
2
PROGRESSION OF SPINAL CORD INJURY
Injury to the spinal cord triggers a cascade of biological events that unfold within seconds and that proceed for months or even years. The events affect three major bodily systems: the nervous system, the immune system, and the vascular system. These systems interact dynamically as they respond to injury. Although some injurious responses heal and promote the recovery of function, others leave a wave of tissue damage that expands well beyond the original site of injury.
The choreography of tightly interwoven responses that lead to dysfunction is known as injury pathophysiology. The final outcome of serious spinal cord injury is shattering: loss of reflexes, loss of sensation, and paralysis (i.e., the loss of control over muscles and movement of the body). Although much has been learned about the progression of spinal cord injuries and the biochemical reactions and pathways that are involved in the process, much remains to be explored. With understanding of injury pathophysiology comes the ability to interfere with its progression, to harness the regenerative potential of the spinal cord, to improve therapies, and to create new ones.
This chapter on the biology of spinal cord injuries provides a broad overview of spinal cord anatomy, injury types, and injury classification. The chapter discusses the cellular and molecular events underlying the body’s response to injury—both pathological and protective—and covers the biological basis of pain, as well as functional losses involving muscles, sensory organs, the bladder, and the bowel. The basic information in this chapter serves as a backdrop for later chapters on therapeutic approaches.
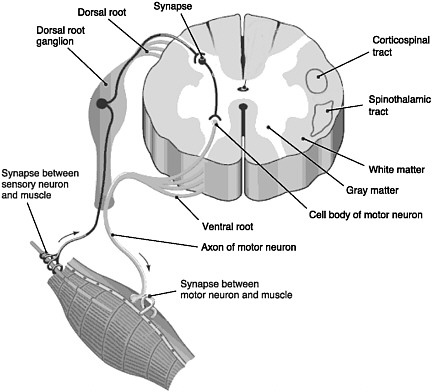
FIGURE 2-1 Cross section of the spinal cord.
SOURCE: Reprinted with permission, from Taber and Thomas, 1997. Copyright 2003 by F. A. Davis Company.
SPINAL CORD, NERVES, MUSCLES, AND THE SENSORY SYSTEM
The spinal cord is the elongated portion of the central nervous system (CNS) that connects the brain to all muscles of the body and most sensory nerves to the brain.1 It is surrounded and protected by vertebrae, or the spinal column. The outer edge of the spinal cord is the white matter (Figure 2-1), which contains the branching portions of nerve cells known as axons. Wrapping around the axons is a fatty whitish substance called myelin, which speeds up the nerve impulses from the brain to the rest of the body. In addition to an axon, each nerve cell (or neuron) has a cell body, which is its control center housing the nerve cell genes and other parts needed to
produce energy and to make proteins. The cell bodies of neurons cluster together in the gray matter of the spinal cord and are not located in the white matter. Two regions of the gray matter are of special interest: the ventral horn and the dorsal horn. The ventral horn contains the cell bodies of motor neurons, which induce muscles to contract. The dorsal horn contains the primary sensory pathways that transmit information from the skin and muscles into the spinal cord and up to the brain. The cell bodies of sensory neurons lie outside the spinal cord in a discrete cluster known as the dorsal root ganglion.
Spinal Cord Anatomy as the Basis for Injury Classification
The term “spinal column” refers to the vertebral bones and discs that collectively encase and protect the soft tissue of the spinal cord. The spinal cord is made up of nerve tracts carrying signals back and forth between the brain and the rest of the body. The spinal cord is traditionally divided into four levels, beginning with the highest (most rostral) portion and ending with the lowest (most caudal) portion: cervical, thoracic, lumbar, and sacral (Figure 2-2). Each of the four levels of the spinal cord controls the functions of a particular region of the body through a defined set of spinal nerves that enter and exit the spinal nerve roots (Figure 2-1) through particular openings in the vertebrae. Injury at one level can often lead to the loss of sensory and motor functions below that level because the injury disrupts nerve conduction to and from the brain (Table 2-1).
On the basis of pathology, there are at least three general types of spinal cord injuries: contusion, laceration, and solid cord injuries (Table 2-2). Contusion injuries, often of the cervical spine, are the most frequent and can be simulated in the most widely used animal models (see Chapter 3). The type of spinal cord injury, as well as its level and severity, dictates its functional impact and prognosis.
In an effort to systematize the classification of spinal cord injuries, the American Spinal Injury Association (ASIA) developed in 1992 a uniform and comprehensive way of assessing the level and extent of injury severity. The ASIA International Standards for Neurological Classification are based on the systematic examination of neurological function to assess any deterioration or improvement in neurological function throughout the course of the injury. The classification, which has prognostic, therapeutic, and research value, has four components: (1) sensory and motor levels, (2) the completeness of the injury, (3) the ASIA Impairment Scale, and (4) the zone of partial preservation for complete injuries (ASIA, 2000).
The sensory and motor levels refer to the spinal location of the injury and indicate the lowest (most caudal) segment with normal function. The sensory level is identified after extensive testing of skin areas via light touch
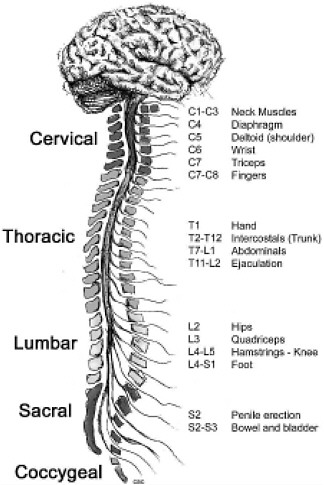
FIGURE 2-2 Functions controlled by nerves at different levels of the spine. Damage at a particular level usually impairs the functions controlled by all nerves at lower levels.
SOURCE: Reprinted with permission, from CRPF, 2002. Copyright 2002 from CRPF.
and pinprick. Predefined regions of the skin covering the whole body, called dermatomes, are each scored as having normal, impaired, or absent sensation. Similarly, the motor level is determined by manual testing and grading of the strengths, on a scale of 0 to 5, of 10 muscle groups that control different motor functions, including limb, bowel, and bladder functions. Although the sensory and motor levels may differ somewhat, they both come under an umbrella “neurological level,” which is defined as the most
TABLE 2-1 Spinal Cord Levels and Areas of Control
|
Spinal Cord Section |
Spinal Cord Levels |
Areas of Controla |
Likely Condition After Injury |
|
Cervical |
C1-C8 |
Head, neck, diaphragm, arms |
Tetraplegic |
|
Thoracic |
T1-T12 |
Chest, abdominal muscles |
Paraplegic |
|
Lumbar |
L1-L5 |
Hips, legs |
Paraplegic |
|
Sacral |
S1-S5 |
Bowel, bladder, groin, calves, buttocks, legs |
Paraplegic |
|
aAn injury at a given level indicates that the portion of the spinal column beneath the site of injury will likely be affected. SOURCES: El Masry et al., 1996; ASIA, 2000; Young, 2002. |
|||
TABLE 2-2 Types of Spinal Cord Injuries
|
Type of Spinal Cord Injury |
Percentage of Total Injuries |
Description |
|
Contusion |
25 to 40 |
Bruising, but not severing, of the spinal cord |
|
Laceration |
25 |
Severing or tearing of the spinal cord and introduction of connective tissue into the spinal cord, typically from gunshot or knife wounds |
|
Solid cord injury |
17 |
Axon injury and demyelination |
|
SOURCES: Bunge et al., 1993, 1997; Harper et al., 1996; Hulsebosch, 2002. |
||
caudal segment of the spinal cord with both normal sensory and motor functions.
The completeness of the injury gives a strong indication and prognosis of the severity of the injury, and it serves as the basis for the ASIA Impairment Scale (described below). A complete injury relies on the detection of any neurological function below the site of the injury (Levi, 2004), especially the loss of motor and sensory functions in the lowest sacral region of the spinal cord (S4 and S5), which supplies nerves to the anal and perineal regions. Few people with a complete spinal cord injury regain the useful function of this region (Levi, 2004). An incomplete injury, on the other
TABLE 2-3 ASIA Impairment Scale
|
ASIA Grade |
Level of Impairment |
|
A |
No motor or sensory function preserved in the lowest sacral segments (S4 and S5) |
|
B |
Sensory but no motor function preserved, including the lowest sacral segments (S4-S5) |
|
C |
Motor function present below the injury, but the strengths of more than half of the key muscles are graded < 3 of 5 |
|
D |
Motor function present below the injury, but the strengths of more than half of the key muscles are graded ≥ 3 of 5 |
|
E |
Motor and sensory functions in key muscles and dermatomes are normal |
|
SOURCE: Reprinted with permission, from ASIA, 2000. Copyright 2000 by ASIA. |
|
hand, leaves a person with some sensory or motor function below the site of injury and in the lowest sacral region. It is important to assess sacral sensation when investigating the completeness of an injury, because there is the potential for partial function to be preserved in this area and this may be the only evidence of neurological function below an injury.
The ASIA Impairment Scale provides clinicians with a standard way of grading the functional severity of a spinal cord injury (Table 2-3). The scale has one grade for complete injuries (ASIA A), three others grades for incomplete injuries (ASIA B through ASIA D), and another for no impairment from the injury (ASIA E). To assign one of the three grades for incomplete injuries (ASIA B through ASIA D), clinicians determine the degree of muscle strength (on a scale from 0 to 5, with 0 being total paralysis and 5 being active movement against full resistance) of the key muscles below the neurological level of the injury. The assignment is based on the extent to which more than half of the key muscles have a muscle strength grade of 3 or higher.
The zone of partial preservation applies only to complete injuries (ASIA A). It refers to the area of the spinal cord that still retains some motor or sensory function above the level of S5 (and below the level of injury). For example, a person might be classified as having a zone of partial preservation at T1 to T3, meaning that he or she has some degree of sensory or motor function at that level of the thoracic spinal cord, even though the injury is complete. A zone of partial preservation is likely due to the presence of intact fiber pathways. About 65 percent of individuals with neuro-
logically complete injuries show some amount of tissue and axonal sparing across the site of the lesion (Bunge et al., 1997).
THREE PHASES OF SPINAL CORD INJURIES
A spinal cord injury immediately injures or kills cells, but it also causes delayed damage and death to cells that survive the original trauma. The biological response to a spinal cord injury is divided into three phases that follow a distinct but somewhat overlapping temporal sequence: acute (seconds to minutes after the injury), secondary (minutes to weeks after the injury), and chronic (months to years after the injury). A general overview of the three phases is presented in Table 2-4. Diverse groups of cells and molecules from the nervous, immune, and vascular systems are involved in each phase. Most participating cells reside in the spinal cord, but others are summoned to the site of injury from the circulatory system (Table 2-5). To carry out many of their functions, the cells depend on changes in gene expression. As would be expected, injury triggers certain cells to up-regulate (increase expression) or down-regulate (decrease expression) genes responsible for a host of proteins involved in inflammation, neurotransmission, regrowth and repair, and other local responses to injury (Bareyre and Schwab, 2003). The final pattern of sensory and motor losses from the
TABLE 2-4 Major Features of the Three Phases of Injury
|
Acute (Seconds after Injury) |
Secondary (Minutes to Weeks) |
Chronic (Months to Years) |
|
|
|
|
SOURCES: Sekhon and Fehlings, 2001; Hulsebosch, 2002. |
||
TABLE 2-5 Cell Types Involved in Spinal Cord Injuries
|
Cell Type |
Function and Description |
|
|
|
|
|
|
|
|
|
|
|
|

|
Cell Type |
Function and Description |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SOURCES: Reprinted with permission, from Lentz, 1971. Copyright 1971 by Elsevier, Inc.; Dammann et al., 2001. Reprinted with permission from Elsevier; Reprinted with permission, from Stimson, 2001. Copyright 2001 by Muscular Dystrophy Association; Reprinted with permission, from Caceci and El-Shafey, 2002. Copyright 2002 by Caceci; Reprinted with permission, from Merck & Co., Inc., 2004. Copyright 2004 by Merck & Co., Inc.; Reprinted with permission, from Shier et al., 2004. Copyright 2004 by McGraw-Hill Higher Education. |
|


three phases of injury depends on which nerve cells and fiber tracts die, which remain intact, and which regenerate or form new branching patterns to compensate for the losses.
The elucidation of the distinct phases of injury—and the cellular and molecular events underlying them—comes largely from animal models. As events unfold, many of the cellular and molecular events designed to heal the injury can paradoxically lead to further neuronal injury or death. The site of injury may spread to adjacent areas of the spinal cord, sometimes extending four spinal segments above and below the initial site (Crowe et al., 1997; Liu et al., 1997). The affected area becomes filled with immune cells, and a “scar” is formed. The details of the pathophysiology continue to evolve.
Acute Phase
The acute phase, which begins within seconds of the injury, is marked by systemic as well as local events (Tator et al., 1998; Hulsebosch, 2002). The foremost systemic event, after a fleeting increase in blood pressure, is a prolonged decrease in blood pressure (hypotension) that sometimes coincides with a decrease in blood volume. Systemic hypoxia, a reduction of the oxygen supply to the tissues, occurs if, during the injury, respiration is compromised by airway obstruction or by paralysis of diaphragm muscles. Failure of the spinal cord to function for the first 2 to 24 hours after injury, a condition known as spinal shock, results from inadequate flow of oxygen and nutrients into the tissue.
Numerous local events within the spinal cord occur immediately after the injury and also contribute to spinal shock. Direct trauma from injury causes necrosis (cell death) to spinal cord neurons and to the endothelial cells lining the blood vessels of the spinal cord. The surviving neurons at the site of injury respond with a procession of electrical impulses, known as action potentials. Because action potentials require the influx and efflux of ions across the neuron’s membrane, the barrage of action potentials creates significant local shifts in ion levels. Higher ion levels are also produced by the mechanical shearing of nerve cells, causing their membranes to rupture and release their contents. Ion buildups can reach levels toxic enough to kill nearby neurons. Similarly, the barrage of action potentials causes the release of excess amounts of neurotransmitters, which then accumulate in the synapses between nerve cells. The accumulation of certain neurotransmitters (e.g., glutamate) can cause the death of nearby neurons through a mechanism called excitotoxicity (Faden and Simon, 1988). Neuron death, by whatever mechanism, contributes to the losses of sensory, motor, and autonomic functions that occur after spinal cord injury.
Direct trauma to the spinal cord causes its small blood vessels to hem-
orrhage, consequently disrupting the blood-spinal cord barrier, which normally helps protect the CNS. Rapid bleeding into the normal fluid-filled spaces of the spinal cord contributes to local edema. As the swelling within the confined space of the vertebral canal continues, it impinges on and further compromises nerve cells. Other important vascular changes are vasospasm, which is a narrowing of the blood vessels that often decreases blood flow by 80 percent (Anthes et al., 1996) and small-vessel thrombosis (Koyanagi et al., 1993). These vascular perturbations result in ischemia (i.e., deprivation of neurons and other cells of the oxygen and the other nutrients that they need to survive). Perhaps because it is more vascularized, the spinal cord’s gray matter (which contains neuron cell bodies) is far more necrotic after injury than the white matter, which contains large tracts of myelinated fibers (axons) that traverse up and down the spinal cord (Wolman, 1965).
Secondary Phase
The secondary phase sets in minutes after injury and lasts for weeks. During this phase the area of injury markedly expands. The secondary phase features a continuation of some events from the acute phase—electrolyte shifts, edema, and necrotic cell death—as well as novel ones, including the formation of free radicals, delayed calcium influx, immune system response (inflammation), and apoptotic cell death.
Formation of Free Radicals
Free-radical formation, usually from oxygen atoms, gives rise to a series of pathological reactions inside cells, including the breakdown of lipids in the cell membrane, a process known as lipid peroxidation. The cell tolerates some degree of lipid peroxidation, but if it is substantial, the cell membrane becomes so disrupted that it bursts and dies. As it dies, the cell spills its contents into the extracellular space, which then threaten neighboring cells. For example, the spillage of the neurotransmitter glutamate can cause the death of nearby cells. If free radical attack does not lyse (burst) the cell membrane, it can invoke other types of damage. Free radicals, for example, can also attack membrane enzymes, distort ion gradients across the cell membrane, and damage genes.
The process of free-radical formation from oxygen begins in the mitochondria, a specialized portion of the cell devoted to converting oxygen into energy-rich molecules. Injury brings an influx of calcium into the cell, which can trigger the process of free-radical formation (Young, 1992). Oxygen atoms lose one of their outermost electrons and become highly reactive. To become more stable, they lure electrons from nearby atoms. In
the case of lipid peroxidation, for example, free radicals draw an electron from a lipid molecule, which in turn becomes less stable, thus launching a chain reaction that ultimately leads to lysis of the membrane and death by necrosis.
Delayed Calcium Influx
Although neurons require some intracellular calcium for their normal function, too much calcium is injurious because it activates damaging enzymes and destructive processes and can trigger the formation of free radicals. Some calcium enters neurons at the time of injury and contributes to the acute phase of damage. An additional influx of calcium is triggered by the acute injury and continues for hours afterwards. A particularly powerful mode of calcium influx within injured axons in white matter involves an initial inward leakage of sodium due to the acute injury, which drives the sodium-calcium exchanger to import damaging levels of calcium; this multistage cascade has been demonstrated within myelinated axons of the optic nerve (Stys et al., 1992b) and the spinal cord (Imaizumi et al., 1997). This delayed calcium influx is an important target for interventions because, by blocking it, it is possible to reduce the degree of secondary damage to myelinated spinal cord axons (Stys et al., 1992a).
Immune System Response
The inflammatory response to injury involves four major categories of immune cells: neutrophils, monocytes, microglia, and T-lymphocytes (Schnell et al., 1999; Bareyre and Schwab, 2003). The neutrophils are the first immune cells to arrive at the site of injury. They are recruited there from the circulatory system, especially by vascular endothelial cells, which up-regulate and express adhesion molecules on their cell membranes to help guide neutrophils to the site of injury. Once the neutrophils have entered the spinal tissue, they remove microbial intruders and tissue debris. This is accomplished in many ways, especially through the release of toxic molecules and antibacterial agents (e.g., myeloperoxidase). Neutrophils also release cytokines, proteases, and free radicals, all of which activate other inflammatory and glial cells for the inflammatory cascade that can ultimately lead to neuron injury or death. Cytokines, which are soluble proteins released by most types of inflammatory cells, act as signals between immune cells and carry out immune functions. Neutrophils are the initial dominant cells involved in the immune response.
Over the next 24 hours, microglia respond in earnest. Monocytes begin to enter from the circulatory system and, after they penetrate the spinal cord tissue, differentiate into macrophages. Microglia, on the other hand,
actually reside within the spinal cord. Once these cells are activated, they too remove degenerating fiber tracts and other tissue debris by phagocytosis. They also secrete numerous cytokines, free radicals, and growth factors, which, in turn, affect nearby cells in positive and negative ways (Lindholm et al., 1992; Schnell et al., 1999; Anderson, 2002). The growth factors are critical for neuron survival and tissue repair. However, free radicals and proinflammatory cytokines contribute to expansion of the lesion, worsening the impact of the injury. Activation of macrophages and microglia is sustained over the course of weeks.
The role of lymphocytes in spinal cord injuries is somewhat controversial. Some argue that one type of lymphocyte (autoreactive T-lymphocytes) have destructive properties: according to this schema they exacerbate injury to axons and induce demyelination, leading to functional loss (Popovich and Jones, 2003). Others argue that this lymphocyte is not pathological but, rather, confers protection to the myelin-insulated neurons (Schwartz and Kipnis, 2001; Kipnis et al., 2002). Protection of myelin also protects the integrity of the axon that it insulates.
Apoptotic Cell Death
During the acute phase, the mechanical trauma to the spinal cord causes cells to die instantaneously by necrosis, a process of cell swelling and then cell membrane rupture. Within hours, however, another type of cell death assumes center stage: apoptosis. This very active form of death afflicts neurons, oligodendrocytes, astrocytes, and other cells of the spinal cord after injury (Liu et al., 1997; Beattie et al., 2000). Apoptosis has been detected in humans (Emery et al., 1998) and lasts for about one month in animal models (Beattie et al., 2000). With apoptosis, cells do not swell before death; rather, they condense and break apart into small fragments in a very orderly process that requires energy and protein synthesis. These fragments of the apoptotic cell are engulfed by other cells in a process that prevents spillage of the dying cells’ contents and avoids elicitation of an inflammatory response. Necrotic cell death, on the other hand, elicits inflammation and spills out neurotransmitters and other contents that build to levels toxic enough to harm or kill nearby cells.
What triggers apoptosis after spinal cord injury? An answer to this question would immediately open up new targets for treatments that could prevent apoptosis from occurring. A major trigger appears to be the injury-induced rush of calcium into cells (Young, 1992). Calcium influx activates key enzymes inside the cell—the caspases and calpain—that break down proteins in the internal cytoskeleton and membrane of the cell (Ray et al., 2003). With the destruction of its structural integrity, the cell dies. Yet, apoptosis of cortical motor neurons can occur after the axons centimeters
away are severed by spinal cord injury, too far for the calcium to diffuse (Hains et al., 2003a). Therefore, besides calcium influx, there are likely other triggers of apoptosis in spinal cord injury.
Chronic Phase
The chronic phase of spinal cord injury sets in over a period of months to years. The chronic phase is marked by the emergence of new types of pathology at both the microlevel and the macrolevel (e.g., the formation of a fluid-filled cavity or a glial scar). At the microlevel, the death of oligodendrocytes has an amplifying effect. Because most oligodendrocytes myelinate (i.e., insulate) about 10 to 40 nerve axons, the loss of one oligodendrocyte can leave many healthy nerve axons without conduction capacity. If nerve conduction is stopped entirely, the spinal cord cannot transmit signals to the brain and body, even though axons may be intact. Axons undergo molecular changes, such as alteration of the ion channels that are normally responsible for propagating electrical impulses through nerves (Waxman, 2001; Hains et al., 2003b). The combination of myelin loss and altered ion channel function, among other changes, can lead to molecular changes in the surviving neurons that can produce chronic pain in animals with experimental spinal cord injuries. At the macrolevel, the lesion site becomes increasingly devoid of normal tissue and begins to form a fluid-filled cavity or a glial scar, or both. The cavity forms within a few weeks of injury in animal models and may extend several segments above and below the site of injury. The cavity creates a physical gap that blocks axon regrowth, whereas the glial scar contains substances that inhibit axon regrowth.
Glial Scar Formation
Glial scarring (also known as reactive gliosis) creates an environment that inhibits axon regeneration. The glial scar is an extracellular matrix that contains astroyctes, microglia, and oligodendrocytes. It grows in size over time, from weeks to months after the injury, but the groundwork is set within hours of the injury. That is when the remnants of the acute phase—myelin debris and damaged axons—begin to accumulate at the site of the injury. The remnants begin to attract an array of different types of glial cells, from oligodendrocytes and their precursors to activated microglia and astrocytes. Astrocytes are most commonly found in the scar, and they are tightly bound to one another (Fawcett and Asher, 1999). If the spinal cord has been penetrated, meningeal cells, which normally form a protective layer around the spinal cord, also accumulate at the lesion site. Each type of cell expresses and/or releases a host of inhibitory molecules (Table 2-6).
TABLE 2-6 Cells and Molecules That Inhibit Axon Regeneration
|
Cell Type |
Inhibitory Molecule |
|
Oligodendrocyte |
NI-250 (Nogo-A) Myelin-associated glycoprotein (MAG) Oligodendrocyte myelin glycoprotein (OMGP) Tenascin-R |
|
Oligodendrocyte precursor |
NG2 (a proteoglycan) DSD-1 or phosphacan (a proteoglycan) Versican (a proteoglycan) |
|
Astrocyte |
Tenascin Brevican (a proteoglycan) Neurocan (a proteoglycan) NG2 (a proteoglycan) |
|
Meningeal cell |
NG2 Semaphorins |
|
Activated microglia |
Free radicals Nitric oxide Arachidonic acid derivatives |
|
SOURCE: Fawcett and Asher, 1999. |
|
The collective action of these inhibitory molecules is the prevention of axon regeneration.
Oligodendrocytes, which are already at the scene because they myelinate axons, express a potent inhibitor of axon growth, Nogo-A, on the exterior surface of the cell membrane (Fournier et al., 2002). The vital importance of Nogo-A was revealed by studies with an animal model that showed that antibodies against this molecule, which block its action, promote some regeneration of severed axons (Schnell and Schwab, 1990). The first glial cells to arrive at the scene, within 3 to 5 days, are thought to be oligodendrocyte precursor cells, although no direct evidence of this has emerged. Oligodendrocyte precursors are immature oligodendrocytes that are destined, with further growth and differentiation, to become mature oligodendrocytes. At the scene they proliferate and release a variety of molecules that block axon growth. Astrocytes also arrive at the injury site and begin to undergo hypertrophy and divide.
Astrocytes form the bulk of the glial scar. In the scar, they are surrounded by an extracellular matrix made up of several types of proteoglycans, which are proteins on the outside of the cell membrane that have sugar moieties attached to them. Proteoglycans are up-regulated and
secreted by astrocytes themselves, and they directly inhibit axon growth (Fawcett and Asher, 1999). The role of astrocytes has perplexed researchers because, in addition to their inhibitory role, they can also play a growth-promoting role under different circumstances (Jones et al., 2003).
Syringomyelia
Syringomyelia is a complication that arises as early as 2 months or as late as 30 years after the injury. It results from the formation of a cyst in the center of the spinal cord. This cyst expands and elongates over time, significantly damaging the center of the spinal cord. About 4 percent of individuals with spinal cord injuries develop syringomyelia (Schurch et al., 1996; Terre et al., 2000). Individuals with syringomyelia can present with multiple symptoms, including pain, weakness, headaches, and stiffness of the limbs and the back. The pathogenesis of syringomyelia, however, is not well understood. It may even lie dormant for many years before symptoms arise. Detection was especially difficult because of the wide range of other complications and sensory deficits that result from spinal cord injuries; however, the advent of magnetic resonance imaging (MRI) technologies has greatly enhanced the ability of clinicians to detect syringomyelia.
SPONTANEOUS HEALING
Often overlooked amid the litany of pathological changes that occur after a spinal cord injury is the natural ability of the spinal cord to heal itself. In fact, most individuals with spinal cord injuries, especially those with incomplete injuries, show some degree of functional recovery, and some show substantial degrees of recovery (Tator et al., 1998). Conventional wisdom had been that although some recovery is possible, it is limited in time and extent. A change of thinking has emerged in recent years, however. It is now well accepted that the spinal cord has the capacity to recover in several unforeseen ways starting at about 24 hours after injury and continuing for years. That capacity has become so well recognized that new treatments are being designed to marshal its potential.
Mature nerve cells lack the capacity to divide once they are injured. Whatever recovery of function that occurs naturally after a spinal cord injury is largely the product of plasticity in the surviving neurons. Plasticity is a generic term that denotes the body’s natural capacity to react to changing conditions in numerous ways, from regrowth to gene up-regulation. The surviving neurons can adapt to compensate for injury; however, for many years it was thought that within limits the axons of neurons in mammals do not spontaneously regrow more than a few millimeters (Raineteau and Schwab, 2001). However, groundbreaking discoveries in
the early 1980s established that CNS axons have the intrinsic capacity for regrowth over long distances, but they are actively inhibited by molecules in their extracellular environment (Aguayo et al., 1982). The extracellular environment may also lack molecules that promote or guide axon regrowth to its correct target site. Thus, CNS axons can regrow if their immediate environment is supportive. By contrast, peripheral nervous system axons can and do regrow spontaneously due to the growth-promoting molecules produced by Schwann cells.
Mechanisms Behind Natural Recovery of Function
After injury, the spinal cord can spontaneously recover to varying degrees through a variety of biological mechanisms (Table 2-7). The degree of recovery depends on numerous factors, including the severity of the injury, the individual’s age, the area of the spinal cord affected, the degree of inhibition by astrocytes and oligodendrocytes, and other factors yet to be identified. Knowledge of the biological basis of functional recovery comes mostly from a host of animal models, although many of the intricate details are still unknown. As described in greater detail in Chapter 5, many strategies are being developed to facilitate these mechanisms of recovery, and these are areas of intense research.
Recovery of function is first apparent within days after injury as a result of recuperation from spinal shock. Some degree of remyelination within the spinal cord can occur and may have a clinical impact. Remyelination can occur in two different ways. The first is by Schwann cells, which are myelinating cells normally found in the peripheral nervous system, but after an injury they are able to migrate directly into the spinal cord, where they can myelinate regrowing axons (Bunge and Wood, 2004). Awareness of the role of Schwann cells has spawned an entire new line of research on therapies involving the transplantation of a variety of cell types (see Chapter 5).
The second means of possible remyelination is by oligodendrocyte precursor cells. Mature oligodendrocytes cannot divide or migrate. Yet, imma-
TABLE 2-7 Mechanisms of Spontaneous Recovery in the Spinal Cord
|
ture oligodendrocytes, already in the spinal cord, migrate short distances to the site of injury, where they can differentiate into mature oligodendrocytes and produce myelin (Gensert and Goldman, 1997). It bears remembering, however, that oligodendrocyte precursor cells can also mature into oligodendrocytes that do the opposite: inhibit axon regrowth through the release of inhibitory substances (see above). What triggers their development into inhibitory cells versus beneficial cells is not yet known.
It is also possible that demyelinated axons within the injured spinal cord may reorganize at the molecular level to acquire the ability to conduct nerve impulses without myelin insulation. This type of recovery is known to occur not only in animal models but also in humans with multiple sclerosis, in whom demyelinated spinal cord axons produce additional sodium channels to support impulse conduction after damage to the myelin (Craner et al., 2004).
Limited regrowth of axons and sprouting of new branches from the tips of existing axons to form new synapses are part of yet another mechanism of functional recovery (Raineteau and Schwab, 2001). The fact that limited regrowth and sprouting do occur reveals that axons possess the capacity for some degree of regrowth, a capacity that can be cultivated with better knowledge of what governs it. Numerous studies with animals have demonstrated the ways in which axonal regrowth from central neurons can be improved, particularly across the area of injury. Research indicates that, after injury, the surviving cells continue to produce certain molecules and release them into the extracellular milieu that bathes the sprouting axons. Some of the molecules are growth factors—members of a family of molecules called neurotrophins (Raineteau and Schwab, 2001). Others are guidance molecules that guide axons to their destination (Walsh and Doherty, 1997; Willson et al., 2002). This area of research is still in its early phases, and much of the information on axonal guidance gained to date involves the developing nervous system. Research is needed to determine if the same or similar mechanisms are involved in axon guidance following injury in the adult CNS.
BIOLOGICAL BASES OF FUNCTIONAL LOSSES
No daily activity can be taken for granted for someone with a spinal cord injury. A range of functions—getting out of bed, walking, dressing, eating, controlling the bladder and the bowel, and breathing—can be severely compromised, and their loss has a staggering effect. To develop the technological or medical means to restore function and to improve quality of life, it is vital to understand the neurological basis of dysfunction. The emphasis in this section is on the nervous system’s role in generating movements and how injury to the spinal cord results in functional loss.
Spinal Cord Injury Disruption of Motor Pathways
The initiation and regulation of movements require a complex set of events that integrate information from many regions of the brain, brain stem, and spinal cord (Figure 2-3). When an action potential is generated in the brain, it travels along axons and down the spinal cord via the corticospinal tract to the motor neurons at speeds upwards of 100 meters per second, resulting in contraction of a muscle and a movement. However, before it reaches the motor neurons, the information is modulated by neurons found in the basal ganglia, cerebellum, and brain stem. When the signals finally reach the motor neurons, these specialized nerve cells provide the final conduit for the transmission of the signals to muscles throughout the body, stimulating muscles to contract. Thus, an injury or disruption to the motor pathways leading to and from the brain could cause a patient to lose motor function.
Differences in Degree of Cortical Control on Motor Function
The circuitry between the primary motor cortex and the motor neurons of the ventral horn of the spinal cord is very complex. Many regions of the CNS, including the basal ganglia, cerebellum, and brain stem, help regulate movements (Figure 2-3). The degree of cortical control varies depending on the motor function. For example, movement of the fingers requires more integration from the brain than gross movement of the legs, which relies more on circuitry confined to the spinal cord. The majority of the signals from the brain are transmitted along bundles of axons that make up the corticospinal tract, which connects the primary motor cortex in the brain to the motor neurons in the ventral horn of the spinal cord. The motor neurons in turn transmit the information from the ventral horn directly to the muscle. Motor control of most other body parts involves additional circuitry, or connections, between the primary motor cortex and the motor neurons. Signals are transmitted either from the primary motor cortex to intermediate layers within the spinal cord to modulate the tone or reflex gain and to cause direct contraction of the muscles or through intermediate processing stages in the midbrain or pons of the brain stem.
In addition to regulating voluntary movements, neurons in the descending motor tracts traveling from the brain down to the spinal cord are also responsible for regulating the smooth muscles of internal organs. Descending motor tracts also contain neurons associated with the autonomic nervous system, which regulates blood pressure, body temperature, and the body’s response to stress.
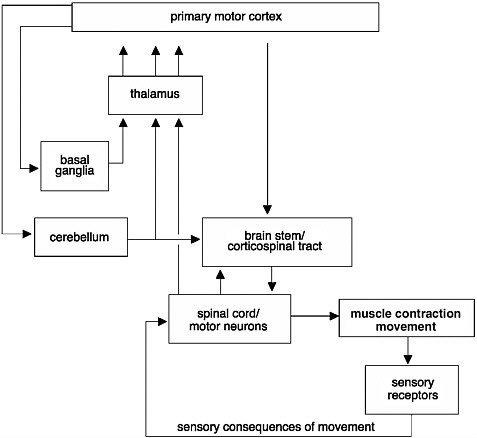
FIGURE 2-3 Initiation and regulation of movements.
Control of movements involves a complex network of connections. Signals commanding the initiation of a movement are generated in the primary motor cortex of the brain. These signals are modulated before they reach the muscle. They are modulated through an intricate circuit in the basal ganglia and thalamus, which regulate the initiation of movements and help coordinate movements. Information from muscle contractions is also transmitted back to the brain through sensory receptors. This information is also used to provide feedback and to modify the movements.
SOURCE: Adapted from Kandel et al., 1991.
Feedback Control of Movements
Critical feedback from sensory nerve endings located on muscles is transferred to the spinal cord via the sensory roots and dorsal horn to the brain, resulting in involuntary modulation of movements. This component of the sensory system is called proprioception. It is responsible for immediately varying the degree of muscle contraction in response to incoming
information regarding external stimuli. When individuals lose their proprioception, they are unable to freely move and interact comfortably with the external environment (see Box 5-1).
A subset of the sensory neurons located in the spinal cord is also responsible for establishing the circuitry that controls simple reflex reactions, such as the knee-jerk reflex that doctors test by tapping a hammer on a patient’s knee. This sensory information bypasses ascending information to the brain and is conveyed directly to lower motor neurons, resulting in involuntary or reflex movements.
Role of the Central Pattern Generator in Humans
Experiments performed by Shik, Severin, and Orlovsky in the 1960s provided evidence of a central pattern generator (CPG), which is a complex circuit of neurons responsible for coordinated rhythmic muscle activity, such as locomotion (Shik et al., 1969). In these experiments, the brain stem of a cat was transected so that no information could travel from the brain to the spinal cord. Surprisingly, following this surgery, cats were still able to stand on their own and could be induced to walk (Box 2-1). Similar results have been observed in rats and mice that have had their spinal cords transected. Therefore, it was concluded that the CPG is located in the spinal cord of these animals and does not require input from the brain.
If the CPG is located in the spinal cord and does not require any input from the brain, why is it that most individuals with spinal cord injuries and a complete transection of the spinal cord cannot walk? The function and control of the CPG in Old World primates and humans may be different from those in animals that walk on four feet, like cats and dogs (i.e., bipeds versus quadrapeds) (Vilensky and O’Connor, 1998). Humans and other bipeds may have more cortical dominance integrated into the locomotor circuitry than quadrapeds (Fulton and Keller, 1932), which may explain why the recovery of rhythmic locomotor activity is not commonly observed in primates and humans with complete spinal cord injuries (Kuhn, 1950; Bussel et al., 1996; Vilensky and O’Connor, 1998). However, because of the limitations of performing invasive experiments with primates and humans, it is difficult to verify the significance of the cortical circuitry. The complexity of the cortical regulation of the CPG in humans and primates compared with that in cats and rodents demonstrates a potential area of concern for the translation of the results from experiments performed with laboratory animals to humans.
Muscle Spasticity as a Result of Altered Activity in Motor Neurons
Spasticity is a state of increased muscular tone, often with heightened stretch reflexes. In severe cases, spasticity causes chronic pain, flexion
|
BOX 2-1 At the turn of the 20th century, Charles Scott Sherrington and T. Graham Brown published two seminal papers that demonstrated the capacity of the spinal cord in cats and dogs to generate rhythmic motor activity (Brown, 1914). Sherrington’s experiment provided evidence that dogs and cats were still able to generate rhythmic movements elicited from their hind limbs weeks after their spinal cords were severed. Later, in the 1960s, further insight was garnered when the work of three Russian scientists, M. K. Shik, F. V. Severin, and G. N. Orlovsky, and one Swedish scientist, Sten Griller, showed that when a portion of the brain stem of a cat was cut across the middle—thus severing any connections between the brain and the spinal cord—the cat was still capable of standing. Furthermore, if a specific region of the brain stem was stimulated, the cats could be induced to walk on a treadmill, and alternating bursts of muscle activity could be recorded in extensors and flexors in conjunction with walking (Shik et al., 1966). These series of experiments led to the conclusion that each limb is controlled by a central pattern generator (CPG) in the spinal cord, which controls rhythmic motor activity, including walking. 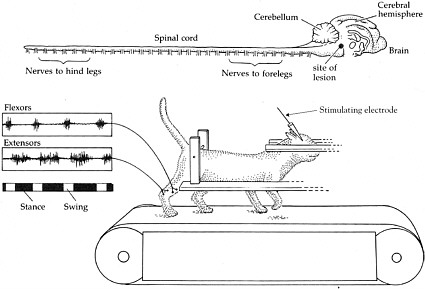 Shik and colleagues experimented with a cat whose brain stem was severed but that was still able to walk on a treadmill when a specific region of the brain stem was stimulated. The top of the figure shows the brain and the spinal cord. The muscle activity recorded from the flexors and extensors demonstrates that they are contracting and relaxing at opposite times from each other, consistent with normal function. SOURCE: Reprinted with permission, from Dowling, 2001. Copyright 2001 by Sinauer Associates, Inc. |
contractures, decubitus ulcers,2 and bone fractures (Nance, 1999). Muscle spasticity frequently occurs after spinal cord injuries, with one study finding 78 percent of individuals experiencing spasticity after they were discharged from the hospital (Maynard et al., 1990).
The precise causes of muscle spasticity are not well understood. Most studies point to the greater excitability of motor neurons, with several possible causes (Burchiel and Hsu, 2001). One is thought to be decreased inhibitory input from the brain to spinal cord motor neurons through direct or indirect (via spinal cord interneuron) connections. For nearly a century, the lower motor neuron has been described as the “final common pathway” to muscles because of the thousands of neurons that converge on it. Some of those neurons are inhibitory, whereas others are excitatory. A single motor neuron can receive a direct or an indirect input from several regions of the brain and from sensory neurons. The array of inputs is critical for the modulation and fine-tuning of motor neuron control of muscles. If inhibitory input to the motor neuron is destroyed or reduced as a result of a spinal cord injury, the balance weighs in favor of heightened excitability and firing of motor neurons.
The spasticity that occurs with a spinal cord injury may also be produced by other mechanisms. One is a by-product of injury-induced sprouting. The new synapses formed by surviving axons (see below) may be too excitatory in nature. They might arise from motor pathways that descend from the brain, from ascending sensory pathways, or from the many synapses between the interneurons that form an intricate local circuitry within the spinal cord. Continual sensory feedback from muscles (such as for the detection of muscle length) is indispensable for the production of graded movements. If stretch reflexes are altered in individuals with spinal cord injuries, the lack of appropriate feedback may lead to spastic muscle contractions.
Spasticity may also be produced by pathological alterations in the electrical properties of the motor neurons themselves, including changes in sodium channel type, number, and distribution (Hiersemenzel et al., 2000) and alterations in neurotransmitter reuptake by glial cells.
Pain and Its Causation
Pain is a common and debilitating outcome of spinal cord injuries. Most studies find that 60 to 80 percent of individuals report chronic pain after a spinal cord injury. More precise estimates have been hindered by a
lack of uniform definitions and a comprehensive classification system (Burchiel and Hsu, 2001). The lack of definitions was addressed in 2000 with the release of a proposed scheme by the International Association for the Study of Pain for characterization of the pain associated with spinal cord injuries. By using those new definitions, a prospective study of 100 people found that 5 years after injury, 81 percent reported pain (of all types), and 58 percent reported that their pain was “severe or excruciating” (Siddall et al., 2003). The impact of chronic pain may be so great—deterioration of quality of life, ability to function, self-image, and care delivery—that depression and thoughts of suicide are common (Cairns et al., 1996).
The new classification system organizes spinal cord injury pain under two broad categories—nociceptive and neuropathic—along with five subclassifications (each of which has further clinical subtypes and possible pathologies; see Table 2-8). Nociceptive pain arises from an external source (e.g., a noxious stimulus and consequent tissue damage), whereas neuropathic pain arises from the pathological changes occurring within sensory neurons or pathways. The two types of nociceptive pain—musculoskeletal and visceral—were reported by 59 and 5 percent of patients, respectively, in the prospective trial cited above (Siddall et al., 2003). Of the three types of neuropathic pain, 41 percent of patients reported at-level neuropathic pain, whereas 34 percent reported below-level neuropathic pain (Siddall et al., 2003).
Nociceptive pain is the dull and aching pains that one encounters when a limb is broken or when one has lower back pain. Painful stimuli are registered by specialized sensory cells known as nociceptors. Nociceptors, which are intact with this type of pain, respond to local damage to nonneural tissues (e.g., bone, muscles, and ligaments).
Neuropathic pain, on the other hand, is produced by direct damage to neural tissue. It is described as a sharp, shooting, burning, or electrical type of pain. Sensory neurons and pathways undergo physiological alterations; they may become exquisitely sensitive, firing off impulses out of proportion to the stimulus (hyperesthesia) or even without an external trigger whatsoever. They may register the light touch of a feather as an unpleasant burning sensation (dysesthesia) instead of a pleasant one.
Nociceptive pain and neuropathic pain have distinct causes and, as a result, distinct treatments. Because nociceptive pain arises from tissue damage and not from nerve pathology, it is often treated with standard therapies, most commonly physical therapy, various pain medications, and surgical therapy. Neuropathic pain is more difficult to treat, partly because its mechanisms are still being uncovered. The distinction between the two types of pain, however, is not always clear-cut (Bryce and Ragnarsson, 2002). Over time, nociceptive pain can lead to the sensitization of spinal
TABLE 2-8 Classification of Pain from Spinal Cord Injury
|
Broad Type (Tier 1) |
Broad System (Tier 2) |
Specific Structure or Pathology (Tier 3) |
|
Nociceptive |
Musculoskeletal |
|
|
Visceral |
|
|
|
Neuropathic |
Above level of injury |
|
|
|
At level of injury |
|
|
|
Below level of injury |
|
|
SOURCE: Vierck et al., 2000. |
||
cord neurons, which leads to neuropathic pain. Sensitization represents an increased response to a standard stimulus, and it is manifest as hypersensitivity to pain (Woolf and Mannion, 1999).
Much of what is known about the pathophysiology of the pain that occurs after a spinal cord injury comes from studies with a host of animal models of different types of injuries. Although much remains to be learned, some of the intensively studied mechanisms underlying spinal cord pain include the following (Yezierski, 2000):
-
loss or disruption of descending pathways from the brain that normally inhibit the sensory input as it enters the spinal cord;
-
increases in pain neurotransmitter3 or receptor levels through up-regulated gene expression;
-
changes in the types or numbers of ion channels in sensory neurons and pathways that render them more excitable (Hains et al., 2003b);
-
sprouting of sensory fibers entering the spinal cord;
-
alterations in post-receptor signal transduction mechanisms; and
-
switching of the identities of sensory fibers from non-pain fibers to pain fibers (Bryce and Ragnarsson, 2002; Hulsebosch, 2002).
Many of these mechanisms also apply to other pain conditions not associated with spinal cord injuries (Woolf and Mannion, 1999).
The brain plays a large role in modulating and interpreting the sensation of pain, so much so that experts describe pain as an “experience” rather than a sensation. Multiple areas of the cerebral cortex process pain information relayed there by a certain tract in the spinal cord, which receives its information from incoming peripheral nerves. The brain, in turn, modulates the incoming messages through several descending pathways from nuclei in the midbrain, including the periaqueductal gray.
Biological Causes of Bladder Dysfunction
Three common types of bladder dysfunction accompany spinal cord injuries, depending on the level of the injury (Kaplan et al., 1991). Understanding of the types of dysfunction first requires some understanding of the anatomy of the bladder and its control by the spinal cord and the brain. Two main muscle groups surrounding the bladder control urination: the detrusor muscle, which controls bladder contraction, and the external sphincter muscles at the base of the bladder, which control bladder outflow. The two muscles normally work reciprocal to one another: the detrusor muscle contracts while the sphincter muscles relax, allowing urine to flow from the bladder. Because each is fed by separate nerves, their coordination—i.e., detrusor muscle contraction with sphincter muscle relaxation—is integrated at a higher level, which, in this case, is performed by the pons region of the brain. That portion of the brain sends its axons to the sacral region of the spinal cord (S2 and S3), which also receives sensory input from the bladder (via the pelvic nerve) about bladder distention. When the pelvic nerve conveys the message that the bladder is full, the information is relayed up to the pons, which then coordinates the motor messages necessary to empty the bladder. This process is called the voiding reflex.
In individuals with complete spinal cord injuries above the level of the sacral cord, disruption of the pathway from the spinal cord to the brain can lead to bladder problems related to the lack of coordination between the detrusor and the sphincter muscles (see below). If the sacral cord or the cauda equina is injured directly, the bladder detrusor muscle becomes flaccid—a condition known as areflexia. The detrusor muscle loses its ability to
contract and can be readily stretched. Large volumes of urine overfill the bladder and back up to the kidneys (Kaplan et al., 1991).
Two common types of bladder conditions occur in individuals with spinal cord injuries at levels above the sacral cord. The first is detrusor hyperreflexia, in which the bladder is overreactive. As the bladder fills with small volumes of urine, the detrusor muscle contracts prematurely, causing frequent urination. Research with animals suggests that part of the pathological process occurs in the sensory nerves coming from the bladder. Sensory fibers normally carrying other types of information actually switch their functioning: they become sensitive to bladder distention and trigger bladder detrusor contraction (de Groat, 1995). This form of sensory plasticity is mediated by changes in electrical properties of C fibers, a particular type of sensory neuron (Yoshimura, 1999).
Less is known about the biological basis of the second type of bladder dysfunction, detrusor-sphincter dyssynergia. This condition is marked by involuntary contractions of the sphincter muscles, which prevent urine from leaving the bladder. It can occur with the loss of the reciprocal relationship between detrusor muscle contraction and sphincter muscle relaxation. One hypothesis is that the condition is related to the reduced activity of the neurotransmitter nitric oxide in sphincter muscles. Nitric oxide is involved in relaxation of the sphincter. Reduced levels would therefore increase sphincter contraction (Mamas et al., 2003). Detrusor-sphincter dyssynergia can also arise from lesions to the pontine reticular nucleus and the reticular formation (Sakakibara et al., 1996).
Bowel Function Disruption
Bowel dysfunction frequently occurs after a spinal cord injury because the brain and spinal cord have major roles in stool elimination. Although the movement of feces down the length of the bowel is partly controlled by independent neurocircuits that reside within the bowel,4 the brain and spinal cord are essential for voluntary control over defecation. Loss of bowel function is so deeply distressing and embarrassing to individuals with spinal cord injuries that it affects their social interactions and their willingness to engage in sexual activities.
The impact of a spinal cord injury on voluntary control of the bowel is known as neurogenic bowel. Neurogenic bowel comes in two types—reflexic and areflexic—depending on the location of the injury. Reflexic
bowel, or upper motor neuron bowel, is the result of injuries above the sacral cord. Reflexic bowel brings constipation and an inability to defecate by conscious effort. The anal sphincter muscle remains tight and can be stimulated manually to induce defecation. Areflexic bowel, or lower motor neuron bowel, results from injuries at or below the sacral cord. It also causes constipation and incontinence. The anal sphincter becomes so flaccid that it is incapable of being manually stimulated to induce defecation. Both types of neurogenic bowel carry the risk of serious complications, including bowel obstruction, colorectal distention, and a life-threatening rise in blood pressure triggered by a distended bladder or bowel.
Control of Sexual Function by the Spinal Cord and Brain
Many aspects of human sexuality are under reflexive control by various centers in the spinal cord, most frequently in the sacral and in the thoracic and lumbar regions. The site and extent of injury are thus key determinants of sexual function. Several brain regions—most notably, the limbic system and the hypothalamus—also contribute to sexual function by exerting some degree of control over neuronal centers located in the spinal cord, especially sexual drive or inhibition. A large proportion of men and women with spinal cord injuries report reduced sexual desire (Alexander et al., 1993; Sipski and Alexander, 1993) and reduced fertility (Elliot, 2002). Male infertility appears to be the result of abnormalities in semen, especially low sperm motility and viability and increased numbers of leukocytes (Randall et al., 2003).
For men, the sexual response includes three separate functions: erection, ejaculation, and orgasm. Erection has two descriptive types, both of which are controlled by distinct spinal cord reflexes. Psychogenic (or mentally induced) erection is controlled by the T11 to L2 segments of the spinal cord, whereas reflexogenic erections are mediated by the sacral cord. Ejaculation is a more complex process, with two stages mediated by the region from T10 to S4, which controls certain sympathetic, parasympathetic, and somatic nerves. A physiological component of an orgasm is rhythmic pelvic floor contractions and other smooth-muscle contractions mediated by sacral regions of the spinal cord. The experience of orgasm as pleasurable depends on processing and interpretation by the brain. Damage to the relevant spinal cord centers or disruption of connections to the brain can thus lead to various types of sexual dysfunction. Dysfunction in the urinary or the gastrointestinal system also has a bearing on ejaculation and orgasm, as does an individual’s mental state, such as depression or anxiety (Elliot, 2002).
For women, the sexual response depends on arousal and orgasm. Sexual arousal involves vaginal lubrication; swelling of the clitoris; and increases
in heart rate, respiratory rate, and blood pressure. Vaginal lubrication has two types, psychogenic or reflexive, which are controlled by the regions of the spinal cord from T10 to L2 and S2 to S5, respectively. Orgasm has been directly investigated in laboratory-based studies with women with spinal cord injuries. Overall, only 52 percent of women with spinal cord injuries were able to stimulate themselves to orgasm, regardless of the nature of their injury (Sipski, 2001). Women with injuries of the sacral cord were significantly less likely to reach orgasm than women with spinal cord injuries at other, higher levels. Researchers therefore postulate that an intact sacral reflex is necessary for orgasm (Sipski, 2001; Benevento and Sipski, 2002).
SUMMARY
Although much progress has been made, especially in the past 25 years, in understanding the basic biology of the nervous system and the complex pathways in the pathophysiology of spinal cord injuries that involve the immune, vascular, and nervous systems, much remains to be learned. As emphasized in the following chapters, this basic research is the underpinning of progress that will be made in developing therapeutic interventions.
Many research avenues remain to be examined to understand the biochemical mechanisms responsible for spinal cord injuries and thus the targets for the development of therapeutic interventions. Research is needed on the processes involved in cellular death and the immediate sequelae of apoptotic and necrotic cell death. The molecular mechanisms that promote and inhibit axonal regeneration need to be further explored, as do the molecular mechanisms that direct axons to their appropriate targets and regulate the formation and maintenance of appropriate and functional synaptic connections and circuitry.
Moving this research forward involves opportunities and challenges that are not isolated to spinal cord injury research. Rather, this research has far-reaching potential to both inform and be informed by many other fields of research and the efforts that are under way to examine other neurological diseases and conditions.
REFERENCES
Aguayo AJ, David S, Richardson P, Bray GM. 1982. Axonal elongation in peripheral and central nervous system transplants. Advances in Cell Neurobiology 3: 215-234.
Alexander CJ, Sipski ML, Findley TW. 1993. Sexual activities, desire, and satisfaction in males pre- and post-spinal cord injury. Archives of Sexual Behavior 22(3): 217-228.
Anderson AJ. 2002. Mechanisms and pathways of inflammatory responses in CNS trauma: Spinal cord injury. Journal of Spinal Cord Medicine 25(2): 70-79.
Anthes DL, Theriault E, Tator CH. 1996. Ultrastructural evidence for arteriolar vasospasm after spinal cord trauma. Neurosurgery 39(4): 804-814.
ASIA (American Spinal Injury Association). 2000. International Standards for Neurological Classification of SCI. Chicago: American Spinal Injury Association.
Bareyre FM, Schwab ME. 2003. Inflammation, degeneration and regeneration in the injured spinal cord: Insights from DNA microarrays. Trends in Neurosciences 26(10): 555-563.
Beattie MS, Farooqui AA, Bresnahan JC. 2000. Review of current evidence for apoptosis after spinal cord injury. Journal of Neurotrauma 17(10): 915-925.
Benevento BT, Sipski ML. 2002. Neurogenic bladder, neurogenic bowel, and sexual dysfunction in people with spinal cord injury. Physical Therapy 82(6): 601-612.
Brown TG. 1914. On the nature of fundamental activity of the nervous centers; together with an analysis of the conditioning of rhythmic activity in procession, and a theory of the evolution of function in the nervous system. Journal of Physiology 48: 18-46.
Bryce TN, Ragnarsson K. 2002. Pain management in persons with spinal cord disorders. In: Lin V, Cardenas DD, Cutter NC, Frost FS, Hammond MC, Lindblom LB, Perkash I, Waters R, eds. Spinal Cord Medicine: Principles and Practice. New York: Demos Medical Publishing. Pp. 441-460.
Bunge MB, Wood PM. 2004. Transplantation of Schwann cells and olfactory ensheathing cells to promote regeneration in the CNS. In: Selzer ME, Clarke S, Cohen LG, Dincan PW, Gage FH, eds. Textbook of Neural Repair and Rehabilitation. Cambridge, United Kingdom: Cambridge University Press.
Bunge RP, Puckett WR, Becerra JL, Marcillo A, Quencer RM. 1993. Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Advances in Neurology 59: 75-89.
Bunge RP, Puckett WR, Hiester ED. 1997. Observations on the pathology of several types of human spinal cord injury, with emphasis on the astrocyte response to penetrating injuries. Advances in Neurology 72: 305-315.
Burchiel KJ, Hsu FP. 2001. Pain and spasticity after spinal cord injury: Mechanisms and treatment. Spine 26(24 Suppl): S146-160.
Bussel B, Roby-Brami A, Neris OR, Yakovleff A. 1996. Evidence for a spinal stepping generator in man. Paraplegia 34(2): 91-92.
Caceci T, El-Shafey S. 2002. Neutrophils. [Online]. Available: http://education.vetmed.vt.edu/Curriculum/VM8054/Labs/Lab6/Examples/exneutro.htm [accessed August 18, 2004].
Cairns DM, Adkins RH, Scott MD. 1996. Pain and depression in acute traumatic spinal cord injury: Origins of chronic problematic pain? Archives of Physical Medicine and Rehabilitation 77(4): 329-335.
Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. 2004. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proceedings of the National Academy of Sciences (U.S.A.) 101(21): 8168-8173.
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. 1997. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nature Medicine 3(1): 73-76.
CRPF (Christopher Reeve Paralysis Foundation). 2002. The Spinal Cord and Muscles Working Together. [Online]. Available: http://www.christopherreeve.org/Research/Research.cfm?ID=178&c=21 [accessed January 11, 2005].
Dammann O, Durum S, Leviton A. 2001. Do white cells matter in white matter damage? Trends in Neurosciences 24(6): 320-324.
de Groat WC. 1995. Mechanisms underlying the recovery of lower urinary tract function following spinal cord injury. Paraplegia 33(9): 493-505.
Dowling JE, 2001. Neurons and Networks: An Introduction to Behavioral Neuroscience. Cambridge, MA: Harvard University Press.
El Masry WS, Tsubo M, Katoh S, El Miligui YH, Khan A. 1996. Validation of the American Spinal Injury Association (ASIA) motor score and the National Acute Spinal Cord Injury Study (NASCIS) motor score. Spine 21(5): 614-619.
Elliot S. 2002. Sexual dysfunction and infertility in men with spinal cord disorders. In: Lin V, Cardenas DD, Cutter NC, Frost FS, Hammond MC, Lindblom LB, Perkash I, Waters R, eds. Spinal Cord Medicine: Principles and Practice. New York: Demos Medical Publishing. Pp. 349-368.
Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD. 1998. Apoptosis after traumatic human spinal cord injury. Journal of Neurosurgery 89(6): 911-920.
Faden AI, Simon RP. 1988. A potential role for excitotoxins in the pathophysiology of spinal cord injury. Annals of Neurology 23(6): 623-626.
Fawcett JW, Asher RA. 1999. The glial scar and central nervous system repair. Brain Research Bulletin 49(6): 377-391.
Fournier AE, GrandPre T, Gould G, Wang X, Strittmatter SM. 2002. Nogo and the Nogo-66 receptor. Progress in Brain Research 137: 361-369.
Fulton JA, Keller AD. 1932. The Sign of Babinski: A Study of the Evolution of Cortical Dominance in Primates. Springfield, IL: Charles C. Thomas.
Gensert JM, Goldman JE. 1997. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 19(1): 197-203.
Hains BC, Black JA, Waxman SG. 2003a. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. Journal of Comparative Neurology 462(3): 328-341.
Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. 2003b. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. Journal of Neuroscience 23(26): 8881-8892.
Harper GP, Banyard PJ, Sharpe PC. 1996. The International Spinal Research Trust’s strategic approach to the development of treatments for the repair of spinal cord injury. Spinal Cord 34(8): 449-459.
Hiersemenzel LP, Curt A, Dietz V. 2000. From spinal shock to spasticity: Neuronal adaptations to a spinal cord injury. Neurology 54(8): 1574-1582.
Hulsebosch CE. 2002. Recent advances in pathophysiology and treatment of spinal cord injury. Advances in Physiology Education 26(1-4): 238-255.
Imaizumi T, Kocsis JD, Waxman SG. 1997. Anoxic injury in the rat spinal cord: Pharmacological evidence for multiple steps in Ca2+-dependent injury of the dorsal columns. Journal of Neurotrauma 14(5): 299-311.
Jones LL, Sajed D, Tuszynski MH. 2003. Axonal regeneration through regions of chondroitin sulfate proteoglycan deposition after spinal cord injury: A balance of permissiveness and inhibition. Journal of Neuroscience 23(28): 9276-9288.
Kandel ER, Schwartz JH, Jessell JH, eds. 1991. Principles of Neural Science. New York: Elsevier.
Kaplan SA, Chancellor MB, Blaivas JG. 1991. Bladder and sphincter behavior in patients with spinal cord lesions. Journal of Urology 146(1): 113-117.
Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. 2002. Neuroprotective autoimmunity: Naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proceedings of the National Academy of Sciences (U.S.A.) 99(24): 15620-15625.
Koyanagi I, Tator CH, Lea PJ. 1993. Three-dimensional analysis of the vascular system in the rat spinal cord with scanning electron microscopy of vascular corrosion casts. Part 1: Normal spinal cord. Neurosurgery 33(2): 277-283.
Kuhn RA. 1950. Functional capacity of the isolated human spinal cord. Brain 73: 1-51.
Lentz TL. 1971. Cell Fine Structure: An Atlas of Drawings of Whole-Cell Structure. Philadelphia: W. B. Saunders Company.
Levi ADO. 2004. Approach to the patient and diagnostic evaluation. In: Winn HR, Youmans JR, eds. Neurological Surgery: A Comprehensive Reference Guide to the Diagnosis and Management of Neurological Problems. Vol. 4. 5th ed. Philadelphia: W. B. Saunders. Pp. 4869-4884.
Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. 1992. Transforming growth factor-beta 1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. Journal of Cell Biology 117(2): 395-400.
Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. 1997. Neuronal and glial apoptosis after traumatic spinal cord injury. Journal of Neuroscience 17(14): 5395-5406.
Mamas MA, Reynard JM, Brading AF. 2003. Nitric oxide and the lower urinary tract: Current concepts, future prospects. Urology 61(6): 1079-1085.
Maynard FM, Karunas RS, Waring WP III. 1990. Epidemiology of spasticity following traumatic spinal cord injury. Archives of Physical Medicine and Rehabilitation 71(8): 566-569.
Merck & Co., Inc. 2004. Drawing of Neuron. [Online]. Available: http://www.merck.com/mrkshared/mmanual_home/illus/i59_3.gif [accessed April 22, 2004].
Nance PW. 1999. Rehabilitation pharmacotherapy: Preface. Physical Medicine & Rehabilitation Clinics of North America 10(2): xv-xvi.
Popovich PG, Jones TB. 2003. Manipulating neuroinflammatory reactions in the injured spinal cord: Back to basics. Trends in Pharmacological Sciences 24(1): 13-17.
Raineteau O, Schwab ME. 2001. Plasticity of motor systems after incomplete spinal cord injury. Nature Reviews Neuroscience 2(4): 263-273.
Randall JM, Evans DH, Bird VG, Aballa TC, Lynne CM, Brackett NL. 2003. Leukocytospermia in spinal cord injured patients is not related to histological inflammatory changes in the prostate. Journal of Urology 170(3): 897-900.
Ray SK, Hogan EL, Banik NL. 2003. Calpain in the pathophysiology of spinal cord injury: Neuroprotection with calpain inhibitors. Brain Research––Brain Research Reviews 42(2): 169-185.
Sakakibara R, Hattori T, Yasuda K, Yamanishi T. 1996. Micturitional disturbance and the pontine tegmental lesion: Urodynamic and MRI analyses of vascular cases. Journal of the Neurological Sciences 141(1-2): 105-110.
Schnell L, Schwab ME. 1990. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343(6255): 269-272.
Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. 1999. Acute inflammatory responses to mechanical lesions in the CNS: Differences between brain and spinal cord. European Journal of Neuroscience 11(10): 3648-3658.
Schurch B, Wichmann W, Rossier AB. 1996. Post-traumatic syringomyelia (cystic myelopathy): A prospective study of 449 patients with spinal cord injury. Journal of Neurology, Neurosurgery & Psychiatry 60(1): 61-67.
Schwartz M, Kipnis J. 2001. Protective autoimmunity: Regulation and prospects for vaccination after brain and spinal cord injuries. Trends in Molecular Medicine 7(6): 252-258.
Sekhon LH, Fehlings MG. 2001. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine 26(24 Suppl): S2-12.
Shier D, Butler J, Lewis R. 2004. Hole’s Human Anatomy & Physiology. 10th ed. Boston: McGraw-Hill Higher Education.
Shik ML, Severin FV, Orlovskii GN. 1966. Control of walking and running by means of electric stimulation of the midbrain. Biofizika 11(4): 659-666.
Shik ML, Severin FV, Orlovsky GN. 1969. Control of walking and running by means of electrical stimulation of the mesencephalon. Electroencephalography & Clinical Neurophysiology 26(5): 549.
Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. 2003. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain 103(3): 249-257.
Sipski ML. 2001. Sexual response in women with spinal cord injury: Neurologic pathways and recommendations for the use of electrical stimulation. Journal of Spinal Cord Medicine 24(3): 155-158.
Sipski ML, Alexander CJ. 1993. Sexual activities, response and satisfaction in women pre-and post-spinal cord injury. Archives of Physical Medicine and Rehabilitation 74(10): 1025-1029.
Stimson D. 2001. From clear-cut endings to complex beginnings: Researchers probe the origins of Charcot-Marie-Tooth disease. Quest 8(1): 1-4.
Stys PK, Ransom BR, Waxman SG. 1992a. Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. Journal of Neurophysiology 67(1): 236-240.
Stys PK, Waxman SG, Ransom BR. 1992b. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na+-Ca2+ exchanger. Journal of Neuroscience 12(2): 430-439.
Taber CW, Thomas CL, eds. 1997. Taber’s Cyclopedic Medical Dictionary. Philadelphia: F.A. Davis.
Tator CH, McCormick PC, Piepmeier JM, Benzel EC, Young W. 1998. Biology of neurological recovery and functional restoration after spinal cord injury. Neurosurgery 42(4): 696-708.
Terre R, Valles M, Vidal J. 2000. Post-traumatic syringomyelia following complete neurological recovery. Spinal Cord 38(9): 567-570.
Vierck CJ Jr, Siddall P, Yezierski RP. 2000. Pain following spinal cord injury: Animal models and mechanistic studies. Pain 89(1): 1-5.
Vilensky JA, O’Connor BL. 1998. Stepping in nonhuman primates with a complete spinal cord transection: Old and new data, and implications for humans. Annals of the New York Academy of Sciences 860: 528-530.
Walsh FS, Doherty P. 1997. Neural cell adhesion molecules of the immunoglobulin superfamily: Role in axon growth and guidance. Annual Review of Cell and Developmental Biology 13: 425-456.
Waxman SG. 2001. Acquired channelopathies in nerve injury and MS. Neurology 56(12): 1621-1627.
Willson CA, Irizarry-Ramirez M, Gaskins HE, Cruz-Orengo L, Figueroa JD, Whittemore SR, Miranda JD. 2002. Upregulation of EphA receptor expression in the injured adult rat spinal cord. Cell Transplantation 11(3): 229-239.
Wolman L. 1965. The disturbance of circulation in traumatic paraplegia in acute and late stages: A pathological study. Paraplegia 59: 213-226.
Woolf CJ, Mannion RJ. 1999. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 353(9168): 1959-1964.
Yezierski RP. 2000. Pain following spinal cord injury: Pathophysiology and central mechanisms. Progress in Brain Research 129: 429-449.
Yoshimura N. 1999. Bladder afferent pathway and spinal cord injury: Possible mechanisms inducing hyperreflexia of the urinary bladder. Progress in Neurobiology 57(6): 583-606.
Young W. 1992. Role of calcium in central nervous system injuries. Journal of Neurotrauma 9 Suppl(1): S9-25.
Young W. 2002. Spinal Cord Injury Levels & Classification. [Online]. Available: http://www.sci-info-pages.com/levels.html [accessed September 30, 2003].



































{kind=link}