation of concerns that have come to FDA’s attention will allow FDA to determine which of these ingredients should move into priority setting.
In keeping with the philosophy that the screening step should be relatively simple and straightforward, answering these questions does not involve evaluation or weighting of the evidence. A single “yes” to any one question is sufficient to move the ingredient to the next step.
Step Two: Priority Setting
The goal of the priority-setting process (Step Two) is to identify those dietary supplement ingredients that require the most immediate attention of FDA for in-depth safety evaluation. The priority-setting process differs from the initial screening process in four fundamental ways:
-
additional factors are considered;
-
additional information is obtained;
-
the evidence of possible risk, as well as the seriousness of potential harm is judged to some degree; and
-
the different factors are weighted differently, based on importance.
Scoring and Sorting the Data
A sorting matrix is proposed to consider the importance of the information available for each key factor and to sort the ingredients accordingly. The available data for each of the four key factors (human data, animal data, biological activity of structurally related and taxonomically related substances, and in vitro data) are examined for all dietary supplement ingredients that were flagged in the screening process. A judgment is made of both the potential seriousness of the physiological effect suggested by the data and the strength of the evidence that the effect may occur. Based on this judgment, the data for each factor are judged and then assigned either a numerical value of 0 to 3, or NAD (no appropriate data), to indicate the evidence of possible risk and the potential seriousness of harm suggested by the data. A score of 3 is assigned for each factor where the data suggest both a potentially serious and very relevant harm and strong
evidence of possible risk, or there is strong evidence suggesting a possible risk of serious drug interaction. A 0 is assigned when there is strong evidence that there is no potential serious harm. Scores of 1 and 2 are not explicitly defined but result from a judgment of relative concern, as described in Chapter 5. For each ingredient, the numerical values for each factor are recorded (see Table ES-3 for a matrix listing fictitious ingredients). The ingredients in the matrix are then sorted into a list in descending priority, based on the scores, as described in the next section.
TABLE ES-3 Matrix of Scores Used in Establishing Relative Priority Among Dietary Supplements
|
Ingredient Name |
Human Data |
Animal Data |
Biological Activity of Structurally Related or Taxonomically Related Substances |
In Vitro Data |
|
Yellow plant extract |
3 |
1 |
2 |
2 |
|
Vitamin X |
2 |
NAD |
2 |
NADa |
|
Animal tissue |
2 |
1 |
1 |
1 |
|
a NAD=no appropriate data. |
||||
Sorting the Ingredients by Scores
After data for an increasing number of dietary supplement ingredients are reviewed and a numerical score is assigned for each of the key factors, the list of ingredients can be sorted into categories of relative priority (called Priority Groups) based on the assigned scores.
In the proposed scheme, priority setting is accomplished through a multi-step sorting mechanism that reflects the importance of the different numerical scores and the hierarchical importance of the different factors when considering the safety of dietary supplement ingredients. Two of the key factors—human data and animal data—are placed at the top of hierarchy of data types. Ingredients with scores of 3 in both of these factors are therefore placed in the highest priority category, Priority Group I, as illustrated in Table ES-4.
Ingredients that were assigned a score of 3 for the human data, but not for the animal data, are categorized as Priority Group II. Ingredients that were assigned a score of 3 in animal data, but not in human data, are categorized as Priority Group III. Priority Group IV includes ingredients that were assigned a score of 3 for either the structure/taxonomy or the in vitro data, but not in the human or animal data. Finally, Priority Group V includes ingredients that did not receive a score of 3 in any of the key factors.
This priority-setting approach of scoring and then sorting into priority groups allows FDA to consider the different factors independently and individually for each ingredient, rather than having to compare them to all the ingredients that are being considered.
Step Three: Critical Safety Evaluation
The screening/flagging and priority-setting steps outlined in the previous sections result in the identification of priority groups of supplement ingredients based on level of priority for in-depth review. The evaluation process begins with completing the data collection, as shown in the flowchart (Figure ES-1). Much data will already have been obtained during the priority-setting step, but efforts should now be expanded to systematically search for relevant information from
TABLE ES-4 Matrix for Priority Establishment Based on Factor Analysis
|
Priority Group |
Human Data |
Animal Data |
Bioactivity of Structurally Related or Taxonomically Related Substances |
In Vitro Data |
Number of Combinations (Total=625) |
Characteristics of Priority Group |
|
I |
3 |
3 |
|
|
25 |
Two 3s in first two factors |
|
II |
3 |
|
|
|
100 |
3 in human data |
|
III |
|
3 |
|
|
100 |
3 in animal data |
|
IV |
|
|
3 |
|
144 |
One or two 3s in structure/ taxonomy or in vitro factors |
|
|
|
|
|
3 |
|
|
|
V |
|
|
|
|
256 |
No 3s in any key factor |
additional sources as well. At this stage in the review, there is sufficient concern about the safety of the ingredient to justify FDA’s request for more information to be volunteered by the industry, including data on safety. All the available information collected should be collated into a “Dietary Supplement Ingredient Safety Review Monograph,” a task that might be appropriate for FDA to contract out if adequate resources are not available to prepare it internally.
After preparing the monograph that describes the available information and where information is missing, FDA should consider the totality of the scientific information collected. FDA should decide, based on the weight of the evidence, whether information in the monograph is conclusive enough to clearly indicate that action or inaction is appropriate. If the data are not sufficiently clear to make action or inaction obvious, or for any other reason FDA deems that external opinions may be valuable, an external advisory committee can be brought in to work on the issue.
After reviewing the information collected in the monograph and obtained from public comment sessions, the external advisory committee should revise the monograph to create a picture of the scientific information available. The advisory committee should evaluate the available scientific information and reach conclusions where possible, describing what is known about the safety of the ingredient based on the weight of the scientific evidence. The advisory committee’s conclusions should include comments about the risk and hazards that may be associated with the general population ingesting the ingredient, as well as risks that may be of particular concern to certain segments of the population. After the advisory committee’s conclusions are shared with FDA, the monograph and the advisory committee’s conclusions should be posted on FDA’s website.
Guiding Principles to Follow in Evaluating Data in the Framework
“Guiding principles” were developed to address the qualitative aspects of data review in the critical safety evaluation and for consideration when scoring during the priority-setting step. These guiding principles are:
-
A credible report of a serious adverse event in humans that is associated with use of a dietary supplement ingredient raises concern about the ingredient’s safety and requires further information gathering and evaluation. A final judgment on the safety of the supplement ingredient, however, will require a consideration of the totality of the evidence. Historical use should not be used as prima facie evidence that the ingredient does not cause harm. It is appropriate, however, to give considerable weight to a lack of adverse events in large, high-quality, randomized clinical trials or cohort studies (prospective or retrospective) that are adequately powered and designed to detect adverse effects.
-
Even in the absence of adverse events in humans, evidence of harm from laboratory animal studies can be indicative of potential harm to humans. This indication may assume greater importance if the route of exposure is similar (e.g., oral), the formulation is similar, more than one species shows the same toxicity, and the general characteristics of good animal studies as described in Chapter 4 are met. Particular weight is placed on evidence of certain types of delayed effects that are less likely to be detected in humans, such as cancer, developmental toxicity (including teratogenicity), and reproductive toxicity.
-
The presence of constituents structurally similar to known toxic or potentially harmful compounds and plants taxonomically related to known toxic plants suggests increased risk, and therefore higher priority, unless there is evidence that the compound is not toxic or harmful, the compound is present in concentrations that will not lead to harm, or there is other evidence supporting the safety of the ingredient.
-
In vitro studies can serve as signals of potential harmful effects in humans, but not as independent indicators of risk of harm unless an ingredient causes an effect that has been associated with harmful effects in animals or humans, and there is evidence that ingredient or its metabolites reach physiological sites where harm may occur. Alone, they should serve only as hypotheses generators and as indicators of possible mechanisms of harm when the totality of the data from the different factors is considered.
Attributes of the Proposed Framework
There are a number of attributes of the framework proposed, and there are also a few limitations. This framework integrates a variety of available evidence about safety, balancing the value of different types of evidence and also integrating usage information to enhance the public-health impact of the work. Using the framework, FDA can be both proactive and reactive, as well as provide an open and transparent process helpful to the general public and the industry.
The proposed framework focuses on how to consider the safety of dietary supplement ingredients rather than offering guidance on how to consider their benefits and role in health. This was a key point of FDA’s request to IOM, and is appropriate since dietary supplements are regulated as foods that must be safe, rather than as drugs requiring a risk-benefit analysis. A strength of the proposed framework is that it allows the incorporation of several different types
of data that may be available, providing a mechanism to evaluate the totality of the available data. The priority-setting step weights the different kinds of data available.
When considering the various types of data, the framework outlines how to consider both the strength of the evidence and the seriousness of harm suggested by the evidence. The evidence of possible risk incorporates both the methodological quality and the quantity of the available evidence, components that are important when considering any scientific data. Considering the potential seriousness of harm enables higher priority to be given to items that are of most concern because of their potential to adversely affect human health.
In addition to the methodology outlined for integrating various types of information, the proposed framework is also practical because it allows FDA to respond to new information in that the categorization of priorities easily changes to reflect new data.
Limitations are also inherent in the proposed framework. By definition, this framework cannot be used to consider the possible benefits of consuming dietary supplements. Another limitation is that, as with any evaluation of dietary supplement ingredients under the current regulatory scheme, this framework’s evaluation of safety depends on publicly available data or data made available voluntarily by industry. A major component of this framework in particular is human data, which unfortunately can be highly variable in quality and quantity.
INGREDIENTS FOR PROTOTYPE MONOGRAPH REVIEWS
The second phase of FDA’s charge to the Committee on a Framework for Evaluating the Safety of Dietary Supplements is, after release of the proposed framework for comment, to develop at least six scientific reviews as prototypes for the system outlined in the framework. The six supplement ingredients selected for the prototype reviews include the following (in no particular order other than alphabetical): chaparral, chromium picolinate, glucosamine, melatonin, saw palmetto, and shark cartilage. These six ingredients were selected to fulfill specific criteria. They include at least one botanical, one vitamin or mineral, one animal product, and one hormonal product. The selected ingredients also include substances for which a range of different types of available information and a range in quality of information available is anticipated. Ingredients included are those that would be expected to be flagged in the screening process and therefore enter the priority-setting step. Based on very preliminary data, it is also expected that this list includes substances that when initially reviewed in the priority-setting step, would not all be placed in the top priority category.
SECOND PHASE OF THIS STUDY
The second phase of this study will be to oversee the preparation of prototype monographs on the six ingredients, following the framework outlined in this report. As outlined in Chapter 6, industry will be requested to provide safety information about the ingredients undergoing in-depth safety evaluation. Panels will be organized to review information included in draft monographs and to arrange for public input on the evidence about safety.
During this phase, comments regarding the proposed framework detailed in this report will be solicited, reviewed, and revisions made as appropriate, followed by release of a revised framework and the six prototype monographs as examples of the safety evaluation envisioned.












