
6
Adequacy of Pediatric Postmarket Surveillance Studies
Today, Esperanza is a 30-year-old mother of two. In 1975, she was a critically ill newborn at the University of California hospital at Irvine. There, in a last-chance attempt to save her life, she became the first infant to be successfully treated with extracorporeal membrane oxygenation (ECMO), a process that allows prolonged cardiopulmonary life support. ECMO had been developed in the early 1970s to support adults with severe respiratory illness, but trial results had been disappointing. Dr. Robert Bartlett, a physician at the hospital where Esperanza was born, had been investigating ECMO with bench and animal testing for 10 years. He thought the procedure might be more successful with infants, who tend to have fewer additional medical complications than adults. By 1985, 10 years after Esperanza’s treatment, death rates for infants with most of the conditions then treated by ECMO had dropped from 90 percent or more to less than 50 percent.
(Bartlett, 1985; Bartlett et al., 2000; University of Michigan, 2005)
The history of ECMO, which involves a complex system of medical devices, is interesting for a number of reasons. As suggested above, it illustrates how patient characteristics and treatment success may vary by age and how the evolution of medical innovations can have unexpected twists and turns. The technology also figured in innovative clinical trials of the device in the 1980s that are still used to illustrate ethical dilemmas in trial design (Truog, 1999). In addition, early on, medical centers using ECMO began a patient registry that has proved useful in a variety of clinical evaluations.
ECMO does not involve a fully implanted device, but patients must have vascular catheters inserted in the major blood vessels of the groin or neck. Its use typically requires days of direct contact between a child’s blood and certain elements of the device system, primarily the artificial lung and the tubing that circulates the blood. As newer, less drastic treatment strategies (e.g., inhaled nitric oxide and high-frequency oscillatory ventilation) for newborn respiratory failure have shown positive short-term results
in randomized clinical trials and other studies, many centers limit ECMO to use as a rescue therapy when the center’s best efforts to ventilate have failed (Truog, 1998; AAP, 2000a). ECMO also continues as a valuable last resort for infants and small children with acute heart failure who require circulatory support until heart function recovers or a heart transplant can be performed.
When successful, innovative medical devices such as those involved in the ECMO procedure can offer dramatic cures, sustain life until another therapy is available, slow the progression of disease, or ease the distress caused by an incurable condition. Long-term and even relatively short-term exposure to a device—and the surgical or other procedures associated with its use—can, however, alter a child’s development in complex ways. Some of the effects may be suspected in advance, but others may be identified only through careful follow-up monitoring and evaluation. Unwanted developmental outcomes may not be evident for a number of years and thus will not be detected by short-term studies.
Chapter 2 introduced the concepts of children’s growth and development and their active lifestyles and then described some of the physical, cognitive, emotional, behavioral, and social characteristics of children that may affect the design, use, and performance of medical devices. It noted that children’s activities pose a risk of traumatic damage to certain implanted or attached devices. In addition, children’s growth and development may affect the performance of a device. For example, growing tissues may put increased stress on some biomechanical devices. Causation may also operate in the reverse direction, that is, certain devices may interfere with children’s growth.
The legislation that called for this report asked for an assessment of whether postmarket surveillance studies last long enough to evaluate the impact of growth and development for the number of years that a child has an implant. It also asked whether such studies are adequate to assess the effects of children’s active lifestyles on implant longevity and failure rates. These questions reflect awareness that children’s developmental characteristics may affect their experience with an implanted device. They likewise show an understanding that short-term studies of safety and effectiveness are not well suited to determine how children’s growth and development may affect the performance of an implant—and vice versa. The committee interpreted its task to involve specifically an assessment of Section 522 Postmarket Surveillance studies, but it also considered other kinds of studies and sources of information.
The next section of this chapter reports the sparse results of the committee’s search for postmarket studies or other information focused on the two child-centered questions identified in the legislation. The discussion then expands to consider more generally strategies for postmarket evalua-
tion of medical devices used with children. The chapter also describes some of the complexities and challenges of conducting medical device research and undertaking studies with children. It concludes with the committee’s reflections and recommendations, including a recommendation that FDA be given authority to order “condition-of-clearance” studies.
FDA-REQUIRED STUDIES AND OTHER INFORMATION
As discussed in Chapter 2, problems with the potential or actual performance of devices in infants, children, and adolescents may be identified in at least three different ways (Table 2.3). They may be identified a priori based on a combination of expert understanding of children’s developmental characteristics and detailed knowledge of the operating characteristics of a particular device as derived from theory, bench testing, simulations, and, perhaps, experience with adult use. In addition, issues or problems may be revealed as side-effects or adverse events during the clinical testing of a device with children. Subsequently, as experience with a device accumulates following its entry into the market, problems may become known through adverse event reports, through case reports or other shared clinical experience associated with normal follow-up care, or through systematic clinical or epidemiological studies, including postmarket studies ordered by the U.S. Food and Drug Administration (FDA).
Systematic studies can accelerate the identification of practices that improve outcomes for children. For example, in the 1960s, clinical observation of infants being treated for hydrocephalus led to the conclusion that cerebrospinal fluid shunt catheters placed in the atrium of the heart should be routinely revised and lengthened. The interval between placement and lengthening depended on age at implantation (e.g., a 4-month interval for shunts placed at 1 month of age and a 32-month interval for shunts placed between 8 and 12 months of age). A well-planned postmarket study that followed children for 24 months after shunt implementation could have detected this considerably earlier than the 11 years it took to accumulate and evaluate observations from clinical practice (Becker and Nulsen, 1968).
Section 522 Postmarket Surveillance Studies
Chapter 5 reported that FDA officials identified only two Section 522 Postmarket Surveillance studies that they had ordered in recent years following the approval or clearance of a medical device. Neither involved pediatric populations as such. Thus, the simple answer to the questions posed to the committee is that there are no relevant Section 522 Postmarket Surveillance studies to assess for length or adequacy. Rather than stop at
this point, the committee expanded its focus to consider condition-of-approval studies and certain other sources of information.
Condition-of-Approval Studies
The committee sought to determine whether any orders for condition-of-approval studies associated with FDA approval of premarket approval applications (PMAs) or Humanitarian Device Exemptions (HDEs) had mentioned children’s lifestyle or growth and development effects. As described in Chapter 5, FDA does not now have a systematic means of identifying and monitoring condition-of-approval studies, including those studies that involve pediatric questions. During initial conversations with the committee, FDA officials reported that they did not know of condition-of-approval studies that involved pediatric questions.
Using FDA’s online database of original PMA approval letters and related materials, the committee reviewed 4 years of original PMA approval letters (2001 to 2004) for a total of 168 letters. Review of these letters yielded one pediatric study associated with the approval of an injectable gel for treatment of vesicoureteral reflux (the flow of urine back from the bladder into the ureters and kidneys) (P000029, FDA, 2001). The postapproval study was to collect 5-year follow-up data on at least 180 children to assess adverse events as well as evaluate treatment outcome (reflux grade) at 3 months, 12 months, and 5 years. These outcomes were to be compared to outcomes reported in the published literature.
Nothing in the brief description of the injectable gel study indicated a specific focus on growth and development or children’s activity levels, but the committee did not have access to the study protocol. The FDA panel that reviewed the PMA application raised a number of concerns about the data submitted in support of the application (e.g., those evaluating treatment outcomes in the two arms of the randomized trial knew the treatment each study subject had received) (Moodie, 2000). The panel also raised concerns about long-term migration of the gel and a slow failure rate, but it did not link these concerns explicitly to questions of growth and development or activity levels. Assuming the manufacturer completes the study, the results would not be expected until 2006.
In addition, the committee found one data collection element involving children in the 2001 letter of approval for a septal occluder device (P000039, FDA, 2001a). Post-approval reports from the manufacturer were to include data on three categories of patients, one of which was children under age 10. The letter mentioned the objective of better characterizing safety and effectiveness but nothing more specific. All other FDA information about the nature or status of the study is confidential, except that a study protocol was approved in a 2002 supplemental PMA (P000039-S001, FDA, 2002).
The committee also identified a few orders for studies that were to follow subjects who had been included in premarket studies. Some of these subjects were children, but the orders did not identify any pediatric questions.
Because supplemental approval letters are usually not accessible online, the committee was not able to systematically review them to determine whether they included provisions for further study of a device. However, while reviewing an article cited by the American Academy of Pediatrics in its statement to the IOM (AAP et al., 2004b), the committee incidentally discovered a required postmarket study associated with approval of a supplemental PMA. The article reported results from a study that FDA required when it approved a small model of a baclofen infusion pump for treatment of patients who could not be treated with a larger model (Albright et al., 2004). The one-paragraph, online summary of the approval statement does not mention this study (see P860004-S042, FDA, 1999).
According to the article reporting the baclofen study, FDA specified that data be collected for 1 year on the first 100 children implanted with the device (Albright et al., 2004). The study included 14 children who had received the implant as part of the premarket evaluation of the pump and 86 who received the implant after approval. The study found four serious system-related complications, all specific to catheters (including two catheters not made by the pump manufacturer). The authors concluded that none of the complications they observed were related to children’s growth.
In addition to reviewing PMA approval letters, the committee also reviewed letters approving HDEs that mentioned use with children. It located one postmarket study involving children that was associated with an HDE for use of a left ventricular assist device with children (H030003, FDA, 2004a). The device, which had been studied with adults but not children, is intended as bridge to heart transplantation. The sponsor is to follow the first 50 children receiving the implant until transplantation, death, or other outcome. The FDA approval letter did not mention growth and development or activity considerations. For the adults implanted with the device during clinical trials, the average duration of pump support was about 3 months.
The committee also learned incidentally about one voluntary postmarket study involving children. In May 2001, when FDA cleared the first automatic external defibrillator system for use with infants and young children who experience cardiac arrest, the sponsor agreed voluntarily to conduct a follow-up study of up to 50 children worldwide to evaluate how well the device performs in actual use (FDA, 2001e). An inquiry to FDA revealed that the study was underway, but FDA would not provide other information on grounds that such details are statutorily protected confidential information (personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, October 21, 2004).
Premarket Clinical Studies
The focus of this report is on postmarket surveillance, but for the certain devices and conditions, the possible value of postmarket studies of growth and development questions may be related to the length and other features of premarket studies. Premarket studies usually focus on short-term outcomes. The summaries of safety and effectiveness published with PMA approval statements may, however, include contraindications or cautions related to growth or development or activity level concerns that are evident even without clinical study. Examples involve implants that are clearly too large for small children or orthopedic devices that will obviously interfere with bone growth.
Conversations with FDA staff and committee review of individual device approvals indicate that clinical studies to support approval or clearance of medical devices generally last 1 to 2 years. Individual research participants may be followed for shorter periods if, for example, patients are entered into the study at different times following diagnosis.
Occasionally, FDA specifically asks sponsors of a PMA to accumulate study data for considerably longer periods than usual. A case in point involves the Vertical Expandable Titanium Rib (VEPTR) implant that recently received a Humanitarian Device Exemption from FDA for use with children suffering from thoracic insufficiency (defined as severe deformities of the chest, spine, and ribs that prevent normal lung development and respiratory function). The implant must be surgically adjusted to accommodate children’s growth approximately every 6 months, which means that a child implanted at age 3 could expect to undergo at least 28 surgeries by age 17. When the sponsor approached FDA about approval for the device, FDA requested long-term safety and effectiveness data on children who received the implant. The sponsor eventually submitted data for a prospective case series of 247 children, some of whom had been followed for 14 years (H030009, FDA, 2004b). The FDA summary of safety and probable benefit did not report the average follow-up period, but an article describing results for 27 of 41 children implanted since 1990 reported an average follow-up period of 5.7 years (range, 2 to 12 years) (Campbell et al., 2004). No condition-of-approval studies were specified by FDA in the HDE approval letter. The sponsor is, however, planning to create a registry and organize a study group (involving the eight hospitals that participated in the multi-center study of the device) to monitor treatment and adverse events and plan prospective studies using the registry (personal communication, Robert Campbell, M.D., Professor of Orthopedics, University of Texas Health Science Center at San Antonio, November 8, 2004).
As described by the primary investigator, the premarket clinical studies
of VEPTR showed expected problems (based on experience with other treatments) related to the need for multiple surgeries (personal communication, Robert Campbell, M.D., Professor of Orthopedics, University of Texas Health Science Center at San Antonio, November 8, 2004). The studies also found migration of the device over longer time periods as a function both of the pressure exerted by the device and the child’s growth. In response to experience during premarket investigation of the device, several changes were made in the design of the device as shown in Figure 6.1. In addition, the study identified the surgical challenges in safely using the device. The first line in the draft professional labeling approved by FDA states “IMPORTANT: Prior to use, the physician should be trained in the surgical procedure recommended for the use of this device” (H030009, FDA, 2004c, p. 1).
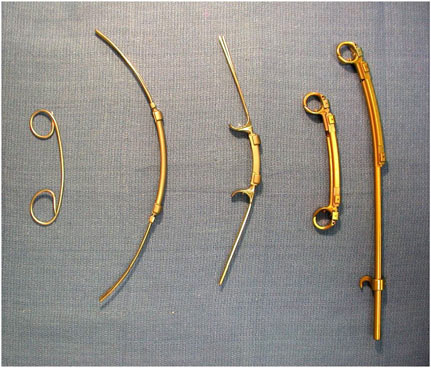
FIGURE 6.1 Evolution of the Vertical Expandable Prosthetic Titanium Rib (VEPTR) showing versions from 1987, 1989, 1991, and 1996. (The rightmost item shows the device in its expanded mode, to its left is the unexpanded device.) (Courtesy of Robert M. Campbell, M.D.)
Safety Advisories and Adverse Event Reports
As an additional step, the committee investigated FDA safety advisories and similar information to determine whether any appeared to have been prompted by adverse event reports associated with children’s activity levels or growth and development on device longevity or performance. The committee found some advisories based at least in part on adverse event reports involving children, but none of the reports obviously involved problems arising from children’s activity levels or growth and development. For example, a public health notification on the risk of bacterial meningitis in children with cochlear implants did not cite developmental considerations explicitly. It stated, however, that it focused on young children “because they account for the majority of known meningitis cases and represent the population that will receive most cochlear implants in the future” (Pressly, 2003, p. 2). The study that investigated the meningitis risk and led to the notification is discussed further below.
Searches of the FDA’s Manufacturer and User Facility Device Experience (MAUDE) database yielded some examples of adverse event reports that might be attributed to children’s activities. For example, use of “basketball” as a search term produced a few potentially relevant reports of incidents involving people playing basketball, although the public database available to the committee did not allow determination of whether these incidents involved children.
The committee concluded that a more systematic search of the MAUDE database was not feasible because the array of possible “active-lifestyle-related” events and possible narrative descriptions of such events is very large, and no recognized nomenclature exists to characterize them. In any case, although FDA has evaluation codes for manufacturers to characterize their evaluation of an adverse event, the agency offers only four very general codes related to use or behavioral factors, for example, “user error caused event” (FDA, 2001j, p. 4). Other patient and device codes are also not specific for lifestyle-related events. In sum, if the narrative for an adverse event report said something like “problems with implant functioning arose after the child jumped off the sofa and bumped her head,” no existing code or feasible search strategy would identify this incident as related to children’s active lifestyle.
Similar difficulties limit the feasibility and value of searching the database for reports that might identify adverse events related to growth and development. For example, reports of a device migration (for which several codes exists) may be related to many factors not related to growth and development. (One device code, 1272, indicates that a device will not support growth [FDA, 2001b].) The committee concluded that further examination of adverse event reports would not be useful in assessing whether
pre- or postmarket studies “last long enough” to identify problems related to active lifestyles or growth and development.
Voluntary Postmarket Studies
In addition to identifying studies required by FDA, the committee made some effort to locate other medical device studies that considered children’s active lifestyles or their growth and development. Unfortunately, the committee found it difficult to identify such investigations. With respect to lifestyles in particular, literature searches and inquiries to clinicians and researchers yielded little—although undiscovered studies of the impact of children’s activities on devices certainly may exist. For example, clinical studies have the potential to identify “active-lifestyle” issues incidentally in the course of investigations that track health and functional outcomes. Unless quite striking, however, the committee concluded that such incidental findings are unlikely to be identified in publication abstracts, key words, or other search aids.
With respect to growth and development, the committee determined that locating relevant clinical studies would, by and large, require device-by-device or condition-by-condition literature searches and device-by-device and condition-by-condition considerations of whether possibly relevant studies are “of long enough duration” to evaluate the impact of child’s growth and development on the performance of an implant or to assess the effects of an implant or other medical device on the way a child grows and develops. Such a search strategy was beyond the committee’s resources. Based on member knowledge, inquiries to pediatric specialists, and literature searches, the committee did identify several relevant studies. Some of these studies are cited in this and other chapters.
DIMENSIONS AND COMPLEXITIES OF MEDICAL DEVICE RESEARCH
Studies of medical devices—especially clinical studies—present challenges both before and after market approval or clearance. In contrast to drugs reviewed by FDA, devices are more often works in progress—subject to minor, modest, or major modifications during both premarket and postmarket clinical studies. Congress has recognized this aspect of device innovation with special provisions to reduce certain regulatory burdens on sponsors (21 USC 360j(g)(6)).1 Depending on the nature of a change during the
course of a study, data may need to be reported and analyzed separately as well as together. Such challenges may be particularly daunting for the small companies that are a more prominent feature of the device industry than the drug industry.
Although specifics will vary depending on the device, premarket testing of medical devices that require clearance or approval may entail a series of evaluations that usually involve nonclinical in vitro testing (also called laboratory or bench testing) and that may extend through tests with animals, possibly cadavers, and then humans. Postmarket studies may also use one or more of these evaluative strategies.
To illustrate the kinds of pre- and postmarket testing that a complex implanted device may undergo, Box 6.1 summarizes the testing of the Amplatzer atrial septal occluder, which, as described earlier, was granted approval for marketing in 2001 (P000039, FDA, 2001a). The device was tested in both children and adults (overall mean age of 18), and results were compared to a nonrandomized, mostly prospectively identified group of individuals (mean age of 6) who were treated surgically. A second small comparison group included patients who were followed through a registry. The FDA summary of safety and effectiveness did not break out study results by age. As described earlier, the approval order for the device included provisions for further postmarket study.
Many devices are proposed for FDA approval as effective for a specific task (e.g., to remove clot from an artery) rather than for a specific therapeutic intervention (e.g., preventing damage to the tissue supplied by the artery). This approach allows a device to become available as a tool with many potential clinical applications and also tends to simplify the premarket evaluation process.
|
|
procedures and conditions that, without requiring an additional approval of an application for an exemption or the approval of a supplement to such an application, permit— (i) developmental changes in the device (including manufacturing changes) that do not constitute a significant change in design or in basic principles of operation and that are made in response to information gathered during the course of an investigation; and (ii) changes or modifications to clinical protocols that do not affect— (I) the validity of data or information resulting from the completion of an approved protocol, or the relationship of likely patient risk to benefit relied upon to approve a protocol; (II) the scientific soundness of an investigational plan submitted under paragraph (3)(A); or (III) the rights, safety, or welfare of the human subjects involved in the investigation. (B) Regulations under subparagraph (A) shall provide that a change or modification described in such subparagraph may be made if— (i) the sponsor of the investigation determines, on the basis of credible information (as defined by the Secretary) that the applicable conditions under subparagraph (A) are met; and (ii) the sponsor submits to the Secretary, not later than 5 days after making the change or modification, a notice of the change or modification. |
|
BOX 6.1 Preclinical study Bench testing for strength and reliability MRI compatibility Corrosion (bench and animal testing) Biocompatibility Sterilization/shelf life Live animal testing in minipigs Premarket clinical study Multi-center, nonrandomized, controlled study to evaluate safety and effectiveness compared to surgical intervention Noncomparative registry study Postmarket condition-of-approval study provisions Five-year follow up of subjects enrolled in phase IIB of the trial Data to be obtained from trial or additional individuals who (1) have device sizes greater than 28 mm or less than 10 mm, (2) residual shunts >2 mm, or (3) were under 10 years of age when the device was implanted. SOURCE: P000039, FDA, 2001b; P000039-S001, FDA, 2002. |
Most of the remainder of this section will consider device research strategies and challenges. Some constraints on research, however, relate less to methodological or technical challenges than to marketing or financial concerns. For example, as is true for other medical products, once a device is approved or cleared, a manufacturer may not be enthusiastic about studying additional uses or populations because such studies could provide negative information that could, in turn, lead to labeling restrictions or even market withdrawal.
If the use of a device is not restricted, medical practitioners can adopt new “unlabeled” uses without oversight by FDA (as described in Chapter 3). Such use is sometimes based on small case series of individual or medical center experiences that are reported at national or international meetings and then followed by diffusion to other centers. For a device with an approval that is not restricted to an adult population, a study with children could generate negative information that might prompt such a restriction. Particularly if the pediatric market is small, a manufacturer might prefer simply to label the device as not indicated for use with children rather than offer or agree to conduct a pediatric study. Given that manufacturers may be reluctant for various reasons to support such studies, other sponsorship and funding for such studies is important (as is the availability of clinicians and other
personnel with sufficient expertise in device studies). The evidence base for practice could benefit from increased clinician involvement in more systematic clinical studies of devices (Moss et al., 2001; Curry et al., 2003).
Even with interest and resources, others barriers to postmarket study may arise. Investigators may find it difficult to recruit participants for clinical trials of devices that are approved (or cleared) and available for use without restrictions. Such trials often make demands on participants (e.g., for extra testing and visits) that go beyond those associated with standard clinical use of the device. In addition, postmarket follow-up studies may be less attractive to research funders and medical journals than studies undertaken for original market approval of innovative device. For these and other reasons, academic investigators may not find involvement in postmarket studies to be professionally appealing.
To cite a practical complication that is not restricted to device studies, the committee understands that manufacturers sometimes have difficulty collecting data for postmarket studies based on professional or institutional concerns about the privacy provisions of the Health Insurance Portability and Accountability Act (HIPAA). For example, some providers may strip patient-identifiers from the information they provide, which makes it difficult or impossible to aggregate information about the same patient from multiple providers. Chapter 4 encouraged FDA to continue its efforts to educate providers about HIPAA and the legality and value of providing information to support postmarket device surveillance, including postmarket studies.
Study Designs and Information Resources for and from Postmarket Devices Studies
A substantial literature exists on the characteristics and merits of various designs for clinical research. These discussions often focus on or assume drugs as the intervention being investigated. Possible constraints on the use of study designs to investigate the safety or effectiveness of medical devices may not be considered. This brief review of study designs is intended to further illustrate the options and challenges of medical device studies. Appendix D discusses in more depth the objectives, characteristics, and limitations of major study designs and data analysis techniques (e.g., data mining).
Some study designs are experimental, which, as used here, means that an investigator controls the use of the intervention(s) being studied.2 Ex-
perimental designs are necessarily prospective, that is, individuals are followed forward in time to assess outcomes. When the investigator does not control the use of the intervention being studied, designs are termed observational. Such studies are sometimes prospective but more often retrospective, that is, based on information already existing in medical records, billing databases, or other sources. The brief overview below starts with experimental studies, not because they are commonly used for postmarket studies of medical devices but because they are considered the benchmark for valid evaluations of clinical care.
Experimental Studies
When designed and implemented properly, the randomized trial is generally held to provide the strongest evidence about the safety and efficacy of a medical intervention.3 In such a trial, investigators use explicit randomization procedures to assign individuals to study groups, for example, an intervention group that will be compared to a group receiving “usual” care or, less often, a group receiving another specific intervention (e.g., an alternative medical device). Randomized trials have occasionally been used to compare medical devices used with children, for example, programmable versus conventional shunts for individuals with hydrocephalus (Pollack et al., 1999).
When entry into study groups is not random, opportunities arise for biases or baseline differences in groups to compromise the validity of study comparisons. Various statistical techniques may help in assessing or controlling for biases, but these are weaker tools than random assignment.
Ideally, to limit bias in the reporting and assessment of outcomes, the assignment of study participants to different groups will be blinded, that is, it will not be known either to the investigators or to individuals being studied. (Unlike such “double-blinded studies, a study is termed “single blinded” if only the evaluator or only the study subject is unaware of the intervention.) The strategy of blinding is difficult if not impossible for many device studies. For example, if use of an implanted device is compared to an accepted medical therapy, individual assignments will usually be obvious to both investigators and study participants. On occasion, investigators have devised “sham” or placebo procedures (e.g., making a shallow surgical incision or arranging exposure to a nonfunctioning device) to limit this source of bias, but such options typically face more ethical and practical barriers than is the case with placebo use in drug studies (Clark and
|
3 |
As described in Chapter 1, the term effectiveness may be used to describe the achievement of desired results in actual practice with the term efficacy reserved for the achievement of such results in controlled studies. |
Leaverton, 1994; Freeman et al., 1999; Albin, 2002). For certain devices, it may be possible to randomize patients to be assessed with the device operating (turned on) or not operating. In general, however, it is necessary to recognize that strategies that may be appropriate and feasible for drug studies may not be appropriate or feasible for device studies.
An example of a randomized, single-blind, long-term postmarket study of device safety is the Children’s Amalgam Trial, a randomized trial of safety that compares silver amalgam dental restorations with a mercury-free restorative material for children aged 6 to 10 (Children’s Amalgam Trial Study Group, 2003). This study, which was not required by FDA, is funded by the National Institute of Dental and Craniofacial Research at the National Institutes of Health (NIH). Originally, intended to last 5 years, the study has been extended to 10 years. The outcomes of interest include neuropsychological development and renal functioning. Those involved in measuring the outcomes do not know the assignment of study participants.
Some studies are prospective and comparative but not randomized. For example, a study might be designed to recruit children for research on an innovative device at one medical center while one or more other institutions might prospectively follow children receiving a common alternative form of care. (Parental permission would be necessary for both groups, and recruitment and other aspects of study design should be as comparable as possible.) This approach, while increasing the opportunity for important non-random differences in study groups and their evaluation, may make sense in certain situations, for example, when the use of a new device involves a significant “learning curve” for clinicians who must master a new surgical technique.
The phenomenon of the learning curve points to another complexity of device trials. Unlike the administration of many drugs in clinical trials, the use of an implant or other device in a trial may depend on a surgical or other procedure that requires new skills or involves unusual elements that must be learned. Differences in skill levels among those “administering” a device intervention can compromise study findings. Thus, some surgical trials have set technical performance standards for participating surgeons (Ferguson et al., 1999). In addition to compromising the validity of study comparisons, differences between clinicians in a trial and clinicians in practice may also limit the generalizability of study findings beyond the research setting.
Often, clinical studies of devices involve neither randomization nor a prospectively followed control group, and such studies can be considered experimental only by the narrow criterion that access is controlled by the investigator. “Single-arm” trials may employ a prospective assessment of a single intervention, access to which is determined by the investigator. Retrospective comparisons are made either to the subject’s status prior to the
intervention or to other individuals (historical controls) for whom information is available in medical records, published materials, or other sources. The former strategy works best when the before-and-after measures for study subjects are standardized and complete, when the natural history of the condition is well-described, and when a “placebo effect” is considered unlikely. The use of historical controls presents many opportunities for bias. For example, depending on the data source for the historical controls, the methods used to collect and record data on key variables may be unstandardized, poorly documented, or unknown. Information that would normally to be used to compare baseline or other differences between groups may simply be missing. Literature reviews suggest that single-arm trials are more likely than comparative controlled trials to conclude that an intervention effect exists (Pocock, 1993).
When FDA approves a device, it may direct a device manufacturer to continue to follow prospectively the individuals who participated in the study that was used to secure FDA approval of the device. If the study originally involved both intervention and control groups, both cohorts would, ideally, be followed.
Some have argued for increased support for pragmatic or practical clinical trials that are designed specifically to answer questions faced by decision makers (see, e.g., Tunis et al., 2003). Such trials would include diverse populations and practice settings and evaluate a range of clinical and functional outcomes. If such practical clinical trials included medical devices used with children, they could help narrow the knowledge gaps that especially characterize pediatric use of many medical devices.
Observational and Other Study Designs
Many device studies involve observational designs, in which access to an intervention or comparison group is not under an investigator’s control. The most ambitious (and, usually, expensive) such studies are prospective and comparative. For example, a study may follow groups (cohorts) of children who—by family choice or other determinant not under the investigator’s control—are treated or are not treated with a device. Long-term, diagnosis-based registries can help investigators identify and monitor children for prospective, comparative observational studies. (Registries are discussed further below.)
When FDA directs a manufacturer to follow the first 50 (or some other number) individuals treated with the device after marketing approval, it is ordering a noncomparative, observational study. The left ventricular assist device study cited above is an example of such a study; it involved a previously unstudied group (children). Although weaker than a controlled experimental study, this kind of planned, prospective observation has the
potential to identify unanticipated safety problems earlier than they would emerge from usual clinical practice.
Most observational studies are retrospective. They depend on existing information (secondary data) to identify study subjects and provide relevant data about their past use of a device and other variables. Again, long-term, diagnosis-based registries may be helpful in identifying individuals for study.
One type of retrospective study design, the case-control study, is particularly useful for investigating rare outcomes. This type of study matches cases (e.g., children with a device who have experienced an adverse event) with controls (e.g., children with a device who have not experienced the event). One component of the postmarket investigation of cochlear implants and meningitis cited above was this kind of case-control study (Reefhuis et al., 2003). Initial sources of information for the study were warranty lists (a limited kind of registry) from implant manufacturers and adverse event reports or other data that identified cases of meningitis in children with cochlear implants. Not all the information was historical. Parents of the children who were identified from warranty lists (which were said to be 95 percent complete) and other sources were contacted for additional information about factors that might have put their child at risk for meningitis (e.g., type of implant, previous diagnosis of meningitis, placement of a ventriculoperitoneal shunt).
Another example of an observational study involving cochlear implants is a study conducted by Waltzman and colleagues that assessed the long-term effects of electrical stimulation (Waltzman et al., 2002). They reviewed the experience of 81 children who had received cochlear implants and had been followed for 5 to 13 years.
Less common and quite different from the prospective and retrospective studies described above are autopsy studies. Such studies can identify physiologic changes not otherwise detectable. Such findings may suggest the need for closer monitoring of certain patient characteristics or adaptations in a device or aspects of its use. Fortunately, child deaths are relatively uncommon, and many critically ill children treated with innovative medical devices survive into adulthood. Nonetheless, despite the stress on families of children who die, future children and families can benefit from sensitive efforts to encourage autopsies.
Parenthetically, although studies of retrieved devices are not clinical studies and although the opportunity for the retrieval of implanted devices is usually not related to a child’s death, it is worth reiterating the argument made in Chapter 4 for systematic study of retrieved devices. Manufacturers and others can accumulate important information from retrieved implants and other devices as part of a comprehensive process of observation, testing, and evaluation once a device has been marketed. This information can
be put to use to improve product reliability or durability and also advise clinicians and patients about the potential for problems that might otherwise not be anticipated.
Registries as Resources for Postmarket Device Studies
As defined in Chapter 1, a registry is a system for collecting information about a class of individuals or patients who have in common a disease, injury, condition, medical procedure or product, or similar characteristic. Registries vary considerably in the amount of information they contain about patients and their care. Manufacturer registries may include only information needed to locate patients in case of a recall, or they may—when designed to support a postmarket study—include considerable clinical information. Although FDA is supportive of other registry studies such as those organized by professional societies, the agency’s primary focus is on registries associated with postmarket studies that they have required or voluntarily negotiated with manufacturers.
Some registries are diagnosis-based and include information about people with a diagnosis who receive certain interventions and people with the diagnosis who do not. Other registries include only individuals who have received a device or intervention. Although a registry managed by a single manufacturer normally would track a single device, an intervention-based registry developed by a professional society or other cooperative group might offer the opportunity to compare different devices or different procedures for using a device. To the extent that centers participating in cooperative intervention-based registries have the most successful programs and the most experienced clinical teams, their results may not be representative of average experience and average complication rates.
An example of a diagnosis-based registry is the pediatric cardiomyopathy registry that is funded by the National Heart Lung and Blood Institute and managed by the New England Research Institute. The registry covers children in the United States and Canada who have been diagnosed with cardiomyopathy since 1990 (NERI, 2004; see also Felker et al., 2000; Harmon et al., 2004). Its aims include estimation of the incidence of the condition (in two geographic regions), better understanding of cardiomyopathies, describing their course and treatment, and identifying correlates of successful patient management. The registry includes 100 clinical sites and records for about 2,500 patients.
An example of an intervention-based registry is the Extracorporeal Life Support Organization (ELSO) registry, which is managed by the University of Michigan. (Extracorporeal Life Support or ECLS is the term that the organization uses for the procedure that historically and earlier this chapter has been referred to as ECMO.) About 110 participating centers in 14
countries submit information to the registry, which includes data on patients until death in hospital or hospital discharge (University of Michigan, 2005). The purpose of the registry is to help define the patient populations treated with the procedure (including specific medical problem), and describe the treatment they received.
The ELSO registry supports retrospective assessments of short-term outcomes and complications as they relate to patient and treatment characteristics. When participating institutions follow or maintain postdischarge contact with a high percentage of their ECMO patients, they can combine the registry data with additional medical record data and new assessments of patients to assess longer term outcomes.
Given the cost and complexity of long-term, multisite studies, it is not surprising that most examinations of longer term ECMO outcomes appear to have involved single centers (see, e.g., Glass et al., 1997; Graziani et al., 1997; Ahmad et al., 1999). In the United Kingdom, however, a multicenter collaborative group is conducting follow-up evaluations of ECMO-treated children at ages 1, 4, and 7 (UK Collaborative ECMO Group, 1998).4
Another intervention-based registry is the Pediatric Heart Transplant Study (PHTS). Until recently, this group was a self-funded collaboration between approximately 20 pediatric heart transplant centers in North America (personal communication, Richard E. Chinnock, M.D., Professor of Pediatrics, Loma Linda University School of Medicine, February 1, 2005; personal communication, Steven A. Webber, M.B.Ch.B., Associate Professor of Pediatrics, University of Pittsburgh School of Medicine, February 1, 2005). The registry began in 2003 but has data extending several years before that. Each center contributed $2,000 to the project and donated the time of personnel to fill out the forms, and the University of Alabama at Birmingham hosted the database. The group has been able to obtain outside funding for some studies, including a study of ventricular assist devices as a bridge to transplantation, but funding for major studies is an ongoing concern.
The experience of the PHTS suggests that the creation of registries, at least for uncommon events, can be initiated without large, direct financial commitments as long as sufficient volunteered expertise is available to
construct and maintain the registry until it can establish itself. The committee encourages greater involvement by professional societies in stimulating and supporting the design and establishment of device-relevant registries, but it also recognizes that the growth of well-designed registry-based studies requires support and encouragement from research funding agencies such as NIH. NIH has, in the committee’s experience, only reluctantly supported registries and then primarily to encourage the orderly transition of a new technology into clinical practice. Ideally, a registry to support clinical studies would be managed by an independent third-party, but the funding for such outside management may only be forthcoming after a registry has been created and shown promise.
Large Automated Patient Databases as Resources for Postmarket Device Studies
Developments in medical informatics have created important, new sources of long-term medical information that make it feasible to monitor device safety—and population health more generally—in ways that were previously not feasible or affordable. These sources include both inpatient databases and databases with inpatient and outpatient information for large populations, for example, health maintenance organizations (HMO) members or Medicare or Medicaid enrollees.
In addition to potential uses in the surveillance of adverse device events and device hazards, electronic inpatient databases may be fruitful for certain postmarket device studies. For example, observational studies based on such databases have investigated common problems with devices to treat hydrocephalus, although the databases have not allowed researchers to distinguish different types of shunts or different manufacturers (see, e.g., Cochrane and Kestle, 2003; Smith et al., 2004).
Some hospitals have cooperated to create multi-institutional automated databases. For example, the Child Health Corporation of America (CHCA, a business alliance of 42 children’s hospitals) has created, among other activities, a Pediatric Health Information System that includes certain clinical and financial information and an extensive data checking process to assure data quality (Fletcher, 2004). The database can support some device safety studies, but the system currently captures only limited information about types of devices (e.g., from itemized bills) and device-related events (e.g., based on ICD-9 codes for certain device-related complications and malfunctions).
Individual hospital databases may include more complete device information, for example, when electronic medical records include data scanned from device and patient armband barcodes in addition to detailed clinical
information. Such databases, especially if expanded to hospital networks with large numbers of children, could provide a useful resource for certain pediatric device safety studies because they would provide greater specificity about device type, more relevant clinical details, larger numbers of children with characteristics of interest, and a denominator for safety and adverse event analyses.
Compared to hospital-only sources, large automated billing and medical record systems maintained by some HMOs and other entities offer the advantage of including data on both inpatient and outpatient care. Unfortunately, as is often the case for inpatient-only databases, detailed information about devices is frequently not available because use of a device is subsumed in another, broader event or transaction such as billing for a medical or surgical procedure. Moreover, as discussed earlier, current device coding options are often insufficient to identify the brand or model of a device implicated in an event. Without such codes, a strategy based on review of computer-based medical records cannot automatically link event reports to specific devices, although the significance of coding limitations varies among devices.
The difficulties for device surveillance and epidemiology created by coding limitations is captured by the following comment by a researcher on problems encountered in determining the specific model of an ultrasound device used for many obstetrical patients, even though investigators knew when and where the procedure occurred. “We needed to contact each obstetrics office and ask a busy person to find and read to us the make and model information, [and] since some offices had more than one model, we couldn’t assign their patients to specific devices” (personal communication, Richard Platt, M.D., Harvard Medical School and Principal Investigator, HMO Research Network CERT, April 8, 2005). This comment reflects experience in the HMO Research Network, which is one of the Centers for Education and Research in Therapeutics (CERTs), a program created by the FDA Modernization Act of 1997 and overseen by the Agency for Healthcare Research and Quality (AHRQ). The network, which includes 14 large HMOs, is conducting several retrospective cohort studies of drug safety and medication errors. Past studies have investigated the association between certain nonsteroidal anti-inflammatory drugs and serious coronary problems (Ray et al., 2002). Device studies have proved substantially more difficult for the Network (personal communication, Richard Platt, M.D., Harvard Medical School and Principal Investigator, HMO Research Network CERT, December 9, 2004; personal communication, James Donahue, Ph.D., Project Scientist, Epidemiology Research Center, Marshfield Clinic Research Foundation, December 9, 2004). In conversations with experts from the Network and other investigators working with large database systems in the CERTs program and elsewhere, the committee found
that none had undertaken a record linkage study involving devices, and all of them reported limitations with existing databases.
In some circumstances, studies involving a medical device used with children could take advantage of ad hoc “hands-on” or manual processes to record device exposures or unusual situations for which the normal data collection process does not suffice (personal communication, Alexander Walker M.D., Dr.P.H., Chief Scientific Officer, Ingenix, March 9, 2005). Thus, children could, in principle, be manually “flagged” as having undergone a procedure or received a device of public policy interest or concern, and then the flagged cohort could be linked to information in automated practice databases. Thereafter, follow-up information would be collected by normal procedures.
AHRQ recently announced that it planned to fund a new CERT that would focus on medical devices (Rundles, 2004). This is a positive step and may focus more attention to the problems identified in this report.
Adequacy of Reporting of Information from Device Studies
Even when clinical studies of medical devices are undertaken, the reporting of key information about the study procedures and outcomes is often too limited to assess the results or the quality of the research procedures that produced them. For example, the summary information of safety and effectiveness published with approvals of PMAs is helpful, but the summaries are not easy to review and they vary in how fully and clearly they describe key aspects of the studies. Some studies may generate more complete descriptions in the peer-reviewed literature, but these must be found through an independent search.
In contrast to the PMA summaries, many peer-reviewed published studies follow a standard reporting format intended to improve the reporting of data and statistical methods for clinical trials. The standard, known as CONSORT (Consolidated Standard of Reporting Trials), has been accepted by many leading medical journals (see, e.g., Begg et al., 1996; Liem et al., 1997; Meinert, 1998; Moher, 1998; Moher et al., 2001). The epidemiology community has been developing a similar set of guidelines, STROBE, which stands for Strengthening the Reporting of Observational Studies in Epidemiology (see, STROBE, 2005).
FDA should consider revisions in the format of PMA summaries to make them more consistent with the CONSORT reporting format. Such a shift would make it much easier for readers to understand the methods used to generate the data submitted to FDA and to evaluate the limitations of these methods and, thus, the limitations of the evidence on which FDA is basing its decisions. A revised format might also direct attention to aspects of device studies that could be improved.
Device Epidemiology as an Underdeveloped Field
Both better data resources and improvements in observational research methods are needed to support epidemiologic studies tailored to the special challenges posed by medical devices. (see, e.g., Shatin et al., forthcoming). The field of device epidemiology can build upon the lessons learned in the related field of pharmacoepidemiology. Over the past 20 years, universities have developed pharmacoepidemology research centers and other programs to offer training and grant degrees. These programs have helped to produce a substantial cadre of research scholars to conduct drug epidemiology studies. As the value of such studies has been recognized, manufacturers and contract research organizations have built their own pharmacoepidemiology units. With this growth, a distinct profession has emerged with its own international professional organization, the International Society for Pharmacoepidemiology (ISPE).
Currently, the committee does not see the same momentum to advance device safety epidemiology. This perpetuates an unfortunate cycle involving an insufficient knowledge base, a shortage of solid research proposals, limited funding for research, and thus, little new knowledge on which to build further research or support for major programs of structured epidemiologic monitoring of device safety.
FDA staff have recognized the importance of device epidemiology and methods development and have contributed in this area. Contributions from other agencies and from university- and industry-based methodologists are important to supplement FDA’s limited resources. One encouraging step is the creation within ISPE of a special interest group for device epidemiology, which may provide a forum to promote the advancement of the field. Another useful step would be for centers with expertise in epidemiology and therapeutics need to offer education in medical device safety studies. In addition, agencies that fund the therapeutics research and education infrastructure (AHRQ, NIH, and the Centers for Medicare and Medicaid Services, in particular) should likewise support the development work necessary to expand the core capacity for epidemiologic research on device safety. Combined with more precise recording of device information in the patient record, these steps should reduce the barriers to needed long-term postmarket studies of medical device safety and effectiveness.
SPECIAL CHALLENGES OF RESEARCH INVOLVING CHILDREN
Beyond the general challenges of conducting ethical, scientifically valid human research and the particular challenges of evaluating medical devices, those interested in developing and testing medical devices to fit children’s
developmental needs and characteristics face some additional constraints (see, e.g., IOM, 2004a; FDA, 2004p). In brief, they include the following.
-
Small populations. Because children are much less likely to suffer ill health than adults, the population of children with a particular health problem may be quite small. As a result, investigators may find it difficult to enroll enough children to make some postmarket research feasible, even more so if one objective is to make statistically valid comparisons of safety or effectiveness in relevant subgroups (e.g., infants and young children or boys and girls). Because enrolling sufficient numbers of children tends to be more difficult than enrolling adequate numbers of adults, pediatric studies are likely to require more study sites and more time. This increases the complexity of planning, funding, and implementing such studies, which may be a particular deterrent to commercial sponsors of research.
-
Special regulatory and ethical protections for child research participants. Certain research that is ethically acceptable and permitted with adults may not be acceptable with children (see, e.g., NHRPAC, 2001; IOM, 2004a). FDA and other regulations of the Department of Health and Human Services (DHHS) limit the level of risk to which children can be exposed in research supported, conducted, or regulated by DHHS (21 CFR 50 and 56; 45 CFR 46). For healthy children, who researchers may want to use as a control group in a study, the level of risk may be no more than minimal. For children who have a medical condition or problem, the level of risk can be only slightly above minimal unless the research has the prospect of benefiting them. These rules limit, for example, the use of certain kinds of invasive studies that are done solely to collect physiologic information.
Under special regulatory provisions, the Commissioner of FDA can approve pediatric studies of special importance that could not otherwise be approved under research protection regulations (21 CFR 50.54). As noted in Chapter 2, the American Thoracic Society provided an example of study that might be appropriate for such consideration. It noted that “[s]tudies of [nebulized] drug deposition to the infant, toddler, or children with tracheostomies are rarely performed in the United States due to the requirement for radiolabeled drug markers” (ATS, 2004b, p. 3). Such investigations are considered to involve more than minimal risk without prospect of benefit to the children being studied. The ATS, however, questioned whether it was ethical to use the devices without allowing testing in infants and children to determine whether the devices were actually delivering therapeutic levels of drugs to these populations. Thus, such studies are candidates for review by the Commissioner.
-
Lack of child-relevant outcome measures and norms. Because mortality rates for children are generally low, other outcomes measures become more important for assessing the quality of pediatric care and the safety and
-
effectiveness of interventions. Sometimes effectiveness requires comparison to normal values of a variable (or usual values for a patient with a particular condition), but these values may not be available for children. An example involves the use of ventilator utilization rates to assess the effectiveness of neonatal intensive care interventions and units. Until recently, reference data were not available to allow risk-adjusted comparisons that take into account differences in the severity of patient condition and baseline risk of needing assisted ventilation and lengthy ventilation (Wilson et al., 2000; see also AAP et al., 2004b).
-
Parent factors. Parents of desperately ill children may see a clinical trial as the last chance for their child. If opportunities exist to obtain an experimental intervention outside a trial—and particularly if the trial has a control arm that involves assignment to a placebo or usual care—then parents may seek out those nonresearch opportunities, perhaps more vigorously than they would for themselves. In addition, with older children in particular, parents and children may be in conflict about enrollment in a study. In most situations, the parent must give permission for child’s inclusion in research, but a child’s assent is not necessary when research presents a prospect of directly benefiting the child. Depending on the circumstances, however, researchers may be reluctant to override a child who does not want to participate, even when a child’s assent is not required.
-
Institutional and professional factors. Many pediatric studies will require multiple centers with a sufficient patient base and a research infrastructure that can support such studies. A model for such multi-institutional studies is the Children’s Oncology Group, which includes over 240 institutions in the United States and other countries and receives support from the National Cancer Institute. Estimates vary but a majority of children with cancer are thought to be enrolled in clinical studies (IOM, 2004a). The model for pediatric cancer studies cannot be moved wholesale to medical devices, which involve a very broad range and diversity of pediatric health problems, devices and associated procedures, and professionals. Nonetheless, the clinical research networks funded by the National Institute for Child Health and Human Development and other NIH units might be better utilized to support the systematic assessment of medical devices used with children. In addition, surgical and device-oriented specialties should be encouraged to place greater value on the systematic evaluation of medical devices and associated procedures used with children.
CONCLUSIONS AND RECOMMENDATIONS
Growth and development issues are significant concerns for clinicians who care for children, including children who have been treated with implants or other medical devices and associated procedures that have the
potential to affect normal growth. The desirable extent and kind of followup evaluation of specific medical devices used with children depends on many factors. Among these factors are the characteristics of the device itself, the nature of the intervention as whole (e.g., whether it involves major surgery), the clinical goals for the intervention, the child’s disease process and other characteristics (e.g., age, activity, and other underlying conditions), and the depth of supporting knowledge about the device and intervention. Because a pathophysiologic event or anatomic or structural change caused by a complex device may not be evident with a child for a number of years, long-term vigilance is important.
With respect to the effects on implants and other devices of children’s active lifestyles and their growth and development, the committee concluded that the systematic consideration of such potential effects should occur during every key stage of device development. Thus, during the conceptualization, development, and preclinical evaluation of a device, manufacturers, FDA staff, and relevant others should ask whether the device may be used with infants, children, or adolescents—and, if so, whether special developmental, growth, activity level, or other issues may be raised by such use that warrant some modification in the design or use of a device.
By the time a device is tested clinically with children (if it is), investigators should have identified certain expected risks and should also be prepared to detect additional unexpected complications. This stage of testing forms a second line of defense against harm to children. Thus, those designing and reviewing clinical trials should consider the significance of growth and development concerns in determining the appropriate length of clinical studies to support the approval of an implant or other relevant device for use with children. They may have to weigh the potential benefits of shorter premarket studies (that allow earlier decisions about approval) against the potential harms of waiting for long-term negative outcomes to be detected after a device is marketed. Depending on the device and other factors, the prospect of longer-term postmarket studies may be considered in the planning of preapproval or preclearance studies. For example, one consideration might be the selection of study sites and investigators (and research participants) who are prepared to continue in a postmarket registry or other study.
Clinician, user facility, manufacturer, and FDA surveillance of device performance after the marketing of a device is a third safeguard for children. Alert and prepared clinicians and provider organizations are the frontline when it comes to identifying apparent device-related problems for reporting as adverse events and for consideration in postmarket evaluations. FDA should be prepared to withdraw approval or clearance, modify device labeling, or otherwise respond if valid follow-up information supports such action.
Because pediatric issues with implants and other medical devices have
not received the same degree of systematic and explicit consideration or analysis that has been devoted to drugs, all parties involved with the initial and continued design and evaluation of medical devices have little formal, evidence-based guidance about differences between children and adults that are relevant for different medical devices. FDA’s recent guidance on premarket assessment of pediatric medical devices, which was discussed in Chapter 2, is a step in the right direction as are the document on neurological devices used with children and the earlier guidance on CT scans with children. The usefulness of additional FDA advice or discussion on pediatric questions should be systematically evaluated in a number of areas, potentially including orthopedics, craniofacial fixation devices, and material biocompatibility.
Recommendation 6.1: FDA should develop additional guidance for its own staff as well as for manufacturers and investigators on the identification and evaluation of pediatric questions or concerns at all stages in the design and evaluation of medical devices used with children.
In developing further pediatric guidance, FDA will want to consider the broad range of variables that may affect device safety. Among these are the environment of use (e.g., hospital, home, or school); operator characteristics (e.g., professionals, patients, or family caregivers); complexity of use (and the associated learning curve); patient developmental status (physical, cognitive, emotional); and use of a device for purposes not systematically studied. To oversee the development of such advice and more generally coordinate attention to concerns involving children, FDA should establish a central point of responsibility for pediatric issues within the Center for Devices and Radiological Health (CDRH) as recommended in Chapter 7.
The promotion and expanding use of electronic patient information systems presents opportunities both to improve surveillance of adverse device events (and hazards) and to undertake more systematic studies of device outcomes. However, this chapter and Chapter 4 have noted several limitations in the utility of such databases for the identification (or prevention) of adverse device events and the systematic study of device safety and effectiveness. In particular, the feasible and useful coding of device information in the medical record has presented many complexities, especially for common “generic” devices such as various kinds of tubing.
Despite the complexities, it is essential that FDA and others continue work on a commonly accepted coding system that allows the more precise identification of specific types and models of devices than is possible with existing coding. This kind of work is critical on multiple fronts, including more efficient and valid identification of device exposure and outcomes in automated databases to support clinical studies and active surveillance of clinical care. When combined with progress in device epidemiology as a
field, such work can help fill gaps in knowledge about safety and effectiveness across the range of postmarket uses of medical devices.
Recommendation 6.2: As part of government and private health informatics initiatives, such as those supporting the electronic medical record, FDA should promote the development and adoption of common device coding and other standards and approaches for capturing and linking use and outcomes data for medical devices. FDA should also work with agencies such as the Agency for Healthcare Research and Quality and university- and industry-based methodologists to strengthen methods and tools for epidemiologic research on medical device safety.
FDA and others could also benefit from more comprehensive information about registries and similar resources that would, in some fashion, track pediatric experience with medical devices. Such information could be provided by a “registry of registries” that included not only registries created as part of FDA postmarket surveillance activities but also relevant registries supported by NIH, professional societies, and others. Such a compilation could provide a basis for evaluating registries and developing guidance for improving registries as a basis for long-term device follow up (Peterson et al., 2004). Although details about FDA-required registries and registry-based studies may now be treated by the agency as confidential, more open access to information about postmarket studies in general should be provided, as recommended in Chapter 5.
Recommendation 6.3: As a resource for itself and others, FDA should create or collaborate with others to create a registry of relevant registries, that is, a database with information about registries that are either device specific or that have the potential to provide information useful in evaluating device safety and effectiveness.
For postmarket device safety to become a more significant focus of attention within FDA, it would be helpful for CDRH to have its own extramural research program similar to that administered by the Center for Drug Evaluation and Research. An extramural program could, for example, support studies using external data sources for postmarket research on device safety questions. These data sources include the HMO Network, Medicare and Medicaid databases, National Electronic Injury Surveillance System (see Chapter 4), and the kinds of registries cited in this chapter. An extramural research program could also help advance the field of device epidemiology, which is, as discussed earlier, an underdeveloped area.
Recommendation 6.4: As part of a public commitment to postmarket surveillance of device safety, the Center for Devices and Radiological Health should have its own extramural research program to support studies using external data sources.
As noted earlier in this report, many complex devices are covered by 510(k) procedures that specify clearance rather than approval of devices before marketing. Some clearance decisions cover devices that involve significant changes from their predecessors (predicate devices). FDA can ask for clinical studies prior to clearing devices, although clinical data are submitted for only a small percentage of devices that go through clearance. FDA cannot, however, order postmarket studies as a condition for clearance. It can (but rarely does) order studies subsequent to clearance through its Section 522 authority. Studies that are ordered subsequent to the approval or clearance of a device are limited to 3 years (which often means a shorter period of evaluation for most individual study subjects). This may be too short a period for certain safety problems or developmental effects to be revealed.
Recommendation 6.5: Congress should amend Section 522 of the Federal Food, Drug, and Cosmetic Act to
-
permit FDA to order postmarket studies as a condition of clearance for the categories of devices for which Section 522 Postmarket Surveillance studies are now allowed and
-
allow FDA to tailor the duration of Section 522 studies of devices likely to have significant pediatric use so that studies can take into account children’s growth and development and, if appropriate, exceed the current 3-year limit on study length.
This recommendation is intended to give FDA more flexibility without prejudging how often the authority would be used. As noted in Chapter 5, the existing Section 522 authority has only been used twice in recent years.
FDA may, of course, encourage long-term studies that it cannot order or is reluctant to order. It can and should work cooperatively with manufacturers to find ways to collect additional, valid information on products that raise concerns about long-term outcomes. It can also encourage pediatric professional societies, academic medical centers, and clinical researchers to get more involved in postmarket device surveillance, for example, by cooperating to create well-designed registries for important medical devices (or device-relevant diagnoses) and undertake registry based-studies that can help answer questions about long-term device effects. FDA can likewise suggest studies for CERTs investigators, including
but not limited to the centers that focus on pediatric topics and medical device questions.
Clearly, many factors influence the long-term safety of medical devices used with children. Scientific and clinical understanding of these factors is limited in many areas, including the underlying biological and physiological phenomena that affect device performance. For example, little is known of the molecular and cellular mechanisms of host-implant interactions that may affect outcomes for pediatric implants and transcutaneous devices. At the behavioral level, strategies that help clinicians, manufacturers, and others communicate successfully with patients and families about the safe and effective use of medical devices at home are little explored.
Given this range of fundamental questions, an opportunity exists for publicly funded national research and research funding organizations to collaborate with industry and FDA to identify priorities for biomedical and bioengineering research to reduce gaps in knowledge about medical device safety and effectiveness. Organizations to involve in such collaboration include not only NIH and AHRQ but also the Centers for Disease Control and Prevention, the National Science Foundation, the Department of Veterans Affairs, the Department of Defense (which operates military health programs), and the National Institute of Standards and Technology.
Recommendation 6.6: FDA should collaborate with the National Institutes of Health, the Agency for Healthcare Research and Quality, and other research funding agencies and interested parties to define a research agenda and priorities for the evaluation of the short- and long-term safety and effectiveness of medical devices used with growing and developing children.
A conference sponsored by funding agencies would be a constructive first step in constructing a research agenda for pediatric device safety and effectiveness. Participants should include researchers and methodologists, engineers, pediatricians and other knowledgeable clinicians, manufacturers, regulators, and representatives of child patients and families. Among the topics considered should be adverse events, including not only catastrophic events but also less dramatic, higher frequency events that may impose significant individual and public health burdens.
Many worthy subjects for further investigation with children can be identified, but resource and feasibility issues inevitably limit what can be done. This committee did not attempt to set priorities for pediatric device research. Some broad objective and subjective criteria can, however, be cited for FDA, NIH, manufacturers, professional societies, and others to consider in targeting topics for investigation. These criteria include
-
prevalence among children of a condition treated with a medical device;
-
presence of significant developmental questions;
-
uncertainty about the short-term or long-term safety and effectiveness of a device to diagnose or treat a condition;
-
known or potential burden (mortality, morbidity, impaired functioning, diminished quality of life) on children and their families;
-
variability in clinical practice;
-
potential of a study to affect clinical practice or regulatory actions; and
-
potential relevance of research findings to other medical conditions or devices.
The recommendations presented above clearly require that many, in addition to FDA, share responsibility for the development of better information about the safety and effectiveness of medical devices used with children. This theme of shared responsibility is reiterated and expanded in the next chapter.






























