
6
Emerging Tools and Technology for Countering Resistance
OVERVIEW
Most efforts to overcome problems resulting from microbial resistance have been aimed at either extending the useful life of current drugs or developing new drugs. But the first approach, which has typically involved developing slightly different chemical derivatives, has provided only marginal gains, and the second approach has in recent decades produced only a single new chemical class of antibiotics. And while many observers suggest that the genomics revolution may be opening promising new avenues for exploration, it has yet to populate the pharmaceutical development pipeline with new classes of compounds.
The focus of this session of the workshop was on examining advances in our understanding of the genetics and biochemistry of a variety of pathogens, and on exploring how this knowledge might lead to novel approaches to developing new drugs and other tools and technologies to help counter the spread of antimicrobial resistance.
Among the antibiotics discussed was the important family of ß-lactams, which include penicillin, methicillin, oxacillin, and the newer cephalosporins. Recent research has revealed at least four strategies by which bacteria develop resistance to these antibiotics. The most common tactic involves certain enzymes, called ß–lactamases, that break apart critical components of the drugs. Knowing the details of these mechanisms may provide scientists with new targets to attack in designing drugs that can circumvent a microbe’s natural defenses.
Another research group is taking a radically different approach to drug design. Since most bacteria enter humans at a mucous membrane site, such as the upper and lower respiratory tract or the intestines, these locations act as reservoirs for many pathogens. However, there currently are no drugs that can kill pathogens on mucous membranes without killing surrounding normal bacteria as well, and physicians therefore must wait for infection to occur systemically before treating the patient. If it were possible to safely deplete this disease reservoir on mucous membranes, then it might be possible to markedly lower the incidence of infections. Toward this end, the scientists have developed a new type of reagent, called lytic enzymes, that can prevent infection by specifically destroying pathogenic bacteria on mucous membranes. The enzymes might prove especially useful in hospitals, nursing homes, daycare centers, and other locations where bacterial infections often run rampant.
Participants also examined how the current regulatory approval process for moving new drug candidates to market might be modified to help reduce problems of antimicrobial resistance. Drug developers now must conduct extensive clinical trials to evaluate whether their agent achieves a clinical cure; that is, whether it frees recipients of symptoms. But this marker may not show whether the drug actually killed all of the pathogens—an important requirement for minimizing the emergence of resistance. One suggestion is to examine the pharmacokinetics and pharmacodynamics of new drugs as a complementary part of the approval process. This technical approach, known as PK/PD, is built on taking regular cultures from a person receiving a therapeutic agent and determining when pathogenic microbes are no longer present. In this way, the technology offers a direct measure of the agent’s ability not only to cure the patient but also to completely eliminate the pathogen.
EVOLUTION OF MULTIPLE MECHANISMS OF RESISTANCE TO ß-LACTAM ANTIBIOTICS
Dasantila Golemi-Kotra, Ph.D., Sergei Vakulenko, M.D., Ph.D., and Shahriar Mobashery, Ph.D.
Departments of Chemistry, Pharmacology and Biochemistry and Molecular Biology Institute for Drug Design, Wayne State University, Detroit, MI
Major discoveries in antibiotics were made in succession from the 1950s through the 1970s, an era that has come to be known as the “Golden Age of Antibiotics.” These accomplishments created a sense of euphoria in the medical community as it perceived that bacterial infections were curable.
Victory over bacteria was declared and financial resources were redirected toward more pressing scientific questions. By the late 1980s to the early 1990s, many pharmaceutical companies stopped research for discovery and development of new types of antibiotics, a trend that was shadowed by federal agencies such as the National Institutes of Health that showed more inclination to support studies of non-microbial systems during that period.
Meanwhile, liberal use of antibiotics in the clinics, in agriculture, in aquaculture, and in animal husbandry was facilitating a quiet revolution in microbial populations. These events resulted in antibiotic-resistant organisms that were perfectly treatable a decade or two earlier. These organisms were included among those defined as re-emerging infectious agents (IOM, 1992; Heymann and Rodier, 2001). While this trend was progressing steadily over the past decades, previously unknown infectious agents were also being discovered (WHO, 1996; Desselberger, 2000).
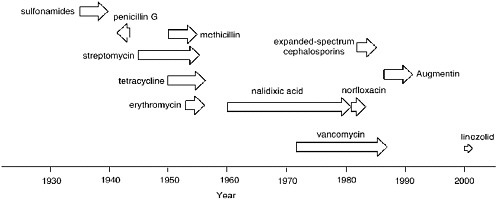
Acquired resistance to antibiotics can develop rapidly. As depicted in Figure 6-1, it takes typically a mere one to four years for clinical resistance to emerge for any antibiotic. The beginning of each arrow in Figure 6-1 indicates the time point when the given antibiotic was introduced to the clinic and the tip of the arrow indicates when resistance to it emerged.
Whereas the data of Figure 6-1 speak for themselves, three examples are worthy of a brief comment. The arrow is in reverse for penicillin G, since the first case of resistance to penicillin was reported two years prior to the first large-scale clinical use of penicillin in 1942 (Abraham and Chain, 1940). The resistant organism harbored a ß-lactamase, an enzyme that to the present day is the major cause of resistance to ß-lactam antibiotics. Figure 6-1 would seem to indicate that resistance to nalidixic acid took roughly 20 years to develop. This is true in part due to the antibiotic glut in the late 1950s into the 1960s. There were so many effective antibiotics that nalidixic acid was not used. Indeed, when quinolone and fluoroquinolone antibiotics, the structural descendants of nalidixic acid, were introduced to the clinic in earnest in the middle to late 1980s, resistance to them developed very rapidly.
The example of the first clinically used oxazolidinone, linezolid (Zyvox; Pharmacia, Inc.), is noteworthy. Linezolid was approved for clinical use in the United States in April 2000. It is an especially active antibiotic against infections caused by multi-drug-resistant gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE) and penicillin-resistant Streptococcus pneumonia (Fung et al., 2001). Linezolid binds the bacterial ribosome reversibly and inhibits initiation of protein biosynthesis by preventing the formation of a ternary complex among tRNAfMet, mRNA, and the ribosome (Swaney et al., 1998). The propensity of emergence of resistance to linezolid in S. aureus and S.
epidermis is less than 10-9 (Kaatz and Seo, 1996). Studies have revealed that single-point mutations clustered in the DNA region encoding the central loop of the domain V of 23S rRNA cause resistance (Swaney et al., 1998; Gonzales et al., 2001; Prystowsky et al., 2001; Tsiodras et al., 2001). Because of the critical importance of the ribosome for survival of any organism, there exists redundancy in the genes for the ribosomal RNA to ensure that mutations in any one gene would not prove fatal. It would appear that mutation of one to three of these genes is necessary for manifestation of resistance to linezolid, and this indeed has happened. Clinical resistance to linezolid emerged in a mere seven months after its introduction to the clinic (Fung et al., 2001).
In principle, the genes that would confer resistance to an antibiotic could be acquired or could evolve. Gene acquisitions could take place as DNA sequences are shared among organisms by conjugation, transformation, and transduction or by classical recombination and transposition (Bennett, 1999). However, the issue of evolution of a resistance determinant is less commonly appreciated (Mobashery and Azucena, 2002). Many stretches of genes encode proteins that serve adventitious functions in addition to their physiological ones. These genes may be duplicated, and the fates of the copies could take them in disparate evolutionary tangents. One would encode the protein for the physiological function and the other may evolve to confer resistance to the antibiotic. Despite the fact that bacteria have enzymes for repair of genetic damage, random mutations do occur in bacteria and the rate is typically 10-10. Considering the fact that the sizes of bacterial genomes are large (e.g., Escherichia coli K-12 has 4.6 × 106 base pairs) and that population sizes of bacteria in infections can also be very large (107 cells/ml for infections of blood and 109 cells/ml for infections of tissues), the opportunities for emergence of mutant variants are plentiful. These three factors taken together would indicate that there may be as many as 105 to 106 mutations per milliliter of bacteria growth, hence these populations are not homogeneous (Mobashery and Azucena, 2002).
These mutations occur at random throughout the genome. Some are silent, others are lethal, yet many may cause an incremental change in the organism that would not result in undue survival hardship to the organism. In the face of the challenge by a given antibiotic, should one or more of these mutant variants be better fit to survive, it would be selected at the expense of the susceptible organisms. The given selected gene for the protein that confers resistance may continue to evolve as a resistance determinant from that point on. For all members of the seven major classes of antibiotics (i.e., ß-lactams, aminoglycosides, fluorquinolones, glycopeptides, macrolides, tetracyclines, and sulfonamides) there are documented mechanisms for resistance. Often, there are multiple known mechanisms for resistance to each.
A full discussion of the various mechanisms of resistance to all known antibiotics is outside the scope of this report, although such reviews have appeared in the literature (Kotra et al., 2000; Walsh, 2000; Poole, 2001; Golemi-Kotra and Mobashery, 2002; Mobashery and Azucena, 2002). In this report we concentrate on the case of the mechanisms of resistance to ß-lactam antibiotics (i.e., penicillins, cephalosporins, carbapenems, etc.). As will be presented below, this has been a fertile field for evolution of resistance, which represents a well-studied microcosm of diversity that must also exist in the microbial world for other antibiotics that await discovery.
There are at least four documented mechanisms for resistance to ß-lactam antibiotics (see Table 6-1). The first and the most common mechanism is the occurrence of ß-lactamases, enzymes that hydrolyze and destroy ß-lactam antibiotics. The reaction of ß-lactamases with a penicillin is given in Figure 6-2. Over 340 distinct ß-lactamases have been identified in bacteria (Bush and Mobashery, 1998; Bush, 1999). These enzymes fall into four classes: A to D. Enzymes of class B are zinc-dependent, and the remaining three classes are enzymes that depend on a catalytic serine in their mechanisms of action. These serine-dependent enzymes experience acylation at the serine by the substrate, and the acyl-enzyme species subsequently under-goes an enzyme-promoted deacylation that liberates the enzyme for additional catalytic cycles and releases the product of the reaction (Bush and Mobashery, 1998; Massova and Mobashery, 1998).
The three classes of ß-lactamases that pursue this strategy all share the acylation event but differ significantly in the deacylation steps. The differences include the direction of the approach of the hydrolytic water and the mechanism of the enzyme-promoted step. In the case of class A enzymes, a glutamate (Glu-166) promotes the hydrolytic water from one face of the acyl-enzyme species (Strynadka et al., 1992; Miyashita et al., 1995; Maveyraud et al., 1998; Mourey et al., 1998). The class C enzymes pro
TABLE 6-1 Four Known Mechanisms of Resistance to ß-lactam Antibiotics
|
1. The catalytic function of ß-lactamases (gram-positive and gram-negative) 2. DD-Transpeptidases that resist acylation by ß-lactam antibiotics (gram-positive; MRSA) 3. Loss of porins or their alteration to reduce penetration into periplasm (gram-negative) 4. Selection of the LD-transpeptidase activity (documented only for E. faecium) |
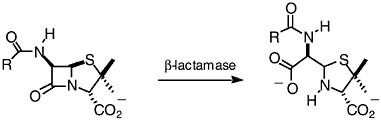
FIGURE 6-2 Hydrolytic reaction of ß-lactamases results in products that lack antibacterial activity.
mote the hydrolytic water from the opposite face and they use a different residue (a tyrosine) along the substrate amine as the basic entities (Bulychev et al., 1995, 1997; Patera et al., 2000). Class D enzymes are different yet. These enzymes, in contrast to the cases of enzymes of classes A and C, pursue a symmetric mechanism for the two-step reaction by having a highly unusual carbamylated lysine that serves the role of a base for both steps of catalysis (Maveyraud et al., 2000; Golemi et al., 2001; Maveyraud et al., 2002). These observations argue for independent evolutions for all four classes of ß-lactamases (Massova and Mobashery, 1998). Structural evidence argues that the family of penicillin-binding proteins (PBPs) is related to that of ß-lactamases (Kelly et al., 1998; Massova and Mobashery, 1998). A sequence alignment of all known classes of ß-lactamases and PBPs has supported the mechanistic assertion for independent evolution of each ß-lactamase. It indicated that evolution of ß-lactamases took place relatively recently on the evolutionary time scale and it did so at branching points for diversification of the biosynthetic PBPs (Massova and Mobashery, 1998).
These enzymes are extremely effective at what they do. It has been shown that class A and class C ß-lactamases have reached catalytic perfection in that they operate at the diffusion limit (Hardy and Kirsch, 1984; Bulychev and Mobashery, 1999). Many others also function at or near the diffusion limit. We have shown that the class D OXA-10 ß-lactamase from Pseudomonas aeruginosa is present in the periplasmic space of clinical strains in concentrations of 4–15 µM (Golemi et al., 2001). This will amount to as many as 1,200 molecules of the enzyme per bacterium. Considering the fact that each molecule of the enzyme turns over 1,500 molecules of cloxacillin (a penicillin) per second, each organism destroys 1.8 million molecules of the drug per second.
The second mechanism for resistance to ß-lactam antibiotics is seen in gram-positive bacteria. The case of methicillin-resistant Staphylococcus aureus (MRSA) exemplifies this category. S. aureus has acquired a penicillin-binding protein (PBP 2a) that has the ability to carry out the functions of the other four PBPs that are normally present in this organism (Pinho et al.,
2001a). However, this PBP resists the inhibitory properties of all clinically used ß-lactam antibiotics. Hence, MRSA survives the treatment by these commonly used antibiotics (Roychoudhury et al., 1994; Pinho et al., 2001b). In light of the fact that MRSA has become resistant to many other classes of antibiotics, it is currently a serious clinical problem. There are a number of projects in the pharmaceutical industry for development of specific cephalosporins that bind to PBP 2a of S. aureus (Chamberland et al., 2001; Hebeisen et al., 2001; Johnson et al., 2002; Wootton et al., 2002).
Antibiotics must penetrate the bacterial envelope to reach their targets. The protective effect of the gram-negative envelope is more than that of the gram-positive organisms in light of the presence of the outer membrane in the former. The outer membrane is a bilayer sheath that encloses the entire organism. Nutrients penetrate this membrane by traversing the porin channels. Porins are integral outer-membrane proteins that create water-filled channels through the outer membrane. ß-lactam antibiotics also need to go through these channels, which set an upper limit of 700–1000 Da for the antibiotic size. It has been known that some organisms may eliminate certain porins or may acquire alterations to the structure of a specific type of porin to limit penetration of the antibiotic into the periplasmic space. This mechanism has been shown to be operative against certain carbapenem antibiotics (Martinez-Martinez et al., 2000; Ochs et al., 2000). It is not very common because the loss of porins or their structural alteration would have implications for potential survival of the organism as well.
The fourth mechanism of resistance to ß-lactam antibiotics is seen only in Enterococcus faecium. Bacteria are required to cross-link the peptidoglycan strands of their cell wall in order to be viable. The cross-linking reaction is carried out by some PBPs referred to as DD-transpeptidases, because the enzymes link one strand of the peptidoglycan to the site of the acyl-D-Ala-D-Ala of the second strand. These enzymes recognize the unusual D-Ala-D-Ala entity of the peptidoglycan structure in their substrates. It would appear that E. faceium has evolved an entirely new PBP that is referred to as LD-transpeptidase. This enzyme attaches the first peptidoglycan strand to the L-amino acid adjacent to the D-Ala-D-Ala moiety, hence it is referred to as the LD-transpeptidase (Mainardi et al., 2000). This is essentially a bypass mechanism, since the LD-transpeptidase is insensitive to ß-lactam antibiotics.
Concluding Remarks
We have disclosed in this report that nature has devised at least four strategies for resistance to ß-lactam antibiotics in bacteria. Of these, the most prevalent is the occurrence of ß-lactamases. Four distinct and independent mechanisms have evolved for the catalytic functions of these en
zymes. Individually, these enzymes have undergone additional diversification. For example, a total of 102 variants of the TEM-1 ß-lactamases from E. coli have been reported as of January 2002. These observations provide strong evidence that random mutation and selection would lead along a different evolutionary tangent depending on when the event takes place, in what organism, and what resources were available to the organism.
USING PHAGE LYTIC ENZYMES TO CONTROL ANTIBIOTIC-RESISTANT PATHOGENIC BACTERIA ON MUCOUS MEMBRANES
Vincent A. Fischetti, Ph.D.*
Rockefeller University, New York, NY
Nearly all bacteria infect at a mucous membrane site (upper and lower respiratory, intestinal, urogenital, and ocular). In addition, the human mucous membranes are a reservoir for many pathogenic bacteria found in the environment (e.g., pneumococci, staphylococci, streptococci) some of which are resistant to antibiotics. In most instances, it is this reservoir that is the focus of infection in the population (Coello et al., 1994; de Lencastre et al., 1999; Eiff et al., 2001). To date, except for polysporin and mupirocin ointments, there are no anti-infectives that are designed to control pathogenic bacteria on mucous membranes; we must first wait for infection to occur before treating. Because of the fear of developing resistance, antibiotics are not indicated to control the carrier state of disease bacteria. It is clear, however, that by reducing or eliminating this human reservoir, the incidence of disease in the community will be markedly reduced. Our laboratory has developed enzymes that are designed to prevent infection by safely and specifically destroying disease bacteria on mucous membranes. For example, enzymes specific for S. pneumoniae and S. aureus may be used nasally to control these organisms in day care centers, hospitals, and nursing homes to prevent or markedly reduce both transmission and serious infections caused by these bacteria.
We accomplish this by capitalizing on the efficient system developed by bacteriophage to kill bacteria. When bacteriophage infect their host bacteria to produce progeny virus particles, they are faced with a problem: to release the progeny phage particles trapped in the bacterium at the end of the replicative cycle. They solve this problem by producing an efficient
enzyme termed lysin that rapidly degrades the cell walls of the infected bacteria to release the phage progeny (Young, 1992).
We have identified and purified these phage-encoded enzymes and found that when applied externally to gram-positive bacteria, the bacteria are killed seconds after contact (Loeffler et al., 2001; Nelson et al., 2001). For example, 107 group A streptococci could be reduced to an undetectable level ten seconds after enzyme addition. To date, except for chemical agents, there is no biological compound known that can kill bacteria this quickly. Such phage lytic enzymes are the culmination of billions of years of development by the bacteriophage during their association with bacteria.
Enzyme Structure
A feature of those phage lytic enzymes that have been characterized so far is their two-domain structure (Diaz et al., 1990; Garcia et al., 1990). Generally, the N-terminal domain contains the catalytic activity of the enzyme that will cleave one of the four major bonds in the peptidoglycan of the bacterial cell wall. This activity may be an endo-beta-N-acetylglucosaminidase or N-acetylmuramidase (lysozyme), both of which act on the sugar moiety, an endopeptidase which acts on the peptide cross bridge, or an N-acetylmuramyl-L-alanine amidase (or amidase) which hydrolyzes the amide bond connecting the sugar and peptide moieties (Young, 1992). Of the phage lytic enzymes that have been reported thus far, the great majority are amidases. The C-terminal domain of phage lytic enzymes has specificity for a cell wall substrate (Garcia et al., 1988; Lopez et al., 1992, 1997). Thus, unless the binding domain binds to its wall substrate the catalytic domain will not cleave, offering specificity to the enzyme. The reason for this specificity was not apparent at first, since it seemed counter-intuitive that the phage would specifically design an enzyme that was lethal for its host organism. However, as we learned more about how these enzymes function, the possible reason for this specificity became apparent (see below, Resistance).
Mode of Action
By thin section electron microscopy of phage enzyme-treated bacteria, it appears that the enzymes exert their lethal effects by digesting the peptidoglycan, forming holes in the cell wall. This results in extrusion of the cytoplasmic membrane and, ultimately, hypotonic lysis (see Figure 6-3). Isolated cell walls treated with lysin are cut into pieces (see Figures 6-4 and 6-5), verifying the results seen with intact bacteria.
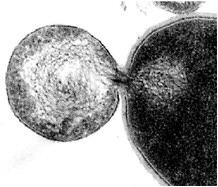
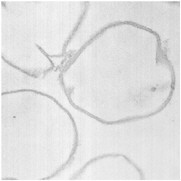

FIGURES6-3–6-5 Effects of lysin on whole bacteria and cell walls. (Fig 6-3) Thin section electron micrographs of whole group A streptococci treated with C1 phage lytic enzyme for 15 seconds. A bacterium is seen in which a hole has been digested in the cell wall allowing the cytoplasmic membrane to become externalized resulting in osmotic lysis and death. Isolated group A streptococcal cell walls pre (Fig 6-4) and post (Fig 6-5) treatment with phage enzyme. Enzyme-treated cells exhibit pieces of wall in this thin section indicating that holes have been digested in the structure.
Targeted Killing
An interesting feature of these enzymes is that they kill the species of bacteria from which they were produced. For instance, enzymes produced from streptococcal bacteriophage kill streptococci, and enzymes produced by pneumococcal bacteriophage kill pneumococci (Loeffler et al., 2001; Nelson et al., 2001). Specifically, the group A streptococcal lysin will kill group A streptococci efficiently, and has a small effect on groups C and G streptococci, but essentially no effect on normal oral streptococci (see Figure 6-6).
Similar results are seen with a pneumococcal-specific lysine; however, in this case, the enzyme was also tested against strains of pneumococci that were resistant to penicillin and the killing efficiency was the same. Unlike antibiotics, which are usually more broad in their spectrum and kill many different bacteria found in the human body, some of which are beneficial, the phage enzymes kill only the disease bacteria with little to no effect on the normal human bacterial flora. Thus, the phage enzymes are molecules
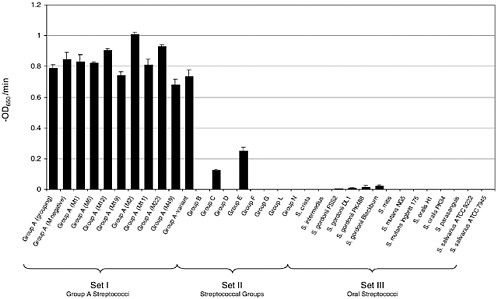
FIGURE 6-6 Representative streptococcal strains were exposed to 250 U of purified lysin and the OD650 was monitored. The activity of lysin for each strain was reported as the initial velocity of lysis, in –OD650/min, based on the time it took to decrease the starting OD by half (i.e., from an OD650 of 1.0 to 0.5). All assays were performed in triplicate and the data are expressed as means standard ± deviations. As can be seen, the enzyme specifically kills group A streptococci, and groups C and G slightly, and has no effect on oral streptococci.
that enable targeted killing of pathogenic bacteria with little effect on the surrounding normal flora. The group A streptococcal enzyme was tested for safety in two animal model systems, one mucosal and the other skin. When enzyme was added to these surfaces daily for 7 days, and the tissues examined both visually and histologically, nothing unusual was observed. This was not surprising since the bonds cleaved by the phage enzymes are only found in bacteria and not mammalian tissues. Thus, it is anticipated that these enzymes will be well tolerated by the human mucous membranes.
In Vivo Experiments
Two in vivo animal models of mucosal colonization were developed to test the capacity for the lysins to kill organisms on these surfaces. An oral colonization model was developed for group A streptococci and a nasal model was developed for pneumococci (Loeffler et al., 2001; Nelson et al., 2001). In both cases, when the animals were colonized with their respective bacteria and treated with a small amount of lysin specific for the colonizing organism, the animals were found to be free of colonizing bacteria two to five hours after lysin treatment (see Figures 6-7 and 6-8). In the group A streptococcal experiment, animals were also swabbed 24 and 48 hours after lysin treatment. During that time most animals remained negative for streptococci but one animal had died and two others showed positive colonies
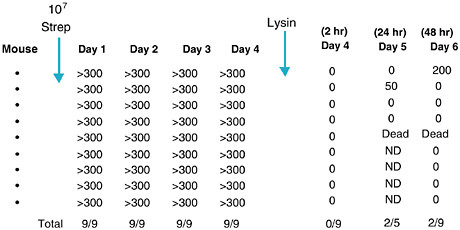
FIGURE 6-7 Elimination of group A streptococci from the mucosal surface of colonized mice. Mice were colonized with streptococci orally followed by oral swab for 4 days. At this time they were treated orally with phage lysin and swabbed 2, 24, and 48 hours later. Figures are colony-forming units of streptococci recovered. ND = not determined.
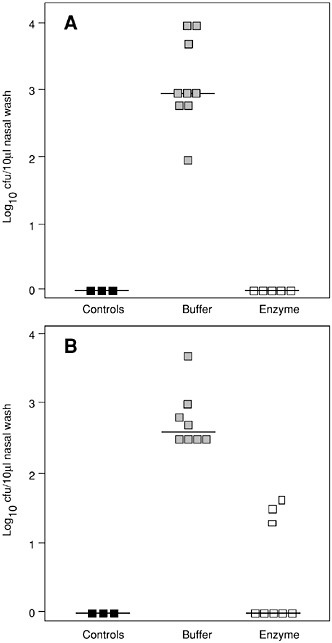
FIGURE 6-8 Killing pneumococci in vivo. Elimination of S. pneumoniae serotype 14 in the mouse model of nasopharyngeal carriage. (A) After nasal and pharyngeal treatment with a total of 1,400 µg of Pal enzyme, no pneumococci were retrieved in the nasal wash, compared to buffer-treated colonized mice (p < 0.001). No pneumococci were isolated from non-colonized control mice (controls). (B) After treatment with a total of 700 µg of Pal enzyme, pneumococci were completely eliminated in 5 of 8 colonized mice (p < 0.001) and overall significantly reduced.
(see Figure 6-7). We interpret these results to mean that the positive animals became infected during the first four days of colonization where some organisms became intracellular. Thus, while the lysin is able to clear organisms found on the surface, it was unable to kill organisms that had initiated an infection. We ruled out the possibility that the organisms that appeared in 24 and 48 hours did so because they became resistant to the lysin by assessing them for their sensitivity to the lysin.
Killing Biowarfare Bacteria
Because phage enzymes are so efficient in killing pathogenic bacteria, they may be a valuable tool in controlling biowarfare bacteria. To determine the feasibility of the approach we identified a lytic enzyme from the gamma phage that is specific for Bacillus anthracis (Watanabe et al., 1975). By cloning the gamma lysin we identified a ~700 bp ORF in the phage genome encoding a 26 kDa product very similar in size and features to a variety of Bacillus, Listeria, and Mycobacteria phage lysins. The gamma lysin, referred to as PlyG, was then purified to homogeneity by a two-step chromatographic procedure and tested for its lethal action on gamma phage sensitive bacilli. Three seconds after contact, as little as 100 units of PlyG (10 µg) mediated a 5,000-fold decrease in viable counts of a ~107Bacillus culture (Schuch et al., 2002). This lethal activity was observed in growth media, phosphate buffer, and even human blood. When the enzyme was then tested against five mutant B. anthracis strains and ten different virulent B. anthracis strains isolated worldwide, it was found to be lethal for them all (Schuch et al., 2002). Although PlyG has no effect on bacillus spores, we discovered that by heat shocking at 60°C and the addition of the germinant L-alanine results in rapid generation of spores which are sensitive to the action of the PlyG enzyme. For example, 108 spores heat-activated for 5 minutes and treated with L-alanine for 5 minutes were reduced in viability by 4-logs after a 5-minute treatment with PlyG.
When 106 bacilli were administered intraperitoneally (IP) to 15 mice all of the animals died of rapidly fatal septicemia within four hours. When a second set of 13 mice was also challenged IP with bacilli but given only 150 µg of PlyG 15 minutes later by the same route, 77 percent of the animals recovered fully, and the remaining died within 6–21 hours (Schuch et al., 2002). We anticipate that higher doses or repeated treatment of enzyme will result in nearly 100 percent protection.
Resistance to Enzymes
Repeated exposure of bacteria grown on agar plates to low concentrations of lysin did not lead to the recovery of resistant strains. Nor were we
able to identify resistant bacteria after several cycles of exposure to low concentrations of enzyme in liquid medium (Loeffler et al., 2001). This may be explained by the fact that the cell wall receptor for the pneumococcal lysin is choline (Garcia et al., 1983), a molecule that is necessary for pneumococcal viability. For group A streptococci, we find that polyrhamnose, a cell wall component of the bacteria, is necessary for lysin binding (Nelson and Fischetti, unpublished data), and polyrhamnose has also been shown to be important for streptococcal growth. While not yet proven, it is possible that during a phage’s association with bacteria over the millennia, to avoid becoming trapped inside the host, the binding domain of their lytic enzymes has evolved to target a unique and essential molecule in the cell wall, making resistance to these enzymes a rare event.
Concluding Remarks
Phage lytic enzymes are new agents for the control of bacterial pathogens. Since this capability has not been previously available, its acceptance may take a while. For the first time we are able to specifically kill pathogens without affecting the surrounding normal bacteria. Whenever there is a need to kill bacteria, and contact can be made with the organism, phage enzymes may be freely utilized. They may not only be used to control pathogenic bacteria on human mucous membranes, but may find utility in the food industry to control disease bacteria without the extensive use of antibiotics in feed or harsh agents to decontaminate. Because of the serious problems of antibiotic-resistant bacteria in hospitals, day care centers, and nursing homes, particularly staphylococci and pneumococci, such enzymes will be of immediate benefit in these environments. The enzymes isolated thus far are remarkably heat stable (up to 60°C) and are very easy to produce in a purified state, resulting in pennies a dose to manufacture. Thus, we may add phage enzymes to our armamentarium against pathogenic bacteria. They are molecules that have been in development for millions of years by bacteriophage in their battle to survive within bacteria. All we have done is exploit them.
ROLES FOR PHARMACOKINETICS AND PHARMACODYNAMICS IN DRUG DEVELOPMENT FOR RESISTANT PATHOGENS
Jerome J. Schentag, Pharm.D. and Alan Forrest, Pharm.D.
University at Buffalo School of Pharmacy, Buffalo, NY
Antimicrobial resistance is a troubling problem, but not all patients have it, and some appear to overcome it even without new antibiotics. As a
consequence, newly marketed antibiotics that cost more are placed on the reserve list, not to be used unless their price either drops to the level of the older generics, or unless an individual patient is failing to respond to other therapies. These issues have made an impact on antibiotic development. When there is no profit from the use of the new antibiotics, the incentive to develop them disappears. It was pointed out that a number of large pharmaceutical companies had either recently exited the antibiotic development business, or were openly contemplating this move in the near future. Disaster loomed large in the audience, because the only solution to the resistance problem that enjoyed unanimous support was a robust pipeline of new antibiotics and many companies involved in their development.
Stakeholders (industry, the Food and Drug Administration [FDA], and academia) need to gather and decide as a group what improvements can be made in the drug development process for antibiotics. The industry needs regulatory assistance to lower antibiotic development costs, and in return they must produce a pipeline containing a wider variety of new chemical or biological entities that deserve use (but only) when they are needed. We cannot expect a wide variety of choices of many chemical classes, unless many companies stay in the hunt for new antibiotics. We believe there are a number of issues that can be addressed in the course of human trials that will lower costs and speed time to approval.
Current and Alternative Means of Developing Antibiotics
The typical New Drug Application (NDA) contains data on antibiotic efficacy and safety in 3,000–5,000 patients. By study design, these NDAs contain no patients with organisms resistant to either the new drug or to existing agents, because under current design specifications for equivalence trials, all patients with an organism resistant to either the new drug or the comparator are excluded from the trials before data analysis. Equivalence cannot be served, after all, if some of the patients on conventional therapy would be expected to fail. The logic of this design has persisted since the 1970s, when marketing an antibiotic required only safety and efficacy equivalent to what was already on the market. The legacy of equivalence models is a large number of closely related compounds with marginal differences, and the need for aggressive sales and marketing efforts to differentiate them after marketing.
The links between strategy and resistance go well beyond the equivalence model. For example, it is widely believed that antibiotics must have only a one-dosage regimen for all patients. This is considered a strategic advantage, because if one is successful in demonstrating efficacy and safety equivalent to the older comparators, one dosage regimen fosters widespread use. Unfortunately, the fixed dose may under- or over-expose some
bacterial pathogens, and this situation in the face of monopolistic use creates maximal opportunities for selection pressure. This combination of factors can bring about a speedy end to the drug because of selection of resistant strains (Schentag, 1995). We shall return to this particular problem using the example of the fluoroquinolones (FQ) in pneumococcal pneumonia (Schentag et al., 2001a; 2001b) later in this paper.
Obviously, the equivalence model is useless if the question is to establish superiority on the basis of greater action against resistant organisms. If the development plan for the new antibiotic calls for replacement of an older one that is losing effectiveness because of resistance, forcing this new antibiotic to become equivalent to this older antibiotic places the new antibiotic at a competitive disadvantage. Patients who would contribute to superiority arguments are usually excluded from consideration in equivalence models, since both comparators must be active in order to retain the patient in the trial database used to determine equivalence.
The alternative study design for this new antibiotic is a superiority trial. When the goal is to show the new antibiotic as being better for the treatment of resistant pathogens, this should become the standard model. Industry suspects that demonstration of clear superiority would result in low usage during the time when most pathogens are at least somewhat susceptible to the older antibiotics. Low use results in low profit, and a longer time to recover the investment in the antibiotic development and marketing. But it appears that equivalence also results in low use in some cases. We encounter this dilemma with the newer gram-positive antibiotics. Clinicians only use the newer gram-positive antibiotics in situations where there is clinical failure of vancomycin, or when bacterial pathogens are resistant to vancomycin (Moise and Schentag, 2000; Schentag, 2001). With equivalence in trials as their only argument, there is low use of the replacement antibiotics because of their high selling price.
So why do a superiority trial, given the risks of use only when needed? Quite simply, if the new agent has any safety problems at all, equivalence to old but widely used antibiotics will no longer be sufficient reason to allow it on the market, especially if the existing market is largely satisfied by the conventional antibiotics. Recently, it is clear that new agents must be superior just to reach the market in the first place.
So how does industry best manage antibiotic regulatory risk in the next few years, assuming they do not withdraw from antibiotic development? Let us first discuss the setting of an antibiotic for community infections, with wide potential to be a “blockbuster” product. Here it seems most prudent to gather antibiotic experience data both for equivalence (i.e., 3,000–5,000 patients) on safety and efficacy and to perform some targeted superiority studies to establish medical need, just in case there might be
safety problems of nearly any degree. Demonstration of the safety of a new agent will ordinarily be a sufficient justification to collect the large numbers, and equivalence might be the goal here on efficacy, so that may be affirmation of the currently accepted procedure. The additional challenge is to manage the targeted superiority trials, which need to be submitted at the time of the NDA. These studies either can be performed on a subset of the safety population or they can be added on to subsets of the equivalence population. A third possibility, in the event that neither of the above will work, is to conduct the superiority trial as a free-standing study. If conducted late, after safety problems arise, the superiority trial must certainly be done free-standing.
What about the antibiotic that is clearly superior, but the market is a niche? The good news on superiority is that if the new antibiotic is truly superior, it does not require large numbers of patients to prove it, especially if you use a microbiological response. If the patient is failing conventional therapy with a resistant organism, and you change to an antibiotic effective against that pathogen, you should be rewarded by a cure (both microbiologically and clinically). This mentality is pervasive in the real world, but is just coming to the attention of the regulatory debates that occur in advisory committee meetings focused on equivalence design and equivalence-based statistical modeling.
The problem we typically encounter is that superiority is only rarely as black and white as the same dose of one drug always being superior to the same dose of the other. Most antibiotics perform differently as the minimum inhibitory concentration (MIC) of the pathogen varies and as you change the dose of the drug (Schentag et al., 1991, 1996, 1997, 1998, 2001b; Schentag, 1999a, 2001). The “one dose for all” strategy (Schentag, 1998) offers many opportunities for low exposure (i.e., low area under the inhibitory time curve [AUIC]) and failure, and for high exposure (renal failure with high blood levels) and resulting toxicity. Therefore, we see a major role for pharmacokinetics/pharmacodynamics [PK/PD] and time to bacterial eradication endpoints in the study of superior responses in failing patients, as well as in the study of antimicrobial resistance selection. The benefits of incorporating PK/PD measurements into superiority trials are clear. Demonstrations of superiority are possible on small numbers of patients (N = 20 per group can yield statistical significance). This does illustrate that for most antibiotics, it is not one dose for all, it is one dose for each. PK/PD will only rarely allow the conclusion of equivalence (i.e., allow defeat); one antibiotic is always better than the other when you have enough information to decide.
Pharmacokinetics and Pharmacodynamics (PK/PD)
When we discuss PK/PD, we typically use the serum AUC measured in the patient being treated versus the MIC of the organism infecting that particular patient. The diagram in Figure 6-9 illustrates the various PK/PD parameters that can be used to describe the interactions between concentration and MIC. We prefer the use of the simple ratio termed AUIC, because it encompasses all the exposure parameters, and has the advantage of being independent of the oscillations created by different divisions of the same 24-hour dose (Schentag et al., 1996, 1997; Schentag, 1999b). The power of considering individual patient response as a direct function of PK as AUC, and PD as the concentration that will kill the organism, becomes apparent in the statistical treatment of superiority. Every regimen has its own unique PK/PD predicted outcome, and very little of the overall clinical or microbiological outcomes is left as “random noise.” This is most clearly apparent when PK/PD is linked to bacterial outcome in bacterially dependent conditions like nosocomial pneumonia (Schentag, 1999b), but it also works if you target the organism rather than the clinical symptoms in mild infections such as acute exacerbation of chronic bronchitis (AECB) (Forrest et al., 1997).
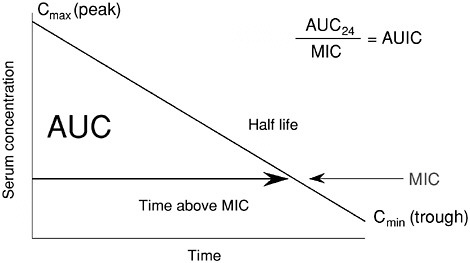
FIGURE 6-9 Diagram illustrating the relationships between antibiotic serum concentrations and MIC. Shown are area under the curve (AUC), peak to MIC, and time above MIC and AUIC, which is the ratio of AUC over 24 hours to MIC. SOURCE: Schentag et al., 2001a.
The most important feature of PK/PD is the use of serial culture to target outcomes such as the rate (speed) of bacterial killing in the individual patient. When expressing individual patient PK/PD as AUIC, it is possible to account for over 80 percent of the variability in (clinical and microbiological) outcomes in a logistic regression model, as well as to determine how fast the organism dies in the patient, as was shown for ciprofloxacin in nosocomial pneumonia in Figure 6-10 (Forrest et al., 1993).
If you collect the data, it becomes possible to use PK/PD to explain selection of a resistant strain when one emerges during therapy, as shown in Figure 6-11 (Thomas et al., 1998). In this study we modeled the day of resistance emergence versus the initial exposure profile (described by AUIC) of the antibiotic regimen (Thomas et al., 1998). Clearly, data of this type explain clinical failure as a failure to eradicate the organism, and link that
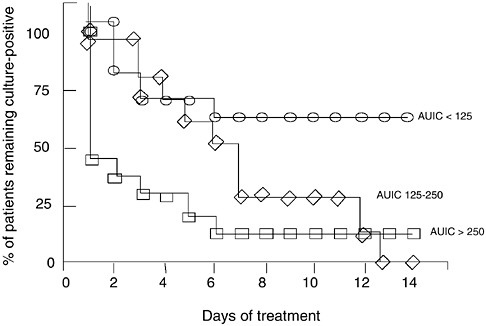
FIGURE 6-10 Relationships between AUIC values and time to bacterial eradication in 74 patients treated with ciprofloxacin for nosocomial lower respiratory tract infection. Each patient had daily tracheal aspirate cultures and the three groups differed significantly from each other. AUIC values >250 produced over 60 percent of the patients culture negative on the first day of treatment. AUIC values of 125– 250 required 6 days to convert 60 percent of the cultures to negative, and AUIC values <125 never eradicated the organism in 70 percent of the patients. These data illustrate concentration-dependent microbial killing in humans. SOURCE: Forrest et al., 1993.
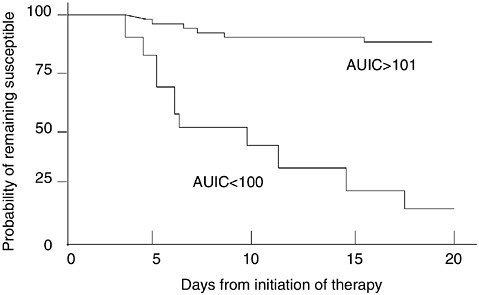
FIGURE 6-11 Relationships between the time cultures remain positive and the initial AUIC value of a group of 127 patients who initially were infected with susceptible organisms. Each patient was followed with serial cultures. Those patients with AUIC <100 demonstrated selection of resistance and overgrowth of these strains progressively over the monitoring period, and most required changes in antimicrobial regimen. If the initial AUIC exceeded 101, only 8 percent of the patients had overgrowth of selected strains resistant to the initial antimicrobial regimen. These data illustrate that it is most dangerous from a resistance selection perspective to have too low an AUIC for too long. SOURCE: Thomas et al., 1998.
occurrence to a low AUIC. The data were among the first to establish a predictability to emergence of resistance caused by selection pressure.
Considering Resistance in Antibiotic Development
If the focus of a new antibiotic becomes superiority to existing alternatives, it is necessary to focus on the microbe that fails conventional antibiotics. Clinical evidence of failure is also important, but you can receive confusing information from failure defined in the absence of knowledge regarding the impact of the antibiotic on the microbe. This is because the impact of antibiotics is on the organism itself, not necessarily the clinical symptoms.
There are many conditions that cause clinical symptoms besides the bacterial pathogen, and there are many things that ablate clinical signs and
symptoms of infection besides successful eradication of the organism. Witness the simple treatment of fever with an antipyretic, as an example of why it is necessary to focus on the antimicrobial effect of the antibiotic. In patients who are severely ill from bacterial infection, it is relatively easy to measure superiority if an antibiotic works better than a comparator that is less active. But more often than not, NDAs powered for equivalence exclude these types of patients, because the organism must be susceptible to both the newer and the older antibiotics to remain in the evaluable population.
The impact of the underlying host defense and its potential to confuse the cause of clinical cure are greatest in modestly ill patients. Host response (in modestly ill outpatients) is more than sufficient to overcome failure of the antibiotic to eradicate a pathogen and even selection of resistance. Many of these patients will not show clinical failure patterns even if they have selected a resistant pathogen from low AUICs.
Measuring AUIC and Correlations with Selection Pressure Resistance
One means of achieving equivalence even though you may have a superior antibiotic is to develop and market it at a marginally low dose. We would like to offer an example of an antibiotic developed at low dosing using equivalence design, now widely used in the community, where the selection pressure resulting from low AUICs is rapidly leading to resistance to the entire class it represents. The example is FQ-resistant Streptococcus pneumoniae, and the antibiotic cited by virtue of frequency is levofloxacin.
The current party line is that there are no resistance problems with older FQs such as ciprofloxacin and levofloxacin regarding S. pneumoniae (Sahm et al., 2000, 2001). Even if this is considered to be a problem, it is argued that all members of the class are the cause, not one compound. The core counter-argument is that if one uses a laboratory breakpoint of 8.0 mcg/ml as a definition of resistance to levofloxacin, resistance went from 0.6 percent to 1.2 percent in the United States in 2000–2001 (Whitney et al., 2000; Doern et al., 2001). Most labs in the United States do not even test levofloxacin against S. pneumoniae unless requested by clinicians who are treating a clinical failure (Thornsberry et al., 2001; Davidson et al., 2002). Fluoroquinolone resistance is widely underappreciated in the laboratories. Testing practices are likely to change quite soon, in the face of a rapidly growing number of case reports of clinical failures worldwide (Ho et al., 1997, 1999a, 1999b, 2001; Davidson et al., 2000; Empey et al., 2001; Schentag et al., 2001b; Urban et al., 2001; Weiss et al., 2001; Davidson et al., 2002; Kays et al., 2002). Our thesis is that this “low” incidence of lab-defined resistance is a product of three problems:
-
The use of a breakpoint of 8.0 mcg/ml, which is well above the peak concentration of the drug in serum; indeed, a false sense of security under the circumstances.
-
Microbiology laboratories seldom test levofloxacin against S. pneumoniae isolates, and even when they do test MICs, the test misses the single step mutants because hetero-resistant MICs of 1–2 mcg/ml are still below the breakpoint of 8.0 mcg/ml (Richardson et al., 2001).
-
Every time levofloxacin is used in patients at its current low dose, its AUIC is below 100, which is the root cause of widespread selection pressure in the community (Schentag et al., 2001a), and MICs continue to rise under the breakpoint of 8.0 mcg/ml.
I should like to briefly illustrate this low AUIC question, focusing on Figure 6-12 (Schentag et al., 2001a; 2001b). First of all, the AUC of levofloxacin (in a volunteer given a dose of 500 mg) is approximately 48
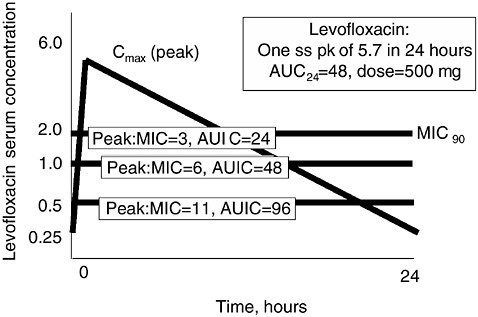
FIGURE 6-12 Relationship between levofloxacin pharmacokinetics and organism MICs of “susceptible” strains. In each case, the AUC of levofloxacin is chosen to be 48, the value typical of normal volunteers and patients with normal renal function. When the MIC is 0.5 mcg/ml (approximately 10 percent of strains in 2001), the AUIC is 96. When the MIC is 1.0 mcg/ml (40–50 percent of strains), the resulting AUIC is 48, and when the MIC is 2.0 mcg/ml (the usual MIC90) for this drug, the AUIC is 24. These calculations illustrate that the AUIC is below 100 in nearly all patients treated with levofloxacin, and illustrate a high risk for selection pressure against S. pneumoniae. SOURCE: Schentag et al., 2001a.
mcg × hr/ml. The MIC90 of Streptococcus pneumoniae to levofloxacin was ~0.5 mcg/ml in 1995 when the drug was marketed, with an initial AUIC of 96. So at the time of marketing, exposure usually resulted in a borderline but probably acceptable AUIC, and assisted by the good host defense in outpatients, the drug appeared to work. The only patients who would be expected to select resistance in the early days were those infected with a strain having an occasional MIC higher than 0.5 mcg/ml or the occasional AUC below 48. Thus, the at-risk population would be younger patients with good renal function or patients who had already been treated with prior levofloxacin and had selected an organism.
As the organism MIC rose from widespread use, extreme selection pressure, and the introduction of one step mutants into the “susceptible” population, all the organisms progressed to intermediately susceptible strains with MICs of 1.0–2.0 mcg/ml. The four-fold loss of activity was not compensated for by increasing the levofloxacin dose four-fold. As the dose remained 500 mg once daily throughout the rise in MICs, all the AUICs became 24–48 (see Figure 6-12) unless the patient had some renal failure, in which case they may have been above 100 on this dose (Ambrose et al., 2001; Drusano et al., 2001; Nicolau and Ambrose, 2001; Owens et al., 2001). The consequences of continued and steady selection pressure include more resistance and eventually clinical failure of a sufficient frequency as to abandon this and other fluoroquinolones in community infections. If we wait until all of the organisms out there are resistant to levofloxacin, we will also lose the rest of the FQs, since most of the double mutants (MICs > 8.0 mcg/ml) selected by levofloxacin are resistant to all of the members of this antimicrobial class, even the newest ones (Urban et al., 2001).
Superiority trials of antibiotics that are active against this newly mutated S. pneumoniae, such as telithromycin or linezolid, ought to be easy to carry out. As the clinicians abandon levofloxacin and the FQs, there will be widespread use of these newer agents for clinical failures of FQs in community-acquired pneumonia.
The first task before contemplating a superiority trial versus S. pneumoniae is to analyze why equivalence trial design did not reveal the differences that are becoming so clearly apparent in retrospect. We believe there are a number of reasons:
-
The organism is changing rapidly in response to widespread use at low AUIC, but this event followed the marketing of levofloxacin for penicillin-resistant S. pneumoniae. The clinical trials themselves did not create enough selection pressure, because use was not monopolistic at that early time.
-
Equivalence design studies which brought the drug to market used cephalosporin comparators, which would logically exclude penicillin-resis
tant S. pneumoniae from the study population used to establish equivalence, and would prevent superiority from being discovered. In fact, one study did conclude superiority (File et al., 1997).
-
Widespread monopolistic use upon marketing, catalyzed by formularies and managed care practices trying to save money, lead to increasing selection pressure in communities with at-risk, reservoir populations.
-
Equivalence designs only measure outcomes at times approximately 14 days post-cessation of the antibiotics. This obscures the other natural phenomenon found when antibiotics begin to fail, which is slower and slower rate of bacterial killing (see Figure 6-13). Two antibiotics which differ as dramatically as A and B in Figure 6-13 are equivalent if your endpoint measurement is taken after host response produces resolution in the case of antibiotic B.
So what could a superiority trial model offer a new antibiotic in this situation, assuming it was highly active against FQ-resistant pneumococci? Certainly, you could not establish superiority in a relatively healthy popula

FIGURE 6-13 Illustration of the relationships between speed of cure on two different antibiotics (A vs. B) and the time that clinical assessment is made in typical clinical trials. This illustrates why equivalence studies are unlikely to show differences between two antibiotics that illustrate different rates of killing, such as a concentration-dependent FQ versus a time-dependent macrolide or beta-lactam. Serial cultures will clarify these early and more rapid cure patterns and illustrate differences between the antibiotics. Adapted with permission from Dr. C.H. Nightingale, Hartford, CT.
tion with host defense if your trial endpoint was test of cure at 10–14 days post-treatment. If the patient population has good host defense and/or signs and symptoms of infection are modest, you show no differences.
However, if you would collect serial assessments of organism viability (serial cultures) and if you have serial assessments of infection signs and symptoms early in therapy, and if you can accurately characterize PK/PD of each patient, superiority is possible with as few as 20 patients per group with an active drug. Furthermore, with AUICs and serial cultures on all patients, you could “probably” differentiate the contribution of the host defense from that of the antibiotic, and could easily separate time-dependent killing from concentration-dependent killing in the infected patient, provided that differences are apparent in vitro.
Endpoints
Killing or inhibiting growth of the bacteria is the only measurable effect of an antibiotic. To further increase the power of discrimination with bacterial killing endpoints, measure how fast bacteria are killed, or how many are killed per unit of time. This technique works in vitro, in animal models and in humans. Furthermore, all three testing models are progressively coming to consensus on the PK/PD characteristics necessary for prediction of outcome.
Any other endpoints, including clinical cure, non-culture proven “micro cure,” and resolution of infection signs and symptoms, are surrogate markers of the results from either presumed or proven bacterial killing. How accurately or successfully the surrogates perform depends on whether the bacteria originally caused the signs and symptoms used to define the disease and its cure.
On the basis of endpoints, it is logical to change the clinical trial endpoint from clinical cure to microbiological eradication. We can further enhance the discriminatory powers of anti-infective trials by using the microbiological results to define parameters like time to eradication (to handle bacteriostatic versus bactericidal, and time-dependent versus concentration-dependent killing in vivo). In addition, resistance can be linked to MICs and achieved serum concentrations and AUICs. This allows prospective definition of when combination therapy is needed, and which antibiotic combinations will be most successful.
Perhaps the most important benefit of expressing antibiotic action as microbiological eradication is that fewer patients are needed for statistical significance in superiority trials, which yields a substantial lowering of drug development costs and produces faster time to NDA approval.
Sample Size, Delta, and Statistical Power on a Dichotomous versus Continuous Endpoint; Tests of Equivalence versus Superiority, and Why Such Large Numbers for Cure versus Failure?
The primary statistical problem of equivalence design and equivalence doctrine is that a large portion of the data in a cure-failure clinical trial are non-contributory to the goal of discriminating the best regimens. All of the following conditions discourage differentiation and force large numbers just to conclude equivalence (Powers et al., 2002):
-
Patients with symptoms but not caused by bacterial pathogens,
-
Patients with self-limiting disease not dependent for cure on antimicrobial actions, and
-
Infected patients where the antimicrobial itself is barely active or inadequate, but there is still some activity of this comparator plus host defense. You conclude clinical cure in these settings regardless of what condition or drug produced it.
The current trend is how many patients do we need to study in order to conclude two antibiotics are equivalent, and yet the new drug is better than placebo, which has led recently to the tightening of the delta to < 10 percent for some infections (Powers et al., 2002). Delta is a measure of statistical differentiation, useful in setting sample size in non-crossover clinical trial designs. As might be surmised, a tighter delta increases the size of the study. A tighter delta does not yield a beneficial return if the goal is to differentiate and the model is superiority, as shown in Table 6-2. However, tighter deltas do not cause an increase in the already small numbers of patients who will show superiority in a well-conducted PK/PD trial (see Table 6-2).
Is Compromise Possible Between PK/PD/Micro Superiority and the Clinical Cure-Based Equivalence Models?
In reality, antibiotics are never equivalent, one is always better. Given the diversity in MICs among bacterial populations, the right endpoint can differentiate almost any antibiotic from any other, with only small numbers of patients. And on the surface, it would seem that both trial designs (equivalence and superiority) have some merit. We have compared the two designs in Table 6-2, extracting some information from recent literature on the topic of delta (Shlaes and Moellering, 2002). The comparison shows that the best features of each can be merged into a development plan that should be less expensive to all concerned.
By way of example, start out with the usual two proof of claim studies
TABLE 6-2 Delta, Power, and Sample Size on a Dichotomous versus Continuous Endpoint
|
|
Dichotomous (cure/failure) |
Continuous (T to Erad versus AUIC) |
|
Power |
90% |
90% |
|
Delta |
10% |
± 1 day |
|
Variability: Type I two tailed error |
- |
SD = 20–40% |
|
Cure Rate (%) |
85–90% (clinical) |
80% (MicroErad) at median Terad 3d |
|
# pts per group to conclude A=B |
1532 |
< 10 to ~ 90 |
|
# pts per group to conclude A>>B |
inf |
< 10 to ~ 90 |
per indication in Table 6-2. In the new mode, these two studies no longer would both be the same design, changing only the comparators but keeping the equivalence mode. Rather, one of the two is equivalence, designed for the therapeutic indication, with a delta even as tight as 10 percent acceptable, since the first study is to gather numbers of patients for safety. You typically want 1,000 per claim and 4 claims is average. The second is designed around the PK/PD and targets superiority for efficacy against microbes (~10 patients per bacteria plus comparator differences at delta 10 percent). The superiority study has a total patient population as few as 40 and as many as 250 patients, depending on how many individual organisms seeking superiority claims.
Beyond the compromise forged between the statistical equivalence and superiority models, the merging of the two types of trials in the same NDA would accomplish some other goals of those who use antibiotics on sick patients. These include data on situations such as:
-
Rare and less encountered indications can be studied with these PK/PD models (e.g., endocarditis, meningitis, pediatric infections).
-
Labeling for unusual or resistant bacteria could be obtained with 10–20 patients per organism if PK/PD targets are used.
-
For antibiotics where there is no active comparator, studies of individual patient AUICs versus the standard AUIC target of 100 could be
carried out. These superiority models (based on speed of bacterial killing or time to eradication) would yield labeling in cases where there is no labeled or active comparator, such as vancomycin-resistant Enterococcus faecium (VREF).
-
We could, early on, vigorously pursue combination regimens with PK/PD; combination therapies are the real world and antibiotic development needs to address this topic very soon.
-
Begin to build these superiority models and study designs into phase II-III rather than waiting for phase IV, since they could lead to claims and will certainly guide use of the new antibiotic in the post-marketing period.
Clearly, if a new drug is better than the old, there is some advantage to proving that for patients using PK/PD. Of course, if it does not differ from the old, it may be better to remove it from trials early, before scarce resources are consumed that are better applied to a follow-on compound.
So what happens if the industry sticks with the equivalence model, and the tighter delta is applied to the community infection models like AECB, sinusitis, otitis media, and others? The prospect is not good, since even with equivalence proven at a now considerably higher clinical trial cost, you might get FDA approval if you show safety, but you still cannot sell the new antibiotic for more. Of course, such cure-failure studies in equivalence populations are primarily valuable for safety, regardless of the statistical treatment. However, if your design will not show a new FQ or a new beta-lactam to be better than an old and largely inactive antibiotic (e.g., cefaclor) in outpatients (Gotfried and Ellison, 1992; Bandak et al., 1999), then you are using the wrong disease and clinical cure is the wrong endpoint. Unfortunately, no improvements in statistical methodology will fix that problem. Somewhere in every antibiotic NDA, there will need to be a demonstration of superiority and medical need, especially to offset any side effects that develop in the course of the trials, and to offset any plans to charge more for the drug when it reaches the market.
Order Out of Chaos?
The current antibiotics development, regulatory, promotion, and clinical use pattern is illogical; the resulting selection pressure from aggressive equivalence-based marketing causes great ecological harm. We advocate changes in the drug development process for antibiotics, so as to achieve the following benefits:
-
A return to a focus on the true target of effect, the bacteria, would greatly improve this situation.
-
New antibiotics should be rapidly developed and marketed precisely for their intended infections, using superiority trials as a basis.
-
Local surveillance could drive use patterns against locally resistant organisms, as well as become the basis for logical dosing and applications of AUIC at the bedside.
-
Public health and microbiology laboratories could gain new relevance in our resistant microbe-enriched world, since resistance is continually evolving in response to local antibiotic use practices.
-
We could finally tackle the important questions of microbial population ecology and evolutionary biology, such as the proper dose and duration to lessen selection pressure.
-
Antibiotics could be developed for use against organisms, or groups of organisms, and health care professionals could be taught to use them when diseases are caused by bacteria, which would finally permit appropriate diagnosis (we could even bring back the bedside gram stain).
-
Industry could run direct to consumer ads teaching patients about the differences between bacteria and viruses, and the rationale for treatment of specific pathogens with effective antibiotics labeled for that purpose.
In summary, we are certain that antibiotic development compromises can be made, and that they should be made. Compromises will speed the development of superior new antibiotics and lower the costs of development and marketing. Absence of compromise will raise antimicrobial development costs and likely aggravate resistance by fostering aggressive marketing and monopolistic use. All of the strategy outlined can be implemented relatively quickly, and further testing of the underlying principles would occur as the superiority trial designs achieve wider use. It is clear there are major problems ahead if we follow the current path. While the new pathways are not free of confrontations and problems, the promise for the future more than outweighs the short-term challenges.
REFERENCES
Abraham EP and Chain E. 1940. An enzyme from bacteria able to destroy penicillin. Nature 146:837–840.
Ambrose PG, Grasela DM, Grasela TH, Passarell J, Mayer HB, Pierce PF. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrobial Agents and Chemotherapy 45:2793–2797.
Bandak SI, Bolzon LD, Turnak MR, Johns D, Henle SK, Allen BS. 1999. Cefaclor af versus amoxycillin/clavulanate in acute bacterial exacerbations of chronic bronchitis: a randomised multicentre study. International Journal of Clinical Practice53:578–583.
Bennett PM. 1999. Integrons and gene cassettes: a genetic construction kit for bacteria. Jour nal of Antimicrobial Chemotherapy43:1–4.
Bulychev A and Mobashery S. 1999. Class C ß-lactamases operate at the diffusion limit for turnover of their preferred cephalosporin substrates. Antimicrobial Agents and Chemo therapy43:1743–1746.
Bulychev A, Massova I, Lerner SA, Mobashery S. 1995. Penem BRL 42715: an effective inactivator for ß-lactamases. Journal of the American Chemical Society117:4797–4800.
Bulychev A, Massova I, Miyashita K, Mobashery S. 1997. Nuances of mechanisms and their implications for evolution of the versatile ß-lactamase activity: from biosynthetic enzymes to drug resistance factors. Journal of the American Chemical Society119:7619– 7625.
Bush K. 1999. ß-lactamases of increasing clinical importance. Current Pharmaceutical Design 5:839–845.
Bush K and Mobashery S. 1998. How ß-lactamases have driven pharmaceutical drug discovery: from mechanistic knowledge to clinical circumvention. Advances in Experimental Medicine and Biology456:71–98.
Chamberland S, Blais J, Hoang M, Dinh C, Cotter D, Bond E, Gannon C, Park C, Malouin F, Dudley MN. 2001. In vitro activities of RWJ-54428 (MC-02,479) against multiresistant gram-positive bacteria. Antimicrobial Agents and Chemotherapy45:1422–1430.
Coello R, Jimenez J, Garcia M, Arroyo P, Minguez D, Fernandez C, Cruzet F, Gaspar C. 1994. Prospective study of infection, colonization and carriage of methicillin-resistant Staphylococcus aureus in an outbreak affecting 990 patients. European Journal of Clini cal Microbiology and Infectious Diseases13:74–81.
Davidson R, Cavalcanti R, Brunton JL, Bast DJ, de Azavedo JC, Kibsey P, Fleming C, Low DE. 2002. Resistance to levofloxacin and failure of treatment of pneumococcal pneumonia. New England Journal of Medicine346:747–750.
Davidson R, de Azavedo J, Bast D, Arbique J, Bethune R, Duncan K, McGeer A, Low DE. 2000. Levofloxacin treatment failure of pneumococcal pneumonia and development of resistance during therapy. Abstract 2103. In: Abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy, Toronto, September 17–20, 2000.
de Lencastre H, Kristinsson KG, Brito-Avo A, Sanches IS, Sa-Leao R, Saldanha J, Sigvaldadottir E, Karlsson S, Oliveira D, Mato R, de Sousa MA, Tomasz A. 1999. Carriage of respiratory tract pathogens and molecular epidemiology of Streptococcus pneumoniae colonization in healthy children attending day care centers in Lisbon, Portugal . Microbial Drug Resistance5:19–29.
Desselberger U. 2000. Emerging and re-emerging infectious diseases. Journal of Infectious Diseases40:3–15.
Diaz E, Lopez R, Garcia JL. 1990. Chimeric phage-bacterial enzymes: a clue to the modular evolution of genes. Proceedings of the National Academy of Sciences87:8125–8129.
Doern GV, Heilmann KP, Huynh HK, Rhomberg PR, Coffman SL, Brueggemann AB. 2001. Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae in the United States during 1999–2000, including a comparison of resistance rates since 1994– 1995. Antimicrobial Agents and Chemotherapy45:1721–1729.
Drusano GL, Preston SL, Owens RC Jr, Ambrose PG. 2001. Fluoroquinolone pharmacodynamics. Clinical Infectious Diseases33:2091–2096.
Eiff CV, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. New England Journal of Medicine344:11–16.
Empey PE, Jennings HR, Thornton AC, Rapp RP, Evans ME. 2001. Levofloxacin failure in a patient with pneumococcal pneumonia. Annals of Pharmacotherapy35:687–690.
File TM Jr, Segreti J, Dunbar L, Player R, Kohler R, Williams RR, Kojak C, Rubin A. 1997. A multicenter, randomized study comparing the efficacy and safety of intravenous and/or oral levofloxacin versus ceftriaxone and/or cefuroxime axetil in treatment of adults with community-acquired pneumonia. Antimicrobial Agents and Chemotherapy41:1965– 1972.
Forrest A, Chodosh S, Amantea MA, Collins DA, Schentag JJ. 1997. Pharmacokinetics and pharmacodynamics of oral grepafloxacin in patients with acute bacterial exacerbations of chronic bronchitis. Journal of Antimicrobial Chemotherapy40 Suppl A:45–57.
Forrest A, Nix DE, Ballow CH, Goss TF, Birmingham MC, Schentag JJ. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrobial Agents and Chemotherapy37:1073–1081.
Fung HB, Kirschenbaum HL, Ojofeitimi BO. 2001. Linezolid: an oxazolidinone antimicrobial agent. Clinical Therapeutics23:356–391.
Garcia E, Garcia JL, Arraras A, Sanchez-Puelles JM, Lopez R. 1988. Molecular evolution of lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Proceeding of the National Academy of Sciences85:914–918.
Garcia P, Garcia E, Ronda C, Tomasz A, Lopez R. 1983. Inhibition of lysis by antibody against phage-associated lysin and requirement of choline residues in the cell wall for progeny phage release in Streptococcus pneumoniae. Current Microbiology8:137–140.
Garcia P, Garcia JL, Sanchez-Puelles JM, Lopez R. 1990. Modular organization of the lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Gene86:81–88.
Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D ß-lactamases. Proceedings of the National Academy of Sciences98:14280–14285.
Golemi-Kotra D and Mobashery S. 2002. Antibiotic resistance. In: Kirk-Othmer Encyclope dia of Chemical Technology (4th ed.). New York: John Wiley. [Online]. Available: http://www.mrw.interscience.wiley.com/kirk/.
Gonzales RD, Schrekenberger P, Graham C, Kelkar S, DenBesten K, Quinn JP. 2001. Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 357:1179–1185.
Gotfried MH and Ellison WT. 1992. Safety and efficacy of lomefloxacin versus cefaclor in the treatment of acute exacerbations of chronic bronchitis. American Journal of Medicine 92(4A):108S–113S.
Hardy LW and Kirsch JF. 1984. Diffusion-limited component of reactions catalyzed by bacil lus cereus ß-lactamase I. Biochemistry23:1275-1282.
Hebeisen P, Heinze-Krauss I, Angehrn P, Hohl P, Page MG, Then RL. 2001. In vitro and in vivo properties of Ro 63-9141, a novel broad-spectrum cephalosporin with activity against methicillin-resistant staphylococci. Antimicrobial Agents and Chemotherapy 45:825–836.
Heymann DL and Rodier GR. 2001. Hot spots in a wired world: WHO surveillance of emerging and re-emerging infectious diseases. Lancet Infectious Diseases5:345–353.
Ho A, Leung R, Lai CK, Chan TH, Chan CH. 1997. Hospitalized patients with community-acquired pneumonia in Hong Kong: a randomized study comparing imipenem/cilastatin and ceftazidime [published erratum appears in Respiration 1997; 64(4):303]. Respira tion64(3):224–228.
Ho PL, Que TL, Tsang DN, Ng TK, Chow KH, Seto WH. 1999a. Emergence of fluoroquinolone resistance among multiply resistant strains of Streptococcus pneumoniae in Hong Kong. Antimicrobial Agents and Chemotherapy43:1310–1313.
Ho PL, Que TL, Tsang DN, Ng TK, Seto WH. 1999b. Characterization of fluoroquinolone-resistant Streptococcus pneumoniae in Hong Kong. Abstract 818. In: Abstracts of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, September 26–29, 1999.
Ho PL, Tse WS, Tsang KW, Kwok TK, Ng TK, Cheng VC, Chan RM. 2001. Risk factors for acquisition of levofloxacin-resistant Streptococcus pneumoniae: a case-control study. Clinical Infectious Diseases32:701–707.
IOM (Institute of Medicine). 1992. Lederberg J and Shope RE (eds). Emerging Infections: Microbial Threats to Health in the United States.Washington, DC: National Academy Press.
Johnson AP, Warner M, Carter M, Livermore DM. 2002. In vitro activity of cephalosporin RWJ-54428 (MC-02479) against multidrug-resistant gram-positive cocci.Antimicrobial Agents and Chemotherapy46:321–326.
Kaatz GW and Seo SM. 1996. In vitro activities of oxazolidinone compounds U100592 and U100766 against Staphylococcus aureus and Staphylococcus epidermidis. Antimicrobial Agents and Chemotherapy40:799–801.
Kays MB, Smith DW, Wack ME, Denys GA. 2002. Levofloxacin treatment failure in a patient with fluoroquinolone-resistant Streptococcus pneumoniae pneumonia. Pharmaco therapy22:395–399.
Kelly JA, Kuzin AP, Charlier P, Fonze E. 1998. X-ray studies of enzymes that interact with penicillins.Cellular and Molecular Life Sciences54:353–358.
Kotra LP, Golemi D, Vakulenko S, Mobashery S. 2000. Bacteria fight back. Chemistry and Industry341–344.
Loeffler JM, Nelson D, Fischetti VA. 2001. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science294:2170–2172.
Lopez R, Garcia E, Garcia P, Garcia JL. 1997. The pneumococcal cell wall degrading enzymes: a modular design to create new lysins?Microbial Drug Resistance3:199–211.
Lopez R, Garcia JL, Garcia E, Ronda C, Garcia P. 1992. Structural analysis and biological significance of the cell wall lytic enzymes of Streptococcus pneumoniae and its bacteriophage. FEMS Microbiology Letters79:439–447.
Mainardi JL, Legrand R, Arthur M, Schoot B, Van Heijenoort J, Gutmann L. 2000. Novel mechanism of ß-lactam resistance due to bypass of DD-transpeptidation in Enterococcus faecium. Journal of Biological Chemistry275:16490–16496.
Martinez-Martinez L, Conejo MC, Pascual A, Hernandez-Alles S, Ballesta S, Ramirez de Arellano-Ramos A, Benedi VJ, Perea EJ. 2000. Activities of imipenem and cephalosporins against clonally related strains of Escherichia coli hyperproducing chromosomal ß-lactamase and showing altered porin profiles. Antimicrobial Agents and Chemotherapy 44:2534–2536.
Massova I and Mobashery S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and ß-lactamases. Antimicrobial Agents and Chemotherapy42:1–17.
Maveyraud L, Golemi D, Ishiwata A, Meroueh, O, Mobashery S, Samama JP. 2002. High-resolution X-ray structure of an acyl-enzyme species for the class D OXA-10 ß-lactamase. Journal of the American Chemical Society124:2461–2465.
Maveyraud L, Golemi D, Kotra LP, Tranier S, Vakulenko S, Mobashery S, Samama JP. 2000. Insights into class D ß-lactamases are revealed by the crystal structure of the Oxa10 enzyme from Pseudomonas aeruginosa.Structure8:1289–1298.
Maveyraud L, Mourey L, Kotra LP, Pedelacq JD, Guillet V, Mobashery S, Samama JP. 1998. Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A ß-lactamases from the antibiotic-resistant bacteria. Journal of the American Chemical Society120:9748–9752.
Miyashita K, Massova I, Taibi P, Mobashery S. 1995. Design, synthesis and evaluation of a potent mechanism-based inhibitor for the TEM ß-lactamase with implications for the enzyme mechanism. Journal of the American Chemical Society117:11055-11062.
Mobashery S and Azucena E. 2002. Bacterial antibiotic resistance. In: Encyclopedia of Life Sciences.Vol. 2. London: Nature Publishing Group. Pp. 472–477.
Moise PA and Schentag JJ. 2000. Vancomycin treatment failures in Staphylococcus aureus lower respiratory tract infections. International Journal of Antimicrobial Agents16 Suppl 1:S31–34.
Mourey L, Miyashita K, Swarén P, Bulychev A, Samama JP, Mobashery S. 1998. Inhibition of the NMC-A ß-lactamase by a penicillanic acid derivative, and the structural bases for the increase in substrate profile of this antibiotic resistance enzyme. Journal of the Ameri can Chemical Society120:9382.
Nelson D, Loomis L, Fischetti VA. 2001. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proceedings of the National Academy of Sciences98:4107–4112.
Nicolau DP and Ambrose PG. 2001. Pharmacodynamic profiling of levofloxacin and gatifloxacin using Monte Carlo simulation for community-acquired isolates of Strepto coccus pneumoniae. American Journal of Medicine111 Suppl 9A:13S–18S; discussion 36S–38S.
Ochs MM, Bains M, Hancock RE. 2000. Role of putative loops 2 and 3 in imipenem passage through the specific porin OprD of Pseudomonas aeruginosa.Antimicrobial Agents and Chemotherapy7:1983–1985.
Owens RC Jr, Tessier P, Nightingale CH, Ambrose PG, Quintiliani R, Nicolau DP. 2001. Pharmacodynamics of ceftriaxone and cefixime against community-acquired respiratory tract pathogens. International Journal of Antimicrobial Agents17:483–489.
Patera A, Blaszczak LC, Shoichet BK. 2000. Crystal structures of substrate and inhibitor complexes with AmpC ß-lactamase: possible implications for substrate-assisted catalysis. Journal of the American Chemical Society122:10504–10512.
Pinho MG, Filipe SR, de Lencastre H, Tomasz A. 2001a. Complementation of the essential peptidoglycan transpeptidase function of penicillin-binding protein 2 (PBP2) by the drug resistance protein PBP2A in Staphylococcus aureus. Journal of Bacteriology183:6525– 6531.
Pinho MG, de Lencastre H, Tomasz A. 2001b. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proceedings of the National Academy of Sciences.98:10886–10891.
Poole K. 2001. Overcoming antimicrobial resistance by targeting resistance mechanisms. Jour nal of Pharmacy and Pharmacology53:283–294.
Powers JH, Ross DB, Brittain E, Albrecht R, Goldberger MJ. 2002. The United States Food and Drug Administration and noninferiority margins in clinical trials of antimicrobial agents. Clinical Infectious Diseases34:879–881.
Prystowsky J, Siddiqui F, Chosay J, Shinabarger DL, Millichap J, Peterson LR, Noskin GA. 2001. Resistance to linezolid: characterization of mutations in rRNA and comparison of their occurrences in vancomycin-resistant enterococci. Antimicrobial Agents and Che motherapy45:2154–2156.
Richardson DC, Bast D, McGeer A, Low DE. 2001. Evaluation of susceptibility testing to detect fluoroquinolone resistance mechanisms in Streptococcus pneumoniae. Antimicro bial Agents and Chemotherapy45:1911–1914.
Roychoudhury S, Dotzlaf JE, Ghag S, Yeh WK. 1994. Purification, properties, and kinetics of enzymatic acylation with ß-lactams of soluble penicillin-binding protein 2a. A major factor in methicillin-resistant Staphylococcus aureus.Journal of Biological Chemistry 269:12067–12073.
Sahm DF, Karlowsky JA, Kelly LJ, Critchley IA, Jones ME, Thornsberry C, Mauriz Y, Kahn J. 2001. Need for annual surveillance of antimicrobial resistance in Streptococcus pneumoniae in the United States: 2-year longitudinal analysis. Antimicrobial Agents and Chemotherapy45:1037–1042.
Sahm DF, Peterson DE, Critchley IA, Thornsberry C. 2000. Analysis of ciprofloxacin activity against Streptococcus pneumoniae after 10 years of use in the United States. Antimicro bial Agents and Chemotherapy44:2521–2524.
Schentag JJ. 1995. Understanding and managing microbial resistance in institutional settings. American Journal of Health-System Pharmacy52(6 Suppl 2):S9–14.
Schentag JJ. 1998. Antibiotic dosing—does one size fit all? [editorial; comment]. Journal of the American Medical Association279:159–160.
Schentag JJ. 1999a. Optimizing antimicrobial therapy in respiratory-tract infections: new agents, new strategies. Introduction. American Journal of Health-System Pharmacy56(22 Suppl 3):S3.
Schentag JJ. 1999b. Antimicrobial action and pharmacokinetics/pharmacodynamics: the use of AUIC to improve efficacy and avoid resistance. Journal of Chemotherapy11:426– 439.
Schentag JJ. 2001. Antimicrobial management strategies for gram-positive bacterial resistance in the intensive care unit. Critical Care Medicine29(4 Suppl):N100–107.
Schentag JJ, Birmingham MC, Paladino JA, Carr JR, Hyatt JM, Forrest A, Zimmer GS, Adelman MH, Cumbo TJ. 1997. In nosocomial pneumonia, optimizing antibiotics other than aminoglycosides is a more important determinant of successful clinical outcome, and a better means of avoiding resistance. Seminars in Respiratory Infections12:278– 293.
Schentag JJ, Gilliland KK, Paladino JA. 2001a. What have we learned from pharmacokinetic and pharmacodynamic theories?Clinical Infectious Diseases32 Suppl 1:S39–46.
Schentag JJ, Gilliland KK, Paladino JA. 2001b. Reply. Clinical Infectious Diseases 33(12):2091–2096.
Schentag JJ, Nix DE, Adelman MH. 1991. Mathematical examination of dual individualization principles (I): Relationships between AUC above MIC and area under the inhibitory curve for cefmenoxime, ciprofloxacin, and tobramycin. DICP: The Annals of Pharma cotherapy25:1050–1057.
Schentag JJ, Nix DE, Forrest A, Adelman MH. 1996. AUIC—the universal parameter within the constraint of a reasonable dosing interval [editorial; comment]. Annals of Pharmaco therapy30:1029–1031.
Schentag JJ, Strenkoski-Nix LC, Nix DE, Forrest A. 1998. Pharmacodynamic interactions of antibiotics alone and in combination. Clinical Infectious Diseases27:40–46.
Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis.Nature418:884–889.
Shlaes DM and Moellering RC. 2002. The United States Food and Drug Administration and the end of antibiotics. Clinical Infectious Diseases34:420–422.
Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. 1992. Molecular structure of the acyl-enzyme intermediate in ß-lactam hydrolysis at 1.7 A resolution. Nature359:700–705.
Swaney SM, Aoki H, Ganoza MC, Shinabarger DL. 1998. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrobial Agents and Chemotherapy 42:3251–3255.
Thomas JK, Forrest A, Bhavnani SM, Hyatt JM, Cheng A, Ballow CH, Schentag JJ. 1998. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrobial Agents and Chemotherapy 42:521–527.
Thornsberry C, Karlowsky JA, Sahm DF, Jorgensen JH, Weigel LM, Swenson JM, Whitney CG, Tenover FC, Ferraro MJ. 2001. Levofloxacin-resistant Streptococcus pneumoniae: second look. Antimicrobial Agents and Chemotherapy45:2183–2184.
Tsiodras S, Gold HS, Sakoulas G, Eliopoulos GM, Wennersten C, Vankataraman L, Moellering RC Jr, Ferraro MJ. 2001. Linezolid resistance in a clinical isolate of Staphy lococcus aureus. Lancet358:207–208.
Urban C, Rahman N, Zhao X, Mariano N, Segal-Maurer S, Drlica K, Rahal JJ. 2001. Fluoroquinolone-resistant Streptococcus pneumoniae associated with levofloxacin therapy. Journal of Infectious Diseases184:794–798.
Walsh CT. 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature 406:775–781.
Watanabe T, Morimoto A, Shiomi T. 1975. The fine structure and the protein composition of gamma phage of Bacillus anthracis. Canadian Journal of Microbiology21:1889–1892.
Weiss K, Restieri C, Gauthier R, Laverdiere M, McGeer A, Davidson RJ, Kilburn L, Bast DJ, de Azavedo J, Low DE. 2001. A nosocomial outbreak of fluoroquinolone-resistant Strep tococcus pneumoniae. Clinical Infectious Diseases33:517–522.
Whitney CG, Farley MM, Hadler J, Harrison LH, Lexau C, Reingold A, Lefkowitz L, Cieslak PR, Cetron M, Zell ER, Jorgensen JH, Schuchat A. 2000. Increasing prevalence of multidrug-resistant Streptococcus pneumoniae in the United States. New England Jour nal of Medicine343:1917–1924.
WHO (World Health Organization). 1996. The World Health Report 1996: Fighting Dis ease, Fostering Development.Geneva: WHO.
Wong KK and Pompliano DL. 1998. Peptidoglycan biosynthesis. Unexploited antibacterial targets within a familiar pathway. Advances in Experimental Medicine and Biology 456:197–217.
Wootton M, Bowker KE, Holt HA, MacGowan AP. 2002. BAL 9141, a new broad-spectrum pyrrolidinone cephalosporin: activity against clinically significant anaerobes in comparison with 10 other antimicrobials. Journal of Antimicrobial Chemotherapy49:535–539.
Young R. 1992. Bacteriophage lysis: mechanism and regulation. Microbiological Reviews 56:430–481.





































