
4
Strengthening the Current Processes to Evaluate New Ingredients for Infant Formulas
ABSTRACT
In the United States the Food and Drug Administration (FDA) is charged with monitoring the safety of infant formulas. If a manufacturer wishes to add a new ingredient to infant formulas, it must either file a Food Additive Petition or declare it a Generally Recognized as Safe (GRAS) substance through a GRAS Notification process. The GRAS Notification has become the route of choice for the introduction of new ingredients because it is scientifically rigorous, far more efficient, and equally or more transparent than the Food Additive Petition route. The committee found that the current regulatory system is limited by the following factors: (1) the complexity of the system has resulted in regulations that are not well understood by the scientific or medical community; (2) the system does not appear to adequately address the uniqueness of infancy and infant nutrition; (3) if a panel of experts is used to show consensus, there is a lack of guidelines for selecting a qualified and unbiased expert panel to evaluate the safety of the proposed new ingredient; and (4) formal guidelines for in-market surveillance do not exist for infant formulas.
The committee recommends that a set of guidelines be developed to provide a hierarchy of decision-making steps for manufacturers wishing to add new ingredients to infant formulas. In addition, elements of the safety assessments of infant formulas need to be standardized. Along with growth and development, other quality factors to be considered for these safety assessments are tolerance, allergenicity, impact of gastrointestinal flora, interference with bioavailability of other nutrients, and possible nutrient imbalances (if ratios or cofactors are important). To review the existing data and identify additional studies needed, the manufacturer should establish balanced, qualified expert panels in consultation with the regulatory agency. Also, the manufacturer should implement an appropriate in-market surveillance strategy that is based on findings from preclinical and clinical studies and the potential for harm to infants.
INTRODUCTION
Existing guidelines and regulations for evaluating the safety of conventional food ingredients (e.g., vitamins and minerals) added to infant formulas have been adequate in the past; however they were not designed to address the unique needs and vulnerabilities of infants and potential new types of ingredients. This chapter provides an overview of the current regulatory system with a discussion of the existing laws; definitions of safety, quality, food, drugs, and dietary supplements; and routes to add new ingredients to infant formulas in the United States. The committee then describes four major limitations of the current system. Finally, an approach is presented to assess the safety of new ingredients to be added to infant formulas, using the current GRAS Notification process as a starting model. The committee suggests that these guidelines could be applied in other countries as regulation parameters.
THE CURRENT U.S. REGULATORY SYSTEM
In the United States infant formulas and ingredients added to infant formulas fall within the purview of the Office of Food Additive Safety and the Office of Nutritional Products, Labeling and Dietary Supplements of FDA’s Center for Food Safety and Applied Nutrition. The safety of new ingredients added to infant formulas is regulated under Section 409 of the Federal Food, Drug and Cosmetic (FD&C) Act (21 U.S.C. §348), which was the primary focus of the committee.
The Infant Formula Acts of 1980 (P.L. 96-359) and 1986 (P.L. 99-570) were incorporated into the FD&C Act as Section 412 (21 U.S.C §350a), which deals with proper manufacturing, formulation, and quality factors of infant formulas. This chapter will focus mainly on safety issues of ingredients (Section 409), but it should be noted that some aspects of Section 412 (quality factors) also play a critical role in the safety assessment process.
Foods, Dietary Supplements, and Drugs as Defined by the Federal Food, Drug and Cosmetic Act
The definition of foods, dietary supplements, and drugs and their respective safety standards are essential to the understanding of the regulatory differences between these classes of bioactive materials. It is beyond the scope of this chapter to delve into the rationale behind the regulations, but the concept of intended use is a critical differentiator between foods, dietary supplements, and drugs, which are defined in the following manner:
-
Foods are “articles or components of articles used for food or drink for humans or animals” (21 U.S.C. §321 (f)(1)).
-
Drugs are “articles intended for the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals” (21 U.S.C. §321 (g)(1)(b)).
-
Dietary Supplements are products “… intended to supplement the diet that bear or contain one or more of the following dietary ingredients: a vitamin; a mineral; an herb or other botanical; an amino acid; a dietary substance for use by man to supplement the diet by increasing the total dietary intake; or a concentrate, metabolite, constituent, extract or combination of any ingredient described above” (21 U.S.C. §321 (ff)(2)).
Table 4-1 provides a comparison of the safety standards for foods, drugs, and dietary supplements. Infant formulas are considered food and, thus, a risk-benefit analysis would be inappropriate because foods are safe for everyone—young or old, male or female, healthy or
TABLE 4-1 Comparison of Safety Standards Included in Federal Regulations
|
Article |
Safety Standard |
Safety Decision |
|
Foods or Food Additives (Federal Food, Drug and Cosmetic Act) |
Reasonable certainty of no harm |
Food Additive Petition—Food and Drug Administration (FDA) must approve that the material is safe Generally Recognized as Safe—Notifier makes safety determination; FDA reviews notification |
|
Drugs (Federal Food, Drug and Cosmetic Act) |
Risk-benefit |
FDA must prove the material is safe and efficacious |
|
Dietary Supplements (Dietary Supplement Health and Education Act) |
Reasonable assurance that ingredient does not present a significant or unreasonable risk of illness or injury |
FDA must prove the material is not safe |
ill. Their purchase and consumption are unsupervised, unlike drugs for which access is carefully controlled and use is supervised by a physician. However because of the special role of infant formulas in the diet of infants, FDA requires that these products meet certain standards developed under the Infant Formula Act.
Quality Factors and Safety Aspects of Infant Formula
Section 412 of the FD&C Act and associated regulations issued by FDA deal with good manufacturing practices, quality control procedures, quality factors, notification requirements, and records and reports for the production of infant formulas. It is important to understand the concept of “quality factors” when considering the safety of ingredients, even though they are not considered as an integral part of the safety assessment of new substances under Section 409.
Quality factors are discussed in the report of the House Committee on Interstate and Foreign Commerce that accompanied the 1980 Infant Formula Act. An ingredient can only be used commercially (incorporated into infant formulas for sale) after the requirements of Section 412 (e.g., nutrient requirements and quality factors) are satisfied. This means that the new ingredient must be incorporated into the formula and the formula must then be tested for each required nutrient (i.e., protein, fat, essential fatty acids, vitamins, and minerals) under 21 U.S.C. 350a, Section 412(i). However the FD&C Act also mandates that the Secretary of the U.S. Department of Health of Human Services establish requirements for quality factors consistent with the scientific knowledge and states that the nutrient and nutrient level required by the Act may be revised. Under current regulations, the concept of quality factors has not been developed; the requirements focus on meeting the level of specific nutrients.
FDA has proposed to revise several aspects of its infant formula regulations, including requirements for quality factors and Good Manufacturing Practices (GMPs). FDA has proposed to revise “Quality Factors for Infant Formulas” (FDA, 1996), the subpart that defines the minimum quality factors for infant formulas. In these proposed regulations, FDA identifies two factors for which there is scientific basis to define them as quality factors, namely that the formula is “capable of supporting normal physical growth of infants” and “protein is of sufficient biological quality to meet the protein requirements of infants.” FDA, in proposed rule Section 106.3(o), has defined quality factors in a manner that encompasses
several basic concepts, including bioavailability and healthy growth (FDA, 1996). The proposed rule states, “The quality factors, therefore provide a means of evaluating whether a nutrient has become less bioavailable than would be expected, so that it is not sufficiently effective to meet its normal nutritive functions, or whether its bioavailability has been enhanced to a level that raises safety concerns” (FDA, 1996, P. 36179). Furthermore, “FDA considers the concept of healthy growth to be broad, encompassing all aspects of physical growth and normal maturational development, including maturation of organ systems and achievement of normal functional development of motor, neurocognitive, and immune systems” (FDA, 1996, P. 36179). These parameters are also considered in the safety evaluation of any new ingredient (Section 409). Thus the safety aspects and the quality factors of the FD&C Act overlap in that they consider the safety of new ingredients by themselves (Section 409) and as part of the matrix (formula) (Section 412).
Routes to Add New Ingredients to Infant Formulas
Infant formulas are regulated as food. If a manufacturer wishes to add a new ingredient to an infant formula, it must follow one of three routes: it may determine a substance is GRAS without formally notifying FDA, it may file a GRAS Notification with FDA, or it may file a Food Additive Petition (see Box 4-1 for the definition of a food additive) with FDA. The GRAS Notification and the Food Additive Petition processes are described below.
GRAS Notification
GRAS status is based on common knowledge about the safety of the ingredient (the substance and its impurities) throughout the scientific community that is knowledgeable in food toxicology and related disciplines specific to the safety and intended use of the ingredient under consideration. A GRAS evaluation through scientific procedures is based on “generally available and accepted scientific data, information, methods, or principles, which ordinarily are published and may be corroborated by unpublished scientific data, information or methods” (FDA, 1997, P. 18960). There must be a “consensus among qualified experts about the safety of the substance for its intended use.”
If the manufacturer believes the potential new ingredient is GRAS, the manufacturer will proceed to make the safety assessment and GRAS determination. Under the proposed GRAS Notification Rule (FDA, 1997), the manufacturer must declare that a substance is GRAS on the basis of scientific consensus by qualified experts. A manufacturer may convene a panel of
|
BOX 4-1 Definition of a Food Additive “… any substance the intended use of which results or may reasonably be expected to result, directly or indirectly, in its becoming a component of food or otherwise affecting the characteristics of any food … if such substance is not generally recognized, among experts qualified by scientific training and experience to evaluate its safety, as having been adequately shown through scientific procedures* to be safe under conditions of its intended use.” *(or, in the case of a substance used in food prior to January 1, 1958, through either scientific procedures or experience based on common use in food.) SOURCE: Federal Food, Drug and Cosmetic Act (21 U.S.C. §301). |
experts. If the manufacturer concludes that the ingredient is safe for its intended use, the manufacturer notifies FDA. FDA reviews the notification (which includes a summary of the scientific evidence and historic use), and if FDA has no questions, it issues a letter of no objection. If FDA has questions concerning the safety of the ingredient, the manufacturer must satisfactorily answer them. In general, the quantity and quality of the data used to support a GRAS determination is comparable with the quantity and quality of the data used to support a Food Additive Petition.
Food Additive Petition
If a potential new ingredient cannot be determined to be GRAS, the manufacturer must file a petition proposing the issuance of a regulation prescribing the conditions under which the proposed additive may be safely used. The manufacturer supplies FDA with all pertinent data, especially safety data. The agency then conducts a comprehensive review of all the safety data and determines if the ingredient is safe for its intended use. In other words, FDA “owns” the safety decision after it makes an exhaustive, rigorous scientific evaluation of all appropriate safety studies. The process is generally lengthy because FDA has limited resources to review all the data and render a decision across a broad spectrum of ingredients and foods. This brief description of FDA’s safety assessment process should not be interpreted to mean the process is simple; quite the contrary, the process is complex and rigorous.
Limitations of the Current Process
The GRAS Notification has become the route of choice for the introduction of new ingredients because it is scientifically rigorous, far more efficient, and equally or more transparent than the Food Additive Petition route. Still, the current process to assess the safety of ingredients new to infant formulas is complex and presents a number of limitations. The committee reviewed the current process and raised a number of issues that need to be addressed. Some are addressed here, some are addressed elsewhere in this report, and some need further study.
The regulatory scheme for the safety assessment of ingredients new to infant formulas is complex and spread out over several sections of the FD&C Act (Sections 409 and 412) and proposed regulations. In reality, the GRAS Notification is a scientifically rigorous and transparent determination of safety. It is flexible enough to accommodate the broad spectrum of issues, yet rigorous enough to ensure safety of the ingredient. However, for infant formulas, the complexity of the system has resulted in regulations that are not well understood by the scientific or medical community and misconceptions about the safety of GRAS ingredients for infant formulas may exist.
A second limitation of the current system is that it does not appear to adequately address the uniqueness of infancy and infant nutrition. Human milk or formulas are the sole source of nutrition for the first 4 to 6 months of life. Animal models to evaluate the safety of food ingredients in the period from birth to weaning are not addressed in FDA’s Toxicological Principles for the Safety Assessment of Direct Food Additives and Color Additives Used in Food (also known as the Redbook1; see Appendix C) (OFAS, 2001, 2003a). Conducting the
types of toxicological studies that are discussed in the Redbook is a significant challenge (see Chapter 5).
Under current regulations, no measures of efficacy (i.e., health benefits) for the substance in question are required. Section 409 deals with safety of food additives independent of efficacy because efficacy has traditionally been considered a property of drugs and inappropriate for foods unless accompanied by an authorized health claim. Interestingly, many of the components of human milk have drug-like effects (e.g., prevention of disease) and are not considered classic nutrients.
The committee’s primary charge was to assess safety of new ingredients added to infant formulas under U.S. regulations, with possible international applications. Safety and efficacy are clearly separated under U.S. regulations, but may not be so clearly delineated elsewhere. The committee was fully cognizant that efficacy is not a consideration of the GRAS process (Section 409) and focused its attention on matters related to safety as delineated in its charge. However safety and efficacy are not always mutually exclusive attributes and, in the case of infant formulas, which serve as a sole source of nutrition for a vulnerable population, there are overlaps of these parameters in Section 412. Although efficacy was outside the charge to the committee and an in-depth discussion was not attempted, the committee recommends consideration be given to convening a scientific expert committee to explore if benefit could be an appropriate requirement under Section 412.
A third limitation is the lack of guidelines for selecting a qualified and unbiased expert panel that may be used to evaluate the safety of the ingredient. The proposed GRAS regulations do not provide guidance about the panel selection or composition for manufacturers that use this mechanism to achieve consensus about the safety of the ingredient under consideration. Since only a few ingredients specifically intended for use in infant formulas have been evaluated by this route, manufacturers may be uncertain about selecting appropriate panel members until they have more experience with the process.
Nearly 25 percent of GRAS Notifications proposed since 1997 were rejected by FDA due to the inability of the notifier to satisfactorily answer FDA’s questions regarding the substance and its health effects (OFAS, 2003b). The expert panel should have the appropriate experts to ask the right questions and form an opinion that is robust and of the highest scientific integrity. Guidelines for selecting a panel early in the process could improve the efficiency and objectivity of the process.
A fourth limitation is that formal guidelines for in-market surveillance do not exist for infant formulas. Infant formula manufacturers routinely conduct passive surveillance via toll-free calls, contact with health care professionals, and reports from their field sales force. Since infants are unable to communicate verbally, any adverse effects must be observed and reported through the parents or caregiver, thus special attention must be paid to detect adverse or unusual reactions. GRAS status is a time- and exposure-dependent judgment that requires the frequent monitoring of toll-free calls during the first 6 to 9 months after introduction of the product to the market. If the original safety determination was properly conducted, adverse outcomes should be rare and it should take a significant period of time to collect sufficient data in order to reaffirm GRAS status of the ingredient. A review of all pertinent data published and unpublished since the GRAS determination should be conducted approximately 2 to 4 years after introduction of the product. (Chapter 7 provides more details on current practices for in-market surveillance.)
In summary, the current GRAS review process was not designed specifically to address possible concerns for new ingredients in infant formulas or for situations where the sole intended use of the substance is in the only dietary product provided to an individual, as is the case for formula-fed infants.
PROPOSED SAFETY ASSESSMENT PROCESS
Use of a Hierarchical Approach
A hierarchical approach is recommended to assist in determining the appropriate level of assessment by considering: (1) the harm (e.g., toxicity), and (2) the potential adverse effects of a new ingredient. This hierarchical approach will guide the level of assessments to be applied to the new ingredient by considering the following factors:
-
the reversibility of potential harmful effects,
-
the severity and consequences of adverse effects,
-
the time of onset of manifestations of the adverse effects,
-
the likelihood that a new ingredient could adversely affect a specific system, and
-
whether the effect would be common or rare.
This approach to evaluating the safety of new ingredients to be added to infant formulas was based on the uniqueness and vulnerability of the infant population. Therefore each step in the process requires empirical evidence from many disciplines and the application of the highest standards, whether using methods of bioassay, nutritional analysis, or basic chemistry. This approach is valuable in determining the relative importance of potential adverse effects for each specific new ingredient by providing generic templates for different steps in the safety assessment process rather than specific recommendations for each compound. It is neither realistic nor desirable to design individual templates for each new ingredient; rather expert panels can refine the generic templates as needed. This approach is designed for a broad spectrum of ingredients and could be applied to new ingredients to be added to infant formulas regardless of the regulatory process used.
The hierarchical approach is graphically presented in Figure 4-1 and by algorithms throughout the report. It is also applied in Appendix D to long-chain polyunsaturated fatty acids and probiotics. Each algorithm is a step-by-step decision tree that depicts the logic of the process, but it does not denote a particular chronology. For example, a manufacturer may initiate several different studies and procedures at the onset of the process, the results of which could be assessed at different steps in the algorithm. Any new ingredient considered for use in infant formulas must be considered in the context of its form, the matrix, and other ingredients with which it may interact.
RECOMMENDATION: Since infants have distinct needs and vulnerabilities, a set of guidelines should be developed to provide a hierarchy of decision-making steps for manufacturers seeking to add new ingredients to infant formulas. Because specific safety assessments will need to be targeted according to the nature of the ingredient, the set of guidelines should allow for flexibility in the approach, while being rigorous and scientifically based (see Figure 4-1).
Proposed Elements of the Safety Assessment
While each submission is unique and a reflection of the ingredient and its intended use, there is an opportunity to standardize the format and approach to the submission that would streamline its preparation and evaluation. The committee recommends amplifying the current approach without being prescriptive. It should be noted that some of the elements of the system proposed here are currently in place. The committee recognizes that some of its recommendations may require statutory changes.
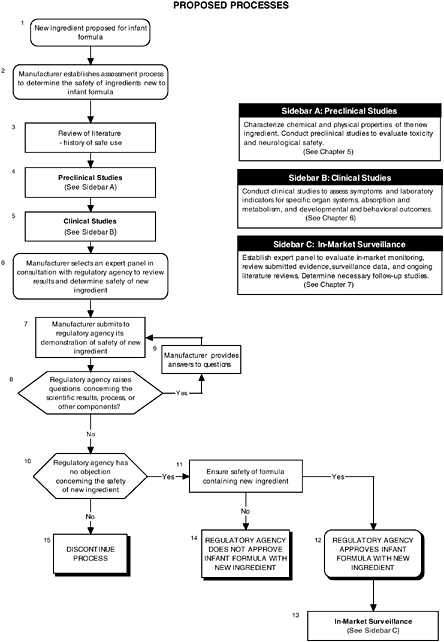
FIGURE 4-1 Proposed process for evaluating the safety of ingredients new to infant formulas algorithm. In-market assessment should be planned in conjunction with preclinical and clinical testing. This algorithm is modeled after the U.S. Generally Recognized as Safe Notification process; similar schemes can be adapted to other regulatory processes. ![]() = a state or condition,
= a state or condition, ![]() = a decision point,
= a decision point, ![]() = an action, sidebar = an elaboration of recommendation or statement.
= an action, sidebar = an elaboration of recommendation or statement.
The committee proposes the following elements, described in detail below, for infant formula-related submissions to the regulatory agency:
-
Introduction
-
Background
-
Chemical or Biological Composition
-
Production Methods
-
Intended Use in Foods
-
Level of Exposure
-
Chemical Structure/Activity Relationships
-
Safety Assessment
-
Other Related Biological Activity or Interactions
-
Hazard Identification and Risk Assessment
-
Expert Panel Findings
-
General Availability of the Data
RECOMMENDATION: Elements of the safety assessments of infant formulas need to be standardized.
Element 1: Introduction
The introduction of a new ingredient submission should include the regulatory background to the submission and a summary of the submission.
Element 2: Background
The special nutritional requirements of infants and the role of formulas as a potential sole source of nutrients should be discussed in the context of the rationale for the addition of the proposed ingredient. A comparison with the composition of human milk should be provided as a reference point. While efficacy and risk-benefit should not be a consideration in the safety determination of an ingredient intended to be used in food, these factors cannot be totally ignored for biologically active substances, as they can be for the more traditional food ingredients (e.g., color, flavor, and preservatives). It must be recognized that it is unlikely that a manufacturer would add an ingredient solely because it is a component of human milk. Thus the rationale for the addition of the ingredient must be provided.
A targeted review of the literature and relevant commercial application within the United States or other countries could be helpful in understanding the novelty of the substance and its use (see Figure 4-1, Box 3). The extent and level of assessments will, in part, be decided upon by an expert panel’s review of the history of use.
Element 3: Chemical or Biological Composition
Many of the substances that can be considered for addition to infant formulas are natural components of human milk, but their source is plants or animals, or they are synthesized. The safety issues therefore often revolve around the purity of the ingredients, as most are components of complex mixtures and may contain other biologically active or possibly toxic substances. The group of active components and the associated impurities must be well characterized (see Figure 4-1, Box 4). If the ingredient is produced synthetically, any by-products and residues must also be well characterized or removed.
Whether natural or synthetic, any complex ingredient must be examined to determine if its other components are toxic or in any way affect the function of the final product; byproducts shown to be of concern should be removed. If appropriate preclinical and clinical evaluations are conducted, this should lessen the safety concern. Sources of new ingredients must be determined to be free of dangerous impurities, such as pesticide residues, heavy metals, toxins, or pathogenic microorganisms. Allergenicity related to the ingredient or byproducts also should be addressed.
For new ingredients not present in human milk, a full review of safety will be required in the submission since there will likely be no history of safe use by infants. Reproducibility of the manufacturing process must be adequately demonstrated and tolerances established for the critical parameters. Complete specifications and analytical methods must be publicly available to allow independent validation of the characterization of the ingredient and associated tolerances. Stability of the ingredient during storage and any subsequent processing should be demonstrated.
Nontraditional sources, such as transgenic products and those produced by new methods, must have programs of evaluation designed to answer concerns related to these methods. For example, a review that includes adequate characterization and documentation of the genetic origins of the starting material and the characteristics of the process should be provided. The active components of the process (e.g., culture, recombinant deoxyribonucleic acid), along with the potential for introducing levels of undesirable compounds (e.g., toxins or allergens), should also be stated. As with other ingredients, the absence of substances that may present a hazard should be demonstrated.
Element 4: Production Methods
A detailed description of the methods used to manufacture the ingredient and the formula into which the ingredient is incorporated must be provided. Trade secret information should be clearly identified. Manufacturing and quality specifications should be established.
Most infant formulas are subjected to some form of heat treatment in order to provide a microbiologically safe product. These may include retorted or overpressure retorted, dried, aseptic, and pasteurized-refrigerated methods. New ingredients may require innovative processing methods to preserve the biological activity for the shelf life of the product. In some cases shelf life may be quite brief, refrigeration may be required, or separate components may need to be mixed at the time of feeding. Any changes in the processing and storage must be evaluated for safety and bioavailability. Individual ingredients have different rates of degradation (e.g., ascorbic acid degrades rapidly). The shelf life of the product depends on the ingredient with the most rapid degradation.
A wide variety of packaging (cans, bottles, composite canisters, pouches) is currently used for infant formulas, and current regulations define methods to assure safety and bioavailability. If new ingredients require novel package forms, the safety assessment must account for this.
GMPs (Good Manufacturing Practices) for infant formulas must also be addressed in this area of the submission to ensure that no unusual safety concerns arise.
Element 5: Intended Use in Foods
The technical, functional, or nutritional rationale for adding the substance to the infant formula should be clearly stated. If label claims will be made (e.g., “improves cognitive
performance,” “lowers cholesterol”), the claim and references to support the claim should be cited. The intended use level should also be provided.
Element 6: Level of Exposure
The estimated daily intake of a substance in the context of the entire diet is of paramount importance in assessing the safety of any food ingredient. In the case of infant formulas, this determination should be straightforward as long as formulas are the sole source of nutrition. When solid food is introduced, a more detailed analysis must be conducted based on dietary records and panel data if the substance naturally occurs in the diet. For example, a careful record (diary) of all food consumed for a period of a few weeks should be kept and then analyzed for the substance in question. Another approach is to analyze available consumption data for foods containing the substance and calculating an estimated level of intake. These values are then combined with the estimated intake from the formula, appropriate safety factors are applied, and a safety determination is conducted.
Element 7: Chemical Structure/Activity Relationships
The analysis of the chemical structure/activity relationships is a useful approach to correlating molecular structure with biological activity or toxicity. In the absence of specific information about the toxicity of a compound, similarity in chemical structure to known toxicants can be used to estimate toxicity. Based on molecular structure, ingredients can be placed in one of the following categories (OFAS, 2001, 2003a):
-
Category A – low potential for toxicity,
-
Category B – intermediate potential for toxicity, or
-
Category C – high potential for toxicity.
Categories, coupled with exposure levels, lead to the classification of ingredients into Concern Levels. Concern Levels are relative measures of the degree to which the use of an ingredient may present a hazard to human health. For example, an ingredient whose structure places it in Category C (high potential) and has a high level of exposure would result in assignment to Concern Level III (high concern), while an ingredient in Category A and low exposure would be placed in Concern Level I. The Concern Levels are used as a starting point to recommend toxicity tests, and the committee recommends using the same criteria for new ingredients added to infant formulas. Table 4-2 summarizes the toxicity tests recommended based on the Concern Levels.
Element 8: Safety Assessment
FDA’s Redbook and the classic principles of toxicology are an excellent starting point for assessing the safety of new ingredients, but blindly applying the Redbook would be clearly inappropriate in the case of infant formulas. Many of the ingredients being considered for addition to infant formulas are naturally occurring nutrients in human milk, yet simply because an ingredient is a component of human milk does not mean it is safe. The matrix effect, ratios of other components, and interactions with other ingredients must be taken into account before a safety conclusion can be drawn for an infant formula. For
TABLE 4-2 Summary of Toxicity Tests Recommended for Different Levels of Concern of Food Ingredients
|
Toxicity Testsa |
Concern Levels |
||
|
I |
II |
III |
|
|
Short-term tests for genetic toxicity |
X |
X |
X |
|
Metabolism and pharmacokinetic studies |
|
X |
X |
|
Short-term toxicity tests with rodents |
Xb |
|
|
|
Subchronic toxicity tests with rodents |
|
Xb |
Xb |
|
Subchronic toxicity tests with nonrodents |
|
Xb |
|
|
Reproduction study with teratology phase |
|
Xb |
Xb |
|
One-year toxicity tests with nonrodents |
|
|
X |
|
Carcinogenicity study with rodents |
|
|
Xc |
|
Chronic toxicity/carcinogenicity study with rodents |
|
|
|
|
aNot including dose range-finding studies, if appropriate. bIncluding neurotoxicity and immunotoxicity screens. cAn in utero phase is recommended for one of the two recommended carcinogenicity studies with rodents, preferably the study with rats. dCombined study may be performed as separate studies. SOURCE: OFAS (2001). |
|||
ingredients that are already a part of human milk, the amount of toxicological testing required should be reduced, but the following must be thoroughly considered:
-
What is the level of addition and interaction with other nutrients, processing, and storage?
-
Are there any differences in absorption, distribution, metabolism, and excretion?
-
Is the added ingredient chemically identical to the substance contained in human milk?
-
Were contaminants introduced via the manufacturing process?
-
Will the ingredient be safe for the shelf life of the product?
In addition to these considerations, the classic issues of subchronic toxicity, repeat-dose target organ toxicity, genotoxicity, carcinogenicity, reproductive and developmental toxicity, and neurotoxicity should be considered and, where appropriate, in vitro and in vivo animal testing should be conducted (see Figure 4-1, Box 4). Animal models must be chosen appropriately to extrapolate results to humans (see Chapter 5). Dose, bioavailability, nutritional requirements, and developmental stage must be considered to prevent confounding the toxicology results.
Currently clinical trials for infant formulas with added new ingredients, although recommended, are not required by FDA, and the manufacturer decides when and if clinical trials are needed. The required tests focus on nutrient requirements, and only in cases of minor changes to the formulation can the manufacturer request a waiver of the tests requirements. However in the proposed rule (Section 106.3(o), subpart E, “Quality Factors for Infant Formulas”), quality factors are defined in terms of growth and development of the infant and therefore would require clinical trials as evidence of normal growth. The committee recommends that those clinical trials that address safety of the ingredient, metabolites, and matrix be included as part of the submission (see Figure 4-1, Box 5).
Further complicating the safety evaluation is the fact that most of the new ingredients are nutrients or biologically-active molecules that supposedly confer health benefits—not classic toxicants. In addition, some of the new ingredients are macronutrients, which require the application of different safety factors, experimental designs, and interpretation of results. In most cases micronutrients fed at the high doses typically used in toxicological testing would be toxic, thus safety factors of 10 or less are more appropriate than safety factors of 100.
Element 9: Other Related Biological Activity or Interactions
Early studies of the isolated ingredient may be helpful to identify biomarkers and to help in the design of more complex products. In addition to identifying any toxic or untoward reactions, preclinical studies may help identify appropriate levels of use. New ingredients should be evaluated in the matrix expected to be used in the final product in order to determine absorption, utilization, and activity when other ingredients are present. For example, soy formulas contain phytate, which can bind zinc, iron, and other divalent cations and make them unavailable. To allow for this, soy formulas are fortified with zinc, iron, and calcium. They also provide relatively large amounts of iron to allow for decreased absorption.
Ingredients are often available in more than one form. For example, selenium may be added to formulas as an inorganic salt or as an organic compound. Selection of the form most desirable for biological availability may present product-development challenges, but clear bioavailability is critical (MacLean and Benson, 1989). For infants, human milk and formulas are the sole source of nutrition for the first 4 to 6 months of life and an important source for the next 8 months. Although it is assumed that many new ingredients will be components of human milk or already accepted as safe for adults and children, the matrix in which the new ingredient is incorporated is very relevant in the safety evaluation (currently covered under Section 412). The importance of the matrix can be simply illustrated by the differences in cow-milk and soy-based formulas. Soy-based formulas may contain phytoestrogens, which could impact on the biological activity of a new ingredient. In addition to biological activity, factors in soy-based formulas also might impact bioavailability. For example, the fatty-acid profile of the matrix may differ, and this could impact on the metabolism of fatty acids added to provide a certain benefit. Since infants are a vulnerable population, the rationale for adding any new ingredient must be clearly stated so that qualified experts can make the safety evaluation with all factors known.
As these examples show, any new ingredient considered for use in infant formulas must be considered in the context of its form, the matrix, and other ingredients with which it may interact. Specific procedures for assuring bioavailability will depend on the compound under consideration.
RECOMMENDATION: Bioavailability is of special importance to infants and should be specifically addressed in any evaluation of the safety of infant formulas. Other factors that should be considered are: tolerance, allergenicity, impact of gastrointestinal flora, and possible nutrient imbalances (if ratios or cofactors are important).
Element 10: Hazard Identification and Risk Assessment
As stated previously, both the subject ingredient and associated contaminants or impurities may be hazardous to the target population, in this case term infants. The risk assessment for the ingredient versus the impurities are likely to be quite different. A no-observed-
adverse-effect level, safety factor, and estimated daily intake need to be determined, taking into account the differences in risk posed by the ingredient and possible contaminants. This will need to be determined for each new ingredient by each new expert panel.
Element 11: Expert Panel Findings
The findings of the expert panel are the heart of the safety evaluation submission. A properly constituted panel must be objective and consist of the appropriate types of scientific experts. In other words, the panel should have the appropriate experts to ask the right questions and form an opinion that is robust and of the highest scientific integrity. The manufacturer, in consultation with the regulatory agency, should determine an appropriate expert panel early in the process (see Figure 4-1, Box 6) to:
-
guard against the theoretical possibility of bias,
-
ensure an efficient process, and
-
minimize the chance of rejection after submission.
The experts serving on the panel need to be free of any conflicts of interest (e.g., former employees or current stockholders of the manufacturer). Although there is no evidence of biased panels in the past, the potential for bias is a theoretical concern that needs to be addressed when selecting panel members.
The committee strongly recommends that in selecting appropriate experts to analyze ingredients new to infant formulas, the expert panel should include a physician with experience in clinical assessment, preferably a pediatrician. The composition of the rest of the panel should be determined in consultation with the regulatory agency and will depend on the nature of the ingredient (e.g., if dealing with probiotics, a microbiologist and immunologist should be included on the panel). A subpanel of specific experts may be needed in certain instances (e.g., when certain levels of in-market surveillance are needed; see Chapter 7). The panel members must be recognized experts in their field of expertise and highly regarded by their peers and the regulatory agency. They must conduct an independent critical evaluation of the information that is publicly available and conclude that they, as well as other experts in the field, would generally recognize that the ingredient is safe for its intended use.
The regulatory agency would review the panel’s safety assessment and other data in the submission and if satisfied, would issue a letter of no objection. If the regulatory agency has questions and the manufacturer does not answer them satisfactorily, the agency can reject the submission, ask the company to withdraw it, or suggest consultation with additional experts qualified to opine on the specific safety concerns.
RECOMMENDATION: In seeking to add new ingredients to infant formulas, manufacturers should establish balanced, qualified expert panels in consultation with the regulatory agency. The panel members should review existing data and may identify a need for additional studies.
Element 12: General Availability of the Data
The requirement for all the pivotal data to be generally available for scrutiny by the scientific community at large makes the process transparent and robust. The data on which the safety determination is based must be published in peer-reviewed scientific journals and could be supplemented by secondary scientific literature, such as review articles and text-
books. For example, FDA’s letter of no objection summarizing the basis for the GRAS Notification is made available on FDA’s website (OFAS, 2003b), and the full notification should be available through the Freedom of Information Act. The responsibility for the safety determination rests with the company filing the notification, and it is the company’s continuing responsibility to ensure that the food ingredients they market are safe and in compliance with all applicable legal and regulatory requirements.
SUMMARY
The safety assessment of infant formulas is complex and not fully standardized. While the current processes that regulate the addition of new ingredients to infant formulas are both flexible and scientifically rigorous, they do not adequately address the uniqueness of infants and infant nutrition. Also, they do not provide either enough guidelines on the selection of an appropriate expert panel that may be used to show consensus or guidelines for in-market surveillance. There is an opportunity to address these limitations and standardize the elements of the safety assessment of ingredients new to infant formulas without being overly prescriptive. The recommendations described here are meant to address the specific needs of infants in improving the regulatory process for this potentially vulnerable population group.
REFERENCES
FDA (Food and Drug Administration). 1996. Current good manufacturing practice, quality control procedures, quality factors, notification requirements, and records and reports, for the production of infant formula. Proposed rule. Fed Regist 61:36153–36219.
FDA. 1997. Substances Generally Recognized as Safe. Proposed rule. Fed Regist 62:18937–18964.
MacLean WC Jr, Benson JD. 1989. Theory into practice: The incorporation of new knowledge into infant formula. Semin Perinatol 13:104–111.
OFAS (Office of Food Additive Safety). 2001. Toxicological Principles for the Safety Assessment of Direct Food Additives and Color Additives Used in Food. Redbook II-Draft. Washington, DC: OFAS, Center for Food Safety and Applied Nutrition, Food and Drug Administration.
OFAS. 2003a. Redbook 2000. Toxicological Principles for the Safety of Food Ingredients. Online. Center for Food Safety and Applied Nutrition, Food and Drug Administration. Available at http://www.cfsan.fda.gov/~redbook/red-toca.html. Accessed November 19, 2003.
OFAS. 2003b. Summary of all GRAS Notices. Online. Center for Food Safety and Applied Nutrition, Food and Drug Administration. Available at http://www.cfsan.fda.gov/~rdb/opa-gras.html. Accessed November 19, 2003.















