
3
Extent and Health Consequences of Chronic Sleep Loss and Sleep Disorders
CHAPTER SUMMARY It is estimated that 50 to 70 million Americans chronically suffer from a disorder of sleep and wakefulness, hindering daily functioning and adversely affecting health and longevity. There around 90 distinct sleep disorders; most are marked by one of these symptoms: excessive daytime sleepiness, difficulty initiating or maintaining sleep, and abnormal events occurring during sleep. The cumulative long-term effects of sleep loss and sleep disorders have been associated with a wide range of deleterious health consequences including an increased risk of hypertension, diabetes, obesity, depression, heart attack, and stroke. After decades of research, the case can be confidently made that sleep loss and sleep disorders have profound and widespread effects on human health. This chapter focuses on manifestations and prevalence, etiology and risk factors, and comorbidities of the most common sleep conditions, including sleep loss, sleep-disordered breathing, insomnia, narcolepsy, restless legs syndrome, parasomnias, sleep-related psychiatric disorders, sleep-related neurological disorders, sleep-related medical disorders, and circadian rhythm sleep disorders.
Sleep loss and sleep disorders are among the most common yet frequently overlooked and readily treatable health problems. It is estimated that 50 to 70 million Americans chronically suffer from a disorder of sleep and wakefulness, hindering daily functioning and adversely affecting health and longevity (NHLBI, 2003). Questions about sleep are seldom asked by physicians (Namen et al., 1999, 2001). For example, about 80 to 90 percent of adults with clinically significant sleep-disordered breathing remain undiagnosed (Young et al., 1997b). Failure to recognize sleep problems not only precludes diagnosis and treatment—it also precludes the possibility of preventing their grave public health consequences.
The public health consequences of sleep loss and sleep-related disorders are far from benign. The most visible consequences are errors in judgment contributing to disastrous events such as the space shuttle Challenger (Walsh et al., 2005). Less visible consequences of sleep conditions are far more prevalent, and they take a toll on nearly every key indicator of public health: mortality, morbidity, performance, accidents and injuries, functioning and quality of life, family well-being, and health care utilization. Some of these consequences, such as automobile crashes, occur acutely within hours (or minutes) of the sleep disorder, and thus are relatively easy to link to sleep problems. Others—for example, obesity and hypertension—develop more insidiously over months and years of chronic sleep problems. After decades of research, the case can be confidently made that sleep loss and sleep disorders have profound and widespread effects on human health.
Although there are around 90 distinct sleep disorders, according to the International Classification of Sleep Disorders (AASM, 2005), most are marked by one of these symptoms: excessive daytime sleepiness, difficulty initiating or maintaining sleep, or abnormal movements, behaviors, and sensations occurring during sleep. The cumulative effects of sleep loss and sleep disorders have been associated with a wide range of deleterious health consequences including an increased risk of hypertension, diabetes, obesity, depression, heart attack, and stroke.
This chapter focuses on the most common sleep conditions, including sleep loss, sleep-disordered breathing, insomnia, narcolepsy, restless legs syndrome (RLS), parasomnias, sleep-related psychiatric disorders, sleep-related neurological disorders, sleep-related medical disorders, and circadian rhythm sleep disorders. The manifestations and prevalence, etiology and risk factors, and comorbidities for each condition are briefly described. There is a large body of data on these disorders, in part because they encompass the most frequently cited sleep disorders or they carry the greatest public health burden. As such, the committee chose to focus primarily on these disorders.
SLEEP LOSS
Manifestations and Prevalence
Sleep loss generally, in adults, refers to sleep of shorter duration than the average basal need of 7 to 8 hours per night. The main symptom of sleep loss is excessive daytime sleepiness, but other symptoms include depressed mood and poor memory or concentration (Dinges et al., 2005). Chronic sleep loss, while neither a formal syndrome nor a disorder, has serious consequences for health, performance, and safety, as described in Chapter 4.
Sleep loss is a highly prevalent problem that continues to worsen in frequency as individuals grow older. Recent studies find that at least 18 percent of adults report receiving insufficient sleep (Liu et al., 2000; Kapur et al., 2002; Strine and Chapman, 2005). Historically, there have been a limited number of nationally representative surveys that provide reliable data on sleep patterns in the population. The National Health Interview Survey (NHIS), run by the Centers for Disease Control and Prevention (CDC) (see Chapter 5), included the following question in the 1977, 1985, 1990 cycles: “On average how many hours of sleep do you get a night (24-hour period)?” The same question was added to the core NHIS questionnaire in 2004. Based on these data, it has been estimated that the percentage of men and women who sleep less than 6 hours has increased significantly over the last 20 years (Figure 3-1) (CDC, 2005). More than 35 years ago, adults reported sleeping 7.7 hours per night (Tune, 1968).
Adolescents also frequently report receiving insufficient sleep. Contrary to public perceptions, adolescents need as much sleep as preteens. A large survey of over 3,000 adolescents in Rhode Island found that only 15 percent reported sleeping 8.5 or more hours on school nights, and 26 percent reported sleeping 6.5 hours or less (Wolfson and Carskadon, 1998). The optimal sleep duration for adolescents, about 9 hours per night, is based on research about alertness, sleep-wake cycles, hormones, and circadian rhythms (Carskadon et al., 2004). Among adolescents, extensive television viewing and growing social, recreational, and academic demands contribute to sleep loss or sleep problems (Wolfson and Carskadon, 1998; Johnson et al., 2004).
Etiology and Risk Factors
The causes of sleep loss are multifactoral. They fall under two major, somewhat overlapping categories: lifestyle/occupational (e.g., shift work,1
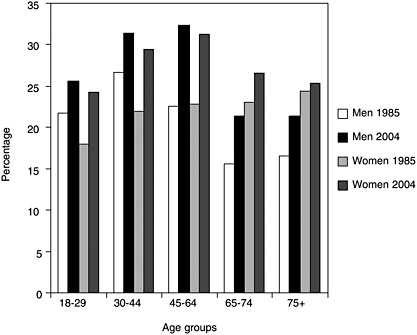
FIGURE 3-1 Percent of adults in the United States who usually slept 6 hours or less a night.
SOURCE: CDC (2005).
prolonged working hours, jet lag, irregular sleep schedules2), and sleep disorders (e.g., insomnia, sleep-disordered breathing, RLS, narcolepsy, and circadian rhythm disorders). Unfortunately, available epidemiological data are not sufficient to determine the extent to which sleep loss is caused by pathology versus behavioral components. The increase in sleep loss is driven largely by broad societal changes, including greater reliance on longer work hours, shift work, and greater access to television and the Internet. About 20 percent of workers are engaged in some kind of shift work (Monk, 2005), of whom there is a growing number of night shift workers suffering chronic sleep loss and disruption of circadian rhythms (Harma et al., 1998; Drake et al., 2004). One indication of the growing trend is the number of adults departing for work between midnight and 5:30 a.m.; that number has grown, over a 10-year period, by 24 percent (United States Census Bureau, 1990). A greater prevalence of insomnia also may contribute to the rise in sleep loss, but probably to a lesser extent than do occupational or lifestyle
changes. Adults are sleeping less to get more work accomplished and are staying up later to watch television or use the Internet (NSF, 2005b).
Sleep Loss Affects Health
In the past 10 or more years, research has overturned the dogma that sleep loss has no health effects, apart from daytime sleepiness. The studies discussed in this section suggest that sleep loss (less than 7 hours per night) may have wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including the following:
-
Obesity in adults and children
-
Diabetes and impaired glucose tolerance
-
Cardiovascular disease and hypertension
-
Anxiety symptoms
-
Depressed mood
-
Alcohol use
Many of the studies find graded associations, insofar as the greater the degree of sleep deprivation, the greater the apparent adverse effect (although the difference may not reach statistical significance). Another common finding is the relationship that adverse effects occur with either short or long sleep duration, as compared to a sleep time of 7 to 8 hours. This type of association is often described as a U-shaped relationship. It should be noted, however, that the majority of these studies are observational in nature, and thus definite causal inferences cannot be made. The associations observed in some studies might be subject to different types of biases, such as temporal (or “reverse causality”) bias, whereby sleep loss might be a manifestation or a symptom of the disease in question. The latter is most likely in cross-sectional studies but could also affect associations observed in cohort studies, particularly when they are relatively short term and/or when the disease under investigation has a long preclinical phase. In the discussion that follows, and wherever possible, potential physiological mechanisms behind epidemiological associations and that support the plausibility of a true causal relationship are noted.
Sleep Loss Is Associated with Obesity
When a person sleeps less than 7 hours a night there is a dose-response relationship between sleep loss and obesity: the shorter the sleep, the greater the obesity, as typically measured by body mass index (BMI)—weight in kilograms divided by height in meters squared. Although most studies were cross-sectional, one prospective study was a 13-year cohort study of nearly
500 adults. By age 27, individuals with short sleep duration (less than 6 hours) were 7.5 times more likely to have a higher body mass index, after controlling for confounding factors such as family history, levels of physical activity, and demographic factors (Hasler et al., 2004). Another study, a large population-based study of more than 1,000 adults, found a U-shaped relationship between sleep duration, measured by polysomnography, and BMI (Figure 3-2). Adults who slept 7.7 hours had the lowest BMI; those with shorter and longer sleep duration had progressively higher BMI. The U-shaped association also applies to other health outcomes, such as heart attacks. The impact of sleep loss diminishes with age. The study also sought to investigate physiological mechanisms behind the relationship between sleep duration and BMI. Measuring two appetite-related hormones, the study found that sleep insufficiency increased appetite. Sleep insufficiency was associated with lower levels of leptin, a hormone produced by an adipose tissue hormone that suppresses appetite, and higher levels of ghrelin, a peptide that stimulates appetite (Taheri et al., 2004). Another study—a small randomized, cross-over clinical trial—also found that sleep restriction was associated with lower leptin and higher ghrelin levels (Spiegel et al., 2004). The findings suggest that a hormonally mediated increase in appetite may help to explain why short sleep is related to obesity. Several mediating mechanisms have been proposed, including effects of sleep deprivation on
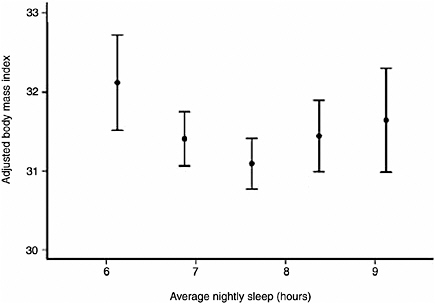
FIGURE 3-2 Curvilinear relationship between BMI and average nightly sleep.
SOURCE: Taheri et al. (2004).
the sympathetic nervous system and/or hypothalamic hormones (Spiegel et al., 2004), which also influence appetite.
Obesity also contributes to obstructive sleep apnea (OSA). This most likely occurs through fat deposition in airways, causing them to narrow. This point is inferred from studies finding that large neck size is a better predictor of OSA than is BMI (Katz et al., 1990) and the finding that central obesity (obesity around the waist) is a better predictor of OSA than total obesity (Grunstein, 2005b). The relationship has been found in well-designed epidemiological studies of young children (Locard et al., 1992; Sekine et al., 2002; von Kries et al., 2002) and adults (Vioque et al., 2000; Kripke et al., 2002; Gupta et al., 2002; Taheri et al., 2004; Hasler et al., 2004).
Taken as a whole, the body of evidence suggests that the serious public health problem of obesity may continue to grow as sleep loss trends continue to worsen. It also suggests that addressing obesity will likely benefit sleep disorders, and treating sleep deprivation and sleep disorders may benefit individuals with obesity (Taheri et al., 2004).
Sleep Loss Is Associated with Diabetes and Impaired Glucose Tolerance
Two large epidemiological studies and one experimental study found an association between sleep loss and diabetes, or impaired glucose tolerance. Impaired glucose tolerance, which is a precursor to diabetes, is manifested by glucose levels rising higher than normal and for a longer period after an intravenous dose of glucose. In the Sleep Heart Health Study, which is a community-based cohort, adults (middle-aged and older) who reported 5 hours of sleep or less were 2.5 times more likely to have diabetes, compared with those who slept 7 to 8 hours per night (Figure 3-3, [Gottlieb et al., 2005]). Those reporting 6 hours per night were about 1.7 times more likely to have diabetes. Both groups were also more likely to display impaired glucose tolerance. Adults with sleep times of 9 hours or more also showed these effects, a finding consistent with the Nurses Health Study. Adjustment for waist girth, a measure of obesity, did not alter the significance of the findings, suggesting that the diabetes effect was independent of obesity.
The relationship between shorter sleep times and impaired glucose tolerance is also supported by an experimental study in which 11 healthy male volunteers were restricted to 4 hours of sleep for a total of six nights (Spiegel et al., 1999). Even after this relatively short period of time, the study found that sleep loss, compared with a fully rested state, led to impaired glucose tolerance. The effect resolved after restoring sleep to normal. Glucose clearance was 40 percent slower with sleep loss than with sleep recovery. Further, mice that have a mutation in a gene that regulates
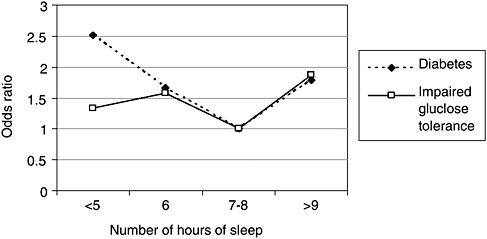
FIGURE 3-3 Sleep duration impacts prevalence of diabetes.
NOTE: Data were adjusted for age, sex, race, waist girth, caffeine, alcohol, smoking, and apnea-hypopnea index.
SOURCE: Gottlieb et al. (2005).
circadian rhythms have metabolic disorders (Turek et al., 2005). The association between sleep loss and diabetes or impaired glucose tolerance may mediate the relationship between sleep loss and cardiovascular morbidity and mortality, as discussed below.
Sleep Loss Is Associated with Cardiovascular Morbidity
Sleep loss and sleep complaints are associated with heart attacks (myocardial infarction) and perhaps stroke, according to several large epidemiological studies (Eaker et al., 1992; Qureshi et al., 1997; Schwartz et al., 1998; Newman et al., 2000; Ayas et al., 2003; Yaggi et al., 2005; Bradley et al., 2005; Caples et al., 2005) and one case-control study (Liu et al., 2002). One of these studies, of incident cases of heart attacks in the Nurses Health Study, was discussed earlier because it also found increased incidence of diabetes (Ayas et al., 2003). The cohort had no coronary heart disease at baseline. Ten years later, in 1996, the likelihood of nonfatal and fatal heart attack was modestly increased for both short and long sleep duration. Five hours of sleep or less was associated with a 45 percent increase in risk (odds ratio [OR] = 1.45, 95% confidence interval [CI], 1.10–1.92), after adjusting for age, BMI, smoking, and snoring. Similarly elevated risks were also found for sleeping 9 hours or more. The effects were independent of a history of hypertension or diabetes because additional adjustment for these
conditions yielded slightly lower, but still significantly elevated, relative risks.
Several potential mechanisms could explain the link between sleep loss and cardiovascular events, including blood pressure increases, sympathetic hyperactivity, or impaired glucose tolerance. Experimental data, showing that acute sleep loss (3.6 hours sleep) for one night results in increased blood pressure in healthy young males, may provide a biological mechanism for the observed associations between sleep loss and cardiovascular disease (Tochikubo et al., 1996; Meier-Ewert et al., 2004).
Sleep Loss, Mood, Anxiety, and Alcohol Use
Sleep loss is associated with adverse effects on mood and behavior. Adults with chronic sleep loss report excess mental distress, depressive symptoms, anxiety, and alcohol use (Baldwin and Daugherty, 2004; Strine and Chapman, 2005; Hasler et al., 2005). A meta-analysis of 19 original articles found that partial sleep deprivation alters mood to an even greater extent that it does cognitive or motor functions (Pilcher and Huffcutt, 1996).
Several studies of adolescents, including one with more than 3,000 high school students, found that inadequate sleep is associated with higher levels of depressed mood, anxiety, behavior problems, alcohol use (Carskadon, 1990; Morrison et al., 1992; Wolfson and Carskadon, 1998), and attempted suicide (Liu, 2004). Nevertheless, it is not clear from cross-sectional studies whether sleep influences mood or anxiety level, or vice versa. On the other hand, a large, 3-year longitudinal study of more than 2,200 middle school students (ages 11 to 14) found that self-reported sleep loss was associated with more depressive symptoms and lower self-esteem over time (Fredriksen et al., 2004). The study measured sleep loss using a single question about sleep duration on school nights and measured depressive symptoms and self-esteem by the Children’s Depressive Inventory and the Self-Esteem Questionnaire, respectively. Therefore, although this study suggests an association, the evidence is still limited.
Sleep Loss and Disease Mortality
Sleep loss is also associated with increased age-specific mortality, according to three large, population-based, prospective studies (Kripke et al., 2002; Tamakoshi et al., 2004; Patel et al., 2004). The studies were of large cohorts, ranging from 83,000 to 1.1 million people. In three studies, respondents were surveyed about their sleep duration, and then they were followed for periods ranging from 6 to 14 years. Deaths in short or long sleepers were compared with those who slept 7 hours (the reference group), after adjusting for numer-
ous health and demographic factors. Sleeping 5 hours or less increased mortality risk, from all causes, by roughly 15 percent. The largest American study, depicted in Figure 3-4, graphically illustrates what has been found in all three studies: a U-shaped curve, showing that progressively shorter or longer sleep duration is associated with greater mortality. Other epidemiological studies suggest that sleep-loss-related mortality is largely from acute heart attacks (Ayas et al., 2003). Potential pathophysiological mechanisms accounting for the relationship, while poorly understood, have become the focus of growing interest and are discussed later in this chapter.
Management and Treatment
Management and treatment of sleep loss are rarely addressed by clinicians, despite the large toll on society (Chapters 4, 5, and 7). There are no formal treatment guidelines in primary or specialty care for dealing with sleep loss (Dinges et al., 1999). The most effective treatment for sleep loss is to sleep longer or take a short nap lasting no more than 2 hours (Veasey et al., 2002), and to have a better understanding of proper sleep habits. Catching up on sleep on the weekends—a popular remedy for sleep loss—does not return individuals to baseline functioning (Szymczak et al., 1993; Dinges et al., 1997; Klerman and Dijk, 2005; Murdey et al., 2005). If extended work hours or shift work cannot be avoided, specific behavioral tips to stay alert are available (NSF, 2005c), as are such wake-promoting
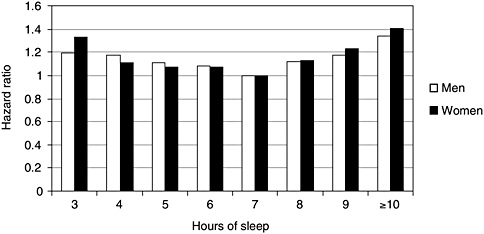
FIGURE 3-4 Shorter or longer sleep duration is associated with greater mortality.
NOTE: Hazard ratio is an individual’s relative risk of dying compared to the general population, based upon average number of hours of sleep per night.
SOURCE: Kripke et al. (2002).
medications as caffeine, modafinil, and sympathomimetic medications (direct and indirect acting), including pemoline and methylphenidate (Mitler and O’Malley, 2005). In a randomized clinical trial caffeine and modafinil showed similar benefits for performance and alertness (Wesensten et al., 2002). Modafinil is the only FDA-approved drug for shift work sleep disorder, although it is not approved for sleep loss. Behavioral approaches developed for insomnia also may be useful for sleep loss, but no formal studies have been undertaken expressly for sleep loss. Furthermore, there have been no large-scale clinical trials examining the safety and efficacy of modafinil, or other drugs, in children and adolescents.
SLEEP-DISORDERED BREATHING
Manifestations and Prevalence
Sleep-disordered breathing refers to a spectrum of disorders that feature breathing pauses during sleep. The most common disorder is characterized by obstructive apneas and hypopneas (White, 2005), where repeated episodes of collapse (apneas) or partial collapse of the pharyngeal airway occur, usually a result of obstruction by soft tissue in the rear of the throat. Snoring, which is produced by vibrations of the soft tissues, is a good marker for OSA (Netzer, et al., 2003). Apneas or hypopneas (a reduction without cessation in airflow or effort) typically result in abrupt and intermittent reduction in blood oxygen saturation, which leads to sleep arousal, often accompanied by loud snorts or gasps as breathing resumes. Episodic interruptions of breathing also frequently cause cortical and brainstem arousals, interrupting sleep continuity, reducing sleep time, and causing increased sympathetic nervous system activation. These broad systemic effects on gas exchange and nervous system activation may lead to a range of systemic effects that affect vascular tone, levels of inflammatory mediators, and hormonal changes. As discussed in the following sections, these in turn may contribute to the development of hypertension, coronary artery disease, congestive heart failure, arrhythmias, stroke, glucose intolerance, and diabetes.
The defining symptom of sleep-disordered breathing is excessive daytime sleepiness. The symptom is likely influenced by sleep fragmentation tied to recurrent arousals that occur in response to breathing pauses. Other symptoms of fragmented sleep include decreased concentration and mood changes. The diagnosis of OSA requires detection, by polysomnography, of at least five or more apneas or hypopneas per hour of sleep (Thorpy, 2005). This rate is expressed as an index, the apnea-hypopnea index (or respiratory disturbance index), which is the average hourly number of apneas plus hypopneas.
OSA is found in at least 4 percent of men and 2 percent of women in the middle-aged workforce, according to the first major United States population-based study of the condition conducted about 15 years ago (Young et al., 1993). Those prevalence figures are based on a cutoff apnea-hypopnea index of 5 or higher, plus a requirement for daytime sleepiness. The prevalence is higher, 9 percent of women and 24 percent of men, with the same apnea-hypopnea index cutoff (Box 3-1), but without the daytime sleepiness requirement. In view of the epidemic increase of obesity (an important determinant of OSA) in recent years, these numbers might underestimate the current prevalence. However, other more recent population-based studies support these prevalence figures (Bixler et al., 1998, 2001).
OSA prevalence appears to increase with age. Adults 65 to 90 years of age had a threefold higher prevalence rate than middle-aged adults (Ancoli-Israel et al., 1991), while the prevalence in children has been reported to be around 2 percent (Ali et al., 1993; Rosen et al., 2003), with higher estimates occurring in ethnic minorities (Gislason and Benediktsdottir, 1995; Redline et al., 1999; Rosen et al., 2003). Underdiagnosis of OSA is common, with between 10 and 20 percent of OSA being diagnosed in adults (Young et al., 1997b). Less than 1 percent of older adults in primary care are referred for polysomnography (Haponik, 1992), although these numbers might have increased in recent years due to increased awareness of the disease. Similarly, children’s OSA often goes undiagnosed too, partly because the implications of snoring are not often recognized by pediatricians. Although OSA can occur in children of any age, it is most common at preschool ages, a time coincident with tonsils and adenoids being largest relative to the underlying airway (Jeans et al., 1981).
Obstructive Sleep Apnea Causes Hypertension
OSA causes chronic elevation in daytime blood pressure (Young et al., 2002a; Young and Javaheri, 2005). The strongest evidence for a rise in systemic hypertension comes from several large, well-designed epidemiological studies, both cross-sectional (Young et al., 1997a; Nieto et al., 2000; Bixler et al., 2000; Duran et al., 2001) and prospective (Peppard et al., 2000). The Wisconsin Sleep Cohort study, a prospective study, tracked adults with sleep-disordered breathing for at least 4 years to determine new onset hypertension and other outcomes. The hypertensive effect was independent of obesity, age, gender, and other confounding factors. Controlling for obesity is especially important because it is a risk factor for hypertension as well as for OSA.
A causal association between OSA and hypertension is supported by evidence of a dose-response relationship; the higher the apnea-hypopnea index, the greater the increase in blood pressure (Peppard et al., 2000; Nieto
|
BOX 3-1 Definitions Impact Disease Prevalence Estimates The metric used most commonly to define obstructive sleep apnea and to quantify its severity is the apnea-hypopnea index, derived by identifying and manually counting each respiratory disturbance (apnea and hypopnea) with subsequent division of the sum by the number of hours slept. Technology for measuring changes in airflow and ventilatory effort has evolved rapidly, with laboratories varying in the implementation of specific sensors and scoring approaches for identifying respiratory events. Variation in event identification has been particularly great for hypopneas (Moser et al., 1994), which requires identification of more subtle changes in airflow than do apneas, and often requires visualization of corroborative changes in oxygen desaturation or evidence of a cortical arousal. Variation in the sensors used to detect breathing changes, the amplitude criteria (from discernible to greater than 50 percent) applied to identify any given reductions in breathing signals as hypopneas, and different uses of corroborative data (associated desaturation and arousal) to discriminate “normal” from “hypopneic” breaths have all contributed to marked laboratory differences in events scored for clinical or research purposes. Likewise, there has been variation in the choice of threshold values for the apnea-hypopnea index considered to define the disease state. An analysis of over 5,000 records from the Sleep Heart Health Study underscores the potential variability introduced by varying either hypopnea definitions or threshold values. This analysis showed that the magnitude of the median apnea-hypopnea index varied 10-fold (i.e., 29.3 when the apnea-hypopnea index was based on events identified on the basis of flow or volume amplitude criteria alone to 2.0 for an apnea-hypopnea index that required an associated 5 percent desaturation with events) (Redline et al., 2000). Using any given definition but varying the threshold to define disease also resulted in marked differences in the percentage of subjects classified as diseased. For example, using an apnea-hypopnea index cutoff value of greater than 15 and an apnea-hypopnea index definition requiring a 5 percent level of desaturation resulted in a prevalence estimate of 10.8 percent. In contrast, almost the entire cohort was identified to be “affected” when sleep-disordered breathing was defined using an apnea-hypopnea index threshold of 5 and when all hypopneas were scored regardless of associated corroborative physiological changes. These data and others have identified the critical need for standardization. As such, at least three efforts led by professional organizations have attempted to develop standards. The latest efforts by the American Academy of Sleep Medicine (2005) have attempted to apply evidence-based guidelines to the recommendations. Unfortunately, the lack of prospective studies that allow various definitions to be compared relative to predictive ability have limited these initiatives, resulting in some recommendations reflecting consensus or expert opinion that may change as further research is developed. |
et al., 2000). Both the Wisconsin Sleep Cohort study and the Sleep Heart Health Study showed dose-response relationships. The Sleep Heart Health Study is a community-based multicenter study of more than 6,000 middle-aged and older adults whose apnea-hypopnea index was measured by polysomnography. The likelihood of hypertension was greater at higher apnea-hypopnea index levels. Case-control studies reveal that approximately 30 percent of patients diagnosed with essential hypertension (hypertension in which the underlying cause cannot be determined) turn out to have sleep apnea (Partinen and Hublin, 2005). Further, evidence from pediatric studies indicate elevations in systemic blood pressure during both wakefulness and sleep in children with sleep apnea (Amin et al., 2004), with additional evidence of left ventricular wall changes by echocardiography.
The causal nature of the relationship between OSA and hypertension is reinforced by randomized controlled clinical trials showing that the most effective treatment for OSA, continuous positive airway pressure (CPAP) therapy, can reduce blood pressure levels. Although findings have been mixed in other studies, a critical review article that evaluated each study’s methodology and results concluded that the trials show convincing decreases in blood pressure in those patients with severe OSA. The benefit is greatest in patients with severe OSA, determined by objective (polysomnography) and subjective (daytime sleepiness) criteria. The review also concluded that there was a lack of benefit in patients who had no daytime sleepiness (Robinson et al., 2004b). However, each of these studies was relatively small (less than 150 individuals), and findings can be considered only tentative.
How does OSA cause sustained hypertension? During the night, the apneas and hypopneas of OSA cause a transient rise in blood pressure (30 mm Hg or more) and increased activity of the sympathetic nervous system (Figure 3-5). Over time, the transient changes become more sustained and are detectable during the daytime, including evidence of sympathetic overactivity (Narkiewicz and Somers, 2003). Studies have found that people with OSA (versus those with similar blood pressure, but no OSA) have faster heart rates, blunted heart rate variability, and increased blood pressure variability—all of which are markers of heightened cardiovascular risk (Caples et al., 2005). The precise pathophysiological steps from transient vascular changes to systemic hypertension are far from clear but may involve oxidative stress, upregulation of vasoactive substances (Caples et al., 2005), and endothelial dysfunction (Faulx et al., 2004; Nieto et al., 2004; Young and Javaheri, 2005).
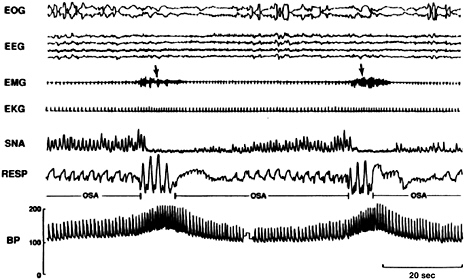
FIGURE 3-5 REM sleep recordings for an individual with OSA.
NOTE: During even the lowest phase, blood pressure during REM was higher than in the awake state. Electrooculogram (EOG), electroencephalogram (EEG), electromyogram (EMG), electrocardiogram (EKG), sympathetic nerve activity (SNA), respiration (RESP), blood pressure (BP).
SOURCE: Somers et al. (1995).
Obstructive Sleep Apnea Is Associated with Cardiovascular Disease and Stroke
Epidemiological studies reveal an association between OSA and cardiovascular disease, including arrhythmias (Guilleminault et al., 1983); coronary artery disease (Andreas et al., 1996) and specifically, myocardial infarction (Hung et al., 1990; D’Alessandro et al., 1990; Mooe et al., 1996a,b; Marin et al., 2005); and congestive heart failure (Javaheri et al., 1998). Most case-control studies detecting a relationship with myocardial infarction found adjusted odds ratios of around 4 (Young et al., 2002a,b). The large, cross-sectional Sleep Heart Health Study of nearly 6,500 (Shahar et al., 2001) found that participants in the highest apnea-hypopnea index quartile (index greater than 11) were 42 percent more likely to self-report cardiovascular disease (coronary heart disease, heart failure, or stroke) than those in the lowest quartile (adjusted OR = 1.42, 95% CI, 1.13–1.78). The adjusted OR for stroke was 1.58 (95% CI, 1.02–2.46). A higher probability of stroke associated with OSA is also supported by other studies (Bassetti and Aldrich, 1999; Parra et al., 2000; Yaggi et al., 2005; Bradley et al., 2005). In the Sleep Heart Health Study, apnea-hypopnea index was deter-
mined by polysomnography, and adjustments were made for a variety of confounding factors, including hypertension. That the hypertension adjustment did not eliminate the effect suggests that hypertension is not the exclusive means by which OSA may lead to cardiovascular disease. A limitation of cross-sectional and case-control analyses is that cause and effect cannot be determined: heart disease may have resulted in OSA or vice versa. However, an observational cohort study of 1,022 individuals, where 68 percent of individuals had OSA (apnea-hypopnea index of 5 or higher), showed that OSA syndrome significantly increased the risk of stroke or death from any cause, and the increase is independent of other risk factors, including hypertension (Yaggi et al., 2005). Other studies have confirmed the risk of OSA syndrome with stroke or death from any cause (Ayas et al., 2003; Gami et al., 2005). Furthermore, other large prospective studies also have shown an association between snoring—a marker for OSA—and incidence of cardiovascular diseases (Jennum et al., 1995; Hu et al., 2000), providing temporal associations in support of OSA playing a causal role in the development of heart diseases. As will be discussed in the next section, OSA is associated with glucose intolerance and diabetes, both of which are independent risk factors for cardiovascular disease.
Studies of the benefits of CPAP further support an association between cardiovascular disease and OSA. Marin and colleagues (2005), in a large, observational study of 10 years’ duration, found that patients with untreated severe OSA (apnea-hypopnea index greater than 30), relative to those receiving CPAP treatment, with similar apnea-hypopnea index severity, had a higher incidence of fatal and nonfatal cardiovascular events. The events included myocardial infarction, stroke, and coronary artery bypass surgery. The untreated patients had refused CPAP but were followed regularly. A second study found an increased mortality rate from cardiovascular disease in individuals who did not maintain CPAP treatment over a 5-year follow-up period (Doherty et al., 2005). However, the number of new cases of cardiovascular disease was independent of CPAP treatment compliance. Although observational evidence of this type is not conclusive proof, because it may be subject to confounding by indication and other biases, it still lends weight to the strength of the association.
Most studies finding elevated cardiovascular disease risk have been conducted in adults. Whether or not children with sleep-disordered breathing are at risk for cardiovascular effects is not known. Children with OSA, as noted previously, do experience changes in blood pressure profiles, heart rate variability, and ventricular wall changes as measured by echocardiography (Marcus et al., 1998; Amin et al., 2005). The paucity of longitudinal data on OSA in children, in whom levels of OSA may vary during growth and development and in whom responses to therapies such as tonsillectomy may be variable (Morton et al., 2001), limits the ability to speculate on the
long-term cardiovascular effects of untreated sleep-disordered breathing in children. Nonetheless, evidence that as many as 20 to 25 percent of children may have persistent OSA even after tonsillectomy underscores the potential importance of OSA as an early childhood risk factor for later cardiovascular diseases (Amin et al., 2005; Larkin et al., 2005).
Obstructive Sleep Apnea Is Associated with Impaired Glucose Tolerance and Diabetes
OSA is associated with impaired glucose tolerance and insulin resistance, according data from several studies (Ip et al., 2002; Punjabi et al., 2002), including the Sleep Heart Health Study (Punjabi et al., 2004). Those outcomes were more prevalent in those with the highest apnea-hypopnea index. The study also found a relationship between sleep-related hypoxemia and glucose intolerance, which has implications for understanding mechanisms behind the OSA-glucose intolerance link (see below). The Sleep Heart Health Study, as noted earlier, was a large, cross-sectional, community-based study that used polysomnography to identify OSA. The analyses adjusted for obesity (BMI and waist circumference), self-reported sleep duration, and other confounding factors. The findings suggest that OSA contributes to the onset of diabetes through the development of glucose intolerance and insulin resistance, which are established pathophysiological processes in diabetes (Martin et al., 1992).
Of studies that have examined diabetes as an outcome measure, the largest was the prospective Nurses’ Health Study. The study found that, after 10 years of follow-up, occasional snoring (versus nonsnoring) was associated with an elevated risk of new onset diabetes in women, and the risk was even greater for regular snoring (Al-Delaimy et al., 2002). Regular or habitual snoring is an indicator of OSA.
The relationship between OSA and metabolic changes that may lead to diabetes is reinforced by studies of the benefits of CPAP. CPAP alleviates glucose intolerance in the short term and long term (Brooks et al., 1994; Harsch et al., 2004). In a separate study of people with type 2 diabetes as well as OSA, CPAP improved glycemic control (Babu et al., 2005). Recent data also indicate that diabetics with OSA have poorer control of glucose levels, with improvement following treatment of OSA with CPAP (Babu et al., 2005).
The mechanisms by which OSA disrupts glucose metabolism are not established. Drawing on human studies and animal models, the biochemical cascade begins with intermittent hypoxia and recurrent sleep arousals (sleep fragmentation). These events stimulate the sympathetic nervous system, hypothalamic-pituitary-adrenal axis, and adipocytes (Punjabi and Beamer, 2005). Their activation, in turn, leads to release of catecholamines, cortisol, and inflammatory cytokines and other vasoactive intermediates,
which may mediate the development of glucose intolerance, insulin resistance, and, ultimately, type 2 diabetes. Because diabetes is also a risk factor for cardiovascular disease, the interrelationships may partly explain why OSA predisposes to cardiovascular disease (Punjabi and Beamer, 2005).
Obstructive Sleep Apnea May Contribute to Obesity
Up to 40 percent of people who are morbidly obese have OSA (Vgontzas et al., 1994). This finding may reflect the role of obesity as a well-established risk factor for the development of OSA. It may also reflect obesity as a consequence of OSA, although the evidence is not yet conclusive (Grunstein, 2005b). Patients with newly diagnosed OSA, compared with controls matched for BMI and percent body fat, show recent weight gain (Phillips et al., 1999). Data from the Wisconsin Sleep Cohort also show that individuals with OSA have reduced levels of physical activity; OSA-related sleepiness may contribute to changes in activity and energy expenditure, and thus contribute to weight gain. OSA-related hormonal changes may also contribute to obesity. In general, patients with OSA have higher levels of leptin, the appetite-suppressing hormone (Phillips et al., 2000; Palmer et al., 2004; Patel et al., 2004) than controls. However, their morning levels are relatively lower than evening levels (Patel et al., 2004). Thus, either via leptin resistance (where high levels of leptin are present, but tissues are poorly responsive to leptin’s action) or because of disturbances in diurnal variability in leptin, individuals with OSA may be predisposed to lower effective levels of appetite suppressing hormones. Although CPAP reduces leptin levels, it is not known whether such effects relate to differences in the effectiveness of leptin’s actions (Chin et al., 2003). Furthermore, obesity also affects the severity of OSA. Significant weight loss in adolescents who underwent gastric bypass surgery (mean, 58 kg) was associated with a dramatic reduction of OSA severity (Kalra et al., 2005).
Etiology and Risk Factors
In simplest terms, OSA is caused by narrowing or collapse of the airway as a result of anatomical and physiological abnormalities in pharyngeal structures. Apnea episodes cause hypoxemia (insufficient oxygen in the blood) and hypercapnia (high concentration of blood carbon dioxide). The episodes also increase the output of the sympathetic nervous system (Narkiewicz and Somers, 2003), the effect of which is to restore pharyngeal muscle tone and reopen the airway. Although increased sympathetic activity is beneficial for restoring normal breathing and oxygen intake over the short term, it has long-term deleterious effects on vascular tone and blood pressure, among other effects (Caples et al., 2005). These early events—which are
mediated by a variety of chemoreceptors in the carotid body and brainstem—trigger pathophysiological changes that occur not only during the obstructive apneas, but also extend into wakeful states during the day. For example, during daytime wakefulness, people with OSA have higher sympathetic activity (Somers, et al., 1995) and heightened chemoreflex sensitivity, which in turn generates an increased ventilatory response (Narkiewicz et al., 1999). The full pathophysiology of OSA remains somewhat elusive, although research is piecing together the relationships between OSA and a range of the previously described long-term health effects. The etiology of central sleep apnea, although also not well understood, is hypothesized to result from instability of respiratory control centers (White, 2005).
There are a number of risk factors for OSA, including:
-
Obesity, male gender, and increasing age (Table 3-1) (Young et al., 1993). It is unclear how incidence changes with older age; some data suggest that snoring and OSA may decline after age 65 years (Young et al., 1993); however, other studies show very high prevalence rates of OSA in elderly individuals (Bliwise et al., 1988; Ancoli-Israel et al., 1993; Foley et al., 2003). The pathophysiological roles of these risk factors are not well understood, although evidence suggests that fat deposition in the upper airways, which is more likely in males, contributes to the physical narrow-
TABLE 3-1 Risk Factors for Obstructive Sleep Apnea
|
Risk Factor |
Reference |
|
Obesity and BMI greater than 25 kg/m2 |
Grunstein et al., 1993 |
|
Male gender |
Strohl and Redline, 1996; Kapsimalis and Kryger, 2002; Shepertycky et al., 2005 |
|
Familial association |
Guilleminault et al., 1995; Pillar and Lavie, 1995; Redline et al., 1995; Buxbaum et al., 2002 |
|
Alcohol consumption |
Taasan et al., 1981 |
|
Cranial facial structure |
Ferguson et al., 1995 |
|
High and narrow hard palate, elongated soft palate, small chin, and abnormal overjet |
|
|
Enlargement of the tonsils |
Behlfelt, 1990 |
|
Lesions of the autonomic nervous system |
Mondini and Guilleminault, 1985; Rosen et al., 2003 |
|
Race: African Americans, Mexican Americans, Pacific Islanders, and East Asians |
Schmidt-Nowara et al., 1990; Redline et al., 1997; Li et al., 2000 |
-
ing that causes OSA (Robinson et al., 2004a). Menopause also increases the risk of OSA (Bixler et al., 2001; Young et al., 2003), possibly through lower levels of progestational hormones that influence the respiratory system through changes in body fat distribution (Vgontzas and Kales, 1999). However, recent studies suggest that there may be a referral bias that results in a lower apparent rate of sleep apnea in females than in males (Kapsimalis and Kryger, 2002; Shepertycky et al., 2005). Epidemiological evidence suggests that hormone replacement therapy lessens the risk of OSA (Shahar et al., 2003). In children, the main risk factor for OSA is tonsillar hypertrophy, although OSA may also occur in children with congenital and neuromuscular disorders and in children who were born prematurely (Rosen et al., 2003). Asthma, a common childhood respiratory illness, is also associated with OSA in children (Sulit et al., 2005).
-
In adolescents, risk factors may be more similar to those seen in adults and include obesity (Redline et al., 1999). Being a minority is a risk factor for both increased prevalence and severity of sleep-disordered breathing in both children and adults (Rosen et al., 1992; Ancoli-Israel et al., 1995; Rosen et al., 2003). The prevalence of sleep-disordered breathing in the United States is approximately three times higher in middle-aged members of minority groups compared to non-Hispanic whites (Kripke et al., 1997). African American children are at increased risk, even after adjusting for obesity or respiratory problems (Redline et al., 1999; Rosen et al., 2003). Familial and probably genetic factors strongly contribute to OSA (Buxbaum et al., 2002; Palmer LJ et al., 2003; Palmer et al., 2004).
-
Patients with cardiovascular disease and diabetes are also at higher risk for developing both OSA and central sleep apnea (Sin et al., 1999).
-
Patients with impaired baroreflexes (e.g., patients with hypertension or heart failure and premature infants) may be especially susceptible to excessive autonomic responses to chemoreflex stimulation during periods of apnea. In these patient groups, bradyarrhythmias, hypoxia, hypoperfusion, and sympathetic activation during apnea may predispose to sudden death (Somers et al., 1988; 1992).
Sleep-Disordered Breathing May Affect Mortality
Limited evidence suggests that sleep-disordered breathing may affect an individual’s mortality (Young et al., 2002a,b; Lavie et al., 2005). Studies of patients at sleep clinics tend to show an association between sleep apnea and mortality (He et al., 1988), but several well-designed, population-based studies failed to find an association (Ancoli-Israel et al., 1996; Lindberg et al., 1998; Kripke et al., 2002), except in one subgroup of patients below age 60 with both snoring and excessive daytime sleepiness. The subgroup experienced twice the risk of mortality (Lindberg et al., 1998). A recent observa-
tional study of a large cohort of sleep apnea patients (n = 403), snorers, and healthy controls who had been followed for an average of 10 years, found a threefold higher risk of fatal cardiovascular events with severe OSA (Marin et al., 2005). An observational follow-up study of the long-term effects of CPAP therapy on mortality found that compared to individuals that began receiving CPAP therapy for at least 5 years (n = 107), individuals that were untreated with CPAP (n = 61) were more likely to die from cardiovascular disease (14.8 percent versus 1.9 percent, log rank test, P = .009) (Yaggi et al., 2005; Doherty et al., 2005).
Treatment
In adults, OSA is most effectively treated with CPAP and weight loss (Strollo et al., 2005; Grunstein, 2005a). Evidence of CPAP’s efficacy for alleviating daytime sleepiness comes from randomized controlled trials and meta-analysis (Patel et al., 2003). The problem is that many patients are noncompliant with CPAP (see Chapter 6). Other options, although less effective, include a variety of dental appliances (Ferguson and Lowe, 2005) or surgery (e.g., uvulopalatopharyngoplasty) (Powell et al., 2005). In children, the first-line treatment for most cases of OSA is adenotonsillectomy, according to clinical practice guidelines developed by the American Academy of Pediatrics (Marcus et al., 2002). Children who are not good candidates for this procedure can benefit from CPAP. Central apnea treatment is tailored to the cause of the ventilatory instability. Commonly used treatments include oxygen, CPAP, and acetazolamide, a drug that acts as a respiratory stimulant (White, 2005).
INSOMNIA
Manifestations and Prevalence
Insomnia is the most commonly reported sleep problem (Ohayon, 2002). It is a highly prevalent disorder that often goes unrecognized and untreated despite its adverse impact on health and quality of life (Benca, 2005a) (see also Chapter 4). Insomnia is defined by having difficulty falling asleep, maintaining sleep, or by short sleep duration, despite adequate opportunity for a full night’s sleep. Other insomnia symptoms include daytime consequences, such as tiredness, lack of energy, difficulty concentrating, and/or irritability (Simon and VonKorff, 1997). The diagnostic criteria for primary insomnia include:
-
Difficulty initiating or maintaining sleep or nonrestorative sleep.
-
Causing clinically significant distress or impairment in social, occupational, or other important areas of functioning.
-
Not occurring exclusively during the course of another sleep disorder.
-
Not due to the direct physiological effects of a substance or a medical condition (APA, 1994).
Insomnia symptoms are remarkably common, affecting at least 10 percent of adults in the United States (Ford and Kamerow, 1989; Ohayon et al., 1997; Simon and VonKorff, 1997; Roth and Ancoli-Israel, 1999). Prevalence is higher among women and older individuals (Mellinger et al., 1985; Ford and Kamerow, 1989; Foley et al., 1995). Severe insomnia tends to be chronic, with about 85 percent of patients continuing to report the same symptoms and impairment months or years after diagnosis (Hohagen et al., 1993; Katz and McHorney, 1998). The comorbidity of sleep disorders with psychiatric disorders is covered later in this chapter.
Etiology and Risk Factors
The precise causes of insomnia are poorly understood but, in general terms, involve a combination of biological, psychological, and social factors. Insomnia is conceptualized as a state of hyperarousal (Perlis et al., 2005). Stress is thought to play a leading role in activating the hypothalamic-pituitary axis and setting the stage for chronic insomnia. A key study showed that adults with insomnia, compared with normal sleepers, have higher levels, over a 24-hr period, of cortisol and adrenocorticotropic hormone (ACTH), which are hormones released by the hypothalamic-pituitary-adrenal axis after stress exposure (Vgontzas et al., 2001). The 24-hour pattern of cortisol and ACTH secretion is different, however, from that in individuals who are chronically stressed. Cognitive factors, such as worry, rumination, and fear of sleeplessness, perpetuate the problem through behavioral conditioning. Other perpetuating factors include light exposure and unstable sleep schedules (Partinen and Hublin, 2005).
Insomnia patients often attribute their difficulty sleeping to an overactive brain. Several lines of evidence, from preclinical to sleep neuroimaging studies in insomnia patients, suggest that there are multiple neural systems arranged hierarchically in the central nervous system that contribute to arousal as well as insomnia complaints. Disturbances in these systems may differ according to the nature of insomnia. Structures that regulate sleep and wakefulness, for example the brainstem, hypothalamus and basal forebrain, are abnormally overactive during sleep in primary insomnia patients (Nofzinger et al., 2004b). In addition, limbic and paralimbic structures that regulate basic emotions and instinctual behaviors such as the amygdala, hippocampus, ventromedial prefrontal cortex and anterior cingulate cortex
have been shown to be abnormally active during sleep in individuals with primary insomnia and secondary insomnias related to depression (Nofzinger et al., 2004a, 2005). Abnormal activity in neocortical structures that control executive function and are responsible for modulating behavior related to basic arousal and emotions has been observed in individuals with insomnias associated with depression (Nofzinger et al., 2004a, 2005).
The two main risk factors of insomnia are older age and female gender (Edinger and Means, 2005). One large, population-based study found that insomnia was nearly twice as common in women than men, although reporting bias cannot be ruled out as a contributing factor (Ford and Kamerow, 1989). The reason behind the apparent higher prevalence in women is not understood. Other risk factors for insomnia include family history of insomnia (Dauvilliers et al., 2005), stressful life styles, medical and psychiatric disorders, and shift work (Edinger and Means, 2005). Although adolescent age is not viewed a risk factor, insomnia has rarely been studied in this age group.
Treatment
Insomnia is treatable with a variety of behavioral and pharmacological therapies, which may be used alone or in combination. While the therapies currently available to treat insomnia may provide benefit, the 2005 NIH State of the Science Conference on the Manifestations and Management of Chronic Insomnia concluded that more research and randomized clinical trials are needed to further verify their efficacy, particularly for long-term illness management and prevention of complications like depression (NIH, 2005). Behavioral therapies appear as effective as pharmacological therapies (Smith et al., 2002), and they may have more enduring effects after cessation (McClusky et al., 1991; Hauri, 1997). Behavioral therapies, according to a task force review of 48 clinical trials, benefit about 70 to 80 percent of patients for at least 6 months after completion of treatment (Morin et al., 1999; Morin, 2005). The therapies are of several main types (Table 3-2). The major problem with current behavioral therapies is not their efficacy; rather it is lack of clinician awareness of their efficacy and lack of providers sufficiently trained and skilled in their use. Other problems are their cost and patient adherence (Benca, 2005a). A specific strategy to improve an individual’s sleep quality is by promoting proper sleep hygiene (Kleitman, 1987; Harvey, 2000).
The most efficacious pharmacological therapies for insomnia are hypnotic agents of two general types, benzodiazepine or nonbenzodiazepine hypnotics (Nowell et al., 1997). Nonbenzodiazepine hypnotics are advantageous because they generally have shorter half-lives, thus producing fewer impairments the next day, but the trade-off is that they may not be as effective at
TABLE 3-2 Psychological and Behavioral Treatments for Insomnia
|
Therapy |
Description |
|
Stimulus control therapy |
A set of instructions designed to reassociate the bed/bedroom with sleep and to reestablish a consistent sleep-wake schedule: Go to bed only when sleepy; get out of bed when unable to sleep; use the bed/bedroom for sleep only (e.g., no reading, watching TV); arise at the same time every morning; no napping. |
|
Sleep restriction therapy |
A method to curtail time in bed to the actual sleep time, thereby creating mild sleep deprivation, which results in more consolidated and more efficient sleep. |
|
Relaxation training |
Clinical procedures aimed at reducing somatic tension (e.g., progressive muscle relaxation, autogenic training) or intrusive thoughts (e.g., imagery training, meditation) interfering with sleep. |
|
Cognitive therapy |
Psychotherapeutic method aimed at changing faulty beliefs and attitudes about sleep, insomnia, and the next-day consequences. Other cognitive strategies are used to control intrusive thoughts at bedtime and prevent excessive monitoring of the daytime consequences of insomnia. |
|
Sleep hygiene education |
General guidelines about health practices (e.g., diet, exercise, substance use) and environmental factors (e.g., light, noise, temperature) that may promote or interfere with sleep. |
|
SOURCE: Morin (2005). |
|
maintaining sleep throughout the night (Morin, 2005; Benca, 2005a). It is still unclear whether hypnotics lead to dependence. It is suggested that they should not be taken for more than 10 days in a row; however, recent studies suggest that hypnotics do not always lead to dependence (Hajak et al., 2003; Walsh et al., 2005; Benca, 2005a). There have been no large-scale trials examining the safety and efficacy of hypnotics in children and adolescents. Other pharmacological classes used for insomnia include sedating antidepressants, antihistamines, and antipsychotics, but their efficacy and safety for treating insomnia have not been thoroughly studied (Walsh et al., 2005).
SLEEP AND PSYCHIATRIC DISORDERS
Manifestations and Prevalence
Sleep disturbances are common features of psychiatric disorders. The most frequent types of sleep disturbances are insomnia, excessive daytime
sleepiness (hypersomnia), and parasomnia. Sleep disturbances are so commonly seen as symptoms of certain psychiatric disorders that they are listed as diagnostic criteria under DSM-IV (APA, 1994). For example, insomnia is a symptom used with others to diagnose major depression. The comorbidity, or coexistence, of a full-blown sleep disorder (particularly insomnia and hypersomnia) with a psychiatric disorder is also common. Forty percent of those diagnosed with insomnia, in a population-based study, also have a psychiatric disorder (Ford and Kamerow, 1989). Among those diagnosed with hypersomnia, the prevalence of a psychiatric disorder is somewhat higher—46.5 percent.
The reasons behind the comorbidity of sleep and psychiatric disorders are not well understood. Comorbidity might be due to one disorder being a risk factor or cause of the other; they might both be manifestations of the same or overlapping physiological disturbance; one might be a consequence of the other. In some cases, the sleep disturbance can be both cause and consequence. In generalized anxiety disorder, for example, the symptoms of fatigue and irritability used to diagnose it are often the result of a sleep disturbance, which itself is also a diagnostic symptom.
Adolescents with major depressive disorders report higher rates of sleep problems and, conversely, those with sleep difficulties report increased negative mood or mood regulation (Ryan et al., 1987). In addition, sleep-onset abnormalities during adolescence have been associated with an increased risk of depression in later life (Rao et al., 1996).
The best studied and most prevalent comorbidity is insomnia with major depression. Insomnia as a symptom of depression is highly common. On the basis of longitudinal studies, insomnia is now established as a risk factor for major depression. Not all people with insomnia have a depression diagnosis; however, studies have found that 15 to 20 percent of people diagnosed with insomnia have major depression (Ford and Kamerow, 1989; Breslau et al., 1996).
Depressed individuals have certain abnormalities detected by polysomnography. One is shorter rapid eye movement (REM) latency (a shorter period of time elapsing from onset of sleep to onset of REM sleep), an effect that persists even after treatment for depression. Other abnormalities include shortened initial REM period, increased REM density, and slow-wave deficits (Benca, 2005a). Shorter REM latency and slow-wave sleep (SWS) deficits tend to run in families; these abnormalities are also found in first-degree relatives of people with major depression, but who are unaffected by depression (Giles et al., 1998). A variety of polysomnographic abnormalities have been found with other psychiatric disorders (Benca, 2005a).
Etiology and Risk Factors
The etiological basis for the comorbidity of sleep disorders and psychiatric disorders is not well understood. Most potential mechanisms for sleep changes in psychiatric disorders deal specifically with insomnia and depression. Possible mechanisms include neurotransmitter imbalance (cholinergic-aminergic imbalance), circadian phase advance, and hypothalamic-pituitary-adrenal axis dysregulation (Benca, 2005a). Recent evidence implicating regions of the frontal lobe has emerged from imaging studies using positron emission tomography. As they progress from waking to non-REM (NREM) sleep, depressed subjects have smaller decreases in relative metabolism in regions of the frontal, parietal, and temporal cortex when compared to individuals who are healthy (Nofzinger et al., 2005). Normally, the transition from waking to NREM sleep is associated with decreases in these frontal lobe regions. What appears to occur with depression is that the decrease is less pronounced. Another finding of the study is that during both waking and NREM sleep, depressed patients show hypermetabolism in the brain’s emotional pathways, including the amygdala, anterior cingulate cortex, and related structures. Because the amygdala also plays a role in sleep regulation (Jones, 2005), this finding suggests that sleep and mood disorders may be manifestations of dysregulation in overlapping neurocircuits. The authors hypothesize that increased metabolism in emotional pathways with depression may increase emotional arousal and thereby adversely affect sleep (Nofzinger et al., 2005).
Treatment
Comorbid psychiatric and sleep disorders are treated by a combination of medication and/or psychotherapy (Krahn, 2005; Benca, 2005a). A major problem is underdiagnosis and undertreatment of one or both of the comorbid disorders. One of the disorders may be missed or may be mistakenly dismissed as a condition that will recede once the other is treated. In the case of depression, for example, sleep abnormalities may continue once the depression episode has remitted (Fava, 2004). If untreated, residual insomnia is a risk factor for depression recurrence (Reynolds et al., 1997; Ohayon and Roth, 2003). Further, because sleep and psychiatric disorders, by themselves, are disabling, the treatment of the comorbidity may reduce needless disability. Insomnia, for example, worsens outcomes in depression, schizophrenia, and alcohol dependence. Treatment of both conditions can improve a patient’s functioning and possibly improve adherence with therapy (Vincent and Hameed, 2003). Another concern is that medication for one disorder might exacerbate the other (e.g., prescription of sedating antidepressants for patients with hypersomnolence). The choice of medica-
tion for psychiatric disorder (or vice versa) should be influenced by the nature of the sleep complaint (e.g., more sedating antidepressant taken at night for insomnia; more alerting antidepressant for excessive daytime sleepiness).
Insomnia and Psychiatric Disorders
As mentioned above insomnia is associated with depression, acting as both a risk factor and a manifestation (Ford and Kamerow, 1989; Livingston et al., 1993; Breslau et al., 1996; Weissman et al., 1997; Chang et al., 1997; Ohayon and Roth, 2003; Cole and Dendukuri, 2003). Several studies done were longitudinal in design, including one that tracked more than 1,000 male physicians for 40 years (Chang et al., 1997). Another study, which followed 1,007 young adults at a health maintenance organization for 3.5 years, found that a history of insomnia at baseline not only predicted new onset depression, but also other psychiatric disorders (any anxiety disorder, alcohol abuse, drug abuse, and nicotine dependence) (Breslau et al., 1996) (Figure 3-6). The adjusted odds of developing a psychiatric disorder were highest for depression (OR = 3.95; 95% CI, 2.2–7.0). This figure is based on 16 percent of the sample who developed depression with a history of insomnia at baseline, as compared with 4.6 percent who developed depression without a history of insomnia. The study’s general findings
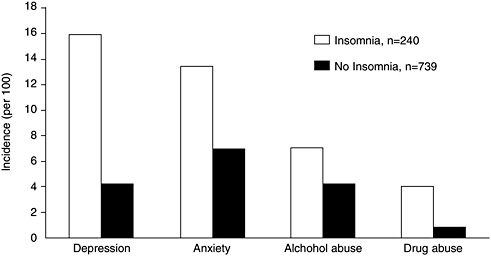
FIGURE 3-6 Incidence of psychiatric disorders during 3.5 years of follow-up of patients with a prior history of insomnia.
SOURCE: Breslau et al. (1996).
are supported by another large study of 10,000 adults by Weissman and colleagues (1997). That study found insomnia to have increased the risk of major depression by a similar magnitude (fivefold) and to have increased the risk of panic disorder (one of the anxiety disorders) even more strikingly, by 20-fold (OR = 20.3, 95% CI, 4.4–93.8). Insomnia is also a predictor of acute suicide among patients with mood disorders (Fawcett et al., 1990).
The striking association between insomnia and depression in so many studies suggests that insomnia is also an early marker for the onset of depression, and the two may be linked by a common pathophysiology. Although the pathophysiological relationship is not known, researchers are focusing on overlapping neural pathways for anxiety, arousal, and/or circadian disturbance (Benca, 2005b). One hypothesis is that common pathways are the amygdala and other limbic structures of the brain (Nofzinger et al., 2005). Another hypothesis is that chronic insomnia increases activity of the hypothalamic-pituitary-adrenal axis, which in turn contributes to depression (Perlis et al., 2005). The close association of insomnia and depression also raises the tantalizing possibility that treating insomnia may prevent some cases of depression (Riemann and Voderholzer, 2003), but limited data are available. The biological basis for the relationship between insomnia and new onset psychiatric disorders (other than depression) is also not known.
NARCOLEPSY AND HYPERSOMNIA
Manifestations and Prevalence
Narcolepsy and idiopathic hypersomnia are characterized by a clinically significant complaint of excessive daytime sleepiness that is neither explained by a circadian sleep disorder, sleep-disordered breathing, or sleep deprivation, nor is it caused by a medical condition disturbing sleep (AASM, 2005). The diagnosis of narcolepsy and hypersomnia is based principally on the Multiple Sleep Latency Test (MSLT), which objectively quantifies daytime sleepiness (Box 3-2) (Carskadon et al., 1986; Arand et al., 2005). Sleep logs or actigraphy (a movement detector coupled with software that uses movement patterns to provide estimate sleep and wake times) can also be used to exclude chronic sleep deprivation as a diagnosis prior to the MSLT. In many cases narcolepsy arises during the mid to late teenage years; however, frequently initial diagnosis is not correct, resulting in delays in diagnosis of 15 to 25 years after the onset of symptoms (Broughton et al., 1997). Onset of narcolepsy can also have a negative impact on school performance (see Chapter 4). Narcolepsy is associated with a number of symptoms (Anic-Labat et al., 1999; Overeem et al., 2002), including the following:
-
Excessive daytime sleepiness, defined as a background of constant sleepiness with sleep attacks leading to unintended napping during the day. In most cases, naps are refreshing, but the rested feeling only lasts a short time. When severe, sleepiness can manifest as automatic behavior, a continuation of activities in a semiautomatic manner when sleepy, with no subsequent memory.
-
Cataplexy, which are sudden and brief bilateral episodes of muscle weakness triggered by a strong emotional stimulus, such as laughing.
|
BOX 3-2 Clinical Laboratory Findings in Narcolepsy and Hypersomnia The Multiple Sleep Latency Test (MSLT) objectively quantifies daytime sleepiness. It consists of five 20 minute daytime naps at 2-hour intervals. The amount of time it takes to fall asleep (sleep latency) and the occurrence of rapid eye movement (REM) sleep is recorded. Mean sleep latency of less than 8 minutes and two or more sleep onset rhythmic eye movement periods is diagnostic for narcolepsy. The MSLT must always be preceded by nocturnal sleep polysomnography to rule out other causes of short MSL or sleep onset rhythmic eye movement periods such as OSA, insufficient sleep, or delayed sleep phase syndrome. At least 6 hours of sleep must have occurred prior to the MSLT. Sleep logs or actigraphy for the preceding 2 weeks can be helpful to exclude chronic sleep deprivation. It must also be conducted after withdrawal of psychotropic medications (generally more than 2 weeks). Antidepressants, most notably, suppress REM sleep and/or may create REM rebound if stopped too recently prior to testing. HLA-DQB1*0602 is the human leukocyte antigen DQB1 subtype associated with narcolepsy. Almost all cases with cataplexy are DQB1*0602 positive; approximately 40 percent of narcolepsy cases without cataplexy are HLA-DQB*0602 positive. The test is not highly predictive of narcolepsy, however, as 25 percent of the population is HLA-DQB1*0602 positive. Idiopathic and recurrent hypersomnia cases are not strongly associated with human leukocyte antigen. Cerebrospinal fluid (CSF) hypocretin-1, also called orexin-A, is a neuropeptide involved in the cause of narcolepsy and cataplexy. It can be measured in the CSF, and this has been used to diagnose narcolepsy. Most cases with cataplexy (and HLA-DQB1*0602) have CSF hypocretin-1 levels below 30 percent of normal control value. This finding is very specific and sensitive for narcolepsy and cataplexy. Low CSF hypocretin is also diagnostic of narcolepsy without cataplexy, but is found in only a small portion of these cases (7 to 40 percent). |
-
Sleep paralysis, or muscle paralysis akin to REM sleep atonia while awake, when falling asleep, or waking up.
-
Hypnagogic/hypnopompic hallucinations, which are dreamlike REM sleep experiences, often frightening, that occur when falling asleep or waking up.
-
Insomnia, typically difficulty maintaining sleep.
-
Autonomic behavior, or continue to function (talking, putting things away, etc.) during sleep episodes but awakens with no memory of performing such activities.
-
REM behavior disorder, characterized by excessive motor activity during REM sleep (Anic-Labat et al., 1999; Overeem et al., 2002).
Narcolepsy can be diagnosed clinically, by using the MSLT, or by measuring cerebrospinal fluid (CSF) hypocretin-1 (Box 3-2).
Idiopathic hypersomnia is classically separated into two subtypes. The first, idiopathic hypersomnia with prolonged sleep time, is a rare disorder and is characterized by the following:
-
Excessive daytime sleepiness occurs, as described above for narcolepsy, but in the typical form naps are unrefreshing.
-
Excessive amounts of daily sleep, typically defined as more than 10 hours of sleep per day, as documented for long periods of time using daily logs and sleep studies.
-
Sleep drunkenness (sometimes referred to as sleep inertia)—difficulty waking up and individual is foggy for long periods of time after wake onset.
The second subtype of idiopathic hypersomnia, idiopathic hypersomnia without long sleep time, is characterized by a complaint of excessive daytime sleepiness and a short mean sleep latency on the MSLT.
In most sleep disorders clinics with experience in this area, approximately one-third of hypersomnia cases are diagnosed with this condition (Aldrich, 1996). The prevalence is estimated to be around 0.01 percent. In contrast, the prevalence of idiopathic hypersomnia without prolonged sleep time may be more substantial, as most patients are likely not diagnosed (Arand et al., 2005).
Recurrent hypersomnia is periodic either in synchrony with menstruation (menstruation-linked periodic hypersomnia) or without any association and mostly in males with Klein-Levin syndrome (Billiard and Cadilhac, 1988; Arnulf et al., 2005a). Klein-Levin syndrome is characterized by recurrent episodes of dramatic hypersomnia lasting from 2 days to several weeks. These episodes are associated with behavioral and cognitive abnormalities, binge eating or hypersexuality, and alternate with long asymptomatic periods that last months or years (Arnulf et al., 2005a).
Narcolepsy and hypersomnia can affect children, adolescents, adults, and older persons. In most cases these disorders begin in adolescence. The prevalence of narcolepsy with definite cataplexy has been documented in adults by numerous population-based studies and occurs in 0.02 to 0.05 percent of the population of Western Europe and North America (Mignot, 1998). In contrast, very little is known about the prevalence of narcolepsy without cataplexy. Recent studies using the MSLT indicate that approximately 3.9 percent of the general population has MSLT score abnormalities consistent with narcolepsy without cataplexy (Singh et al., 2005).
Secondary cases of narcolepsy or hypersomnia are also common, but the overall prevalence is not known (Table 3-3). These can occur in the context of psychiatric disorders, for example depression; central nervous system tumors, most notably in the hypothalamus; neurodegenerative disorders, such as Parkinson’s disease; inflammatory disorders, such as multiple sclerosis or paraneoplastic syndromes; traumatic disorders, such as head trauma; vascular disorders, such as those that are attributed to median thalamic stroke; and genetic disorders, including myotonic dystrophy or Prader-Willi syndrome (Billiard et al., 1994; Mignot et al., 2002a).
Etiology and Risk Factors
Similar to other sleep disorders, little is known about the pathophysiology and risk factors for narcolepsy and hypersomnia. Most of the knowledge in this area pertains to narcolepsy with cataplexy, which affects males and females equally. Symptoms usually arise during adolescence. Many contributing factors influence an individual’s susceptibility, including both genetic and environmental factors (Mignot, 1998, 2001).
Virtually all individuals who suffer narcolepsy with cataplexy carry the haplotype HLA-DQB1*0602 and have severe neuronal loss in regions of the brain that are responsible for regulating the sleep-wake cycle. Approximately 70,000 hypothalamic neurons that are responsible for producing the neuropeptide hypocretin (orexin) are lost in individuals with narcolepsy with cataplexy (Thannickal et al., 2000; Peyron et al., 2000). Hypocretin is an excitatory neuropeptide that regulates the activity of other sleep regulatory networks. Consequently, in some cases low levels of hypocretin-1 in the CSF, may be used to diagnose narcolepsy (Kanbayashi et al., 2002; Krahn et al., 2002; Mignot et al., 2002a) (Table 3-3). The cause of hypocretin cell loss is unknown but it may be autoimmune due to the association with the HLA-DQB1*0602 (Juji et al., 1984; Mignot, 2001).
Less is known regarding the pathophysiology of narcolepsy without cataplexy. The etiology is likely heterogeneous. An unknown portion may be caused by partial or complete hypocretin deficiency (Kanbayashi et al., 2002; Krahn et al., 2002; Mignot et al., 2002a). However, it has been
TABLE 3-3 International Classification of Sleep Disorders: Definitions, Prevalence, and Pathophysiology of Narcolepsy and Hypersomnias
hypothesized that some individuals with partial cell loss may have normal CSF hypocretin-1 (Mignot et al., 2002a; Scammell, 2003).
The pathophysiology of idiopathic hypersomnia is unknown. When the disorder is associated with prolonged sleep time, it typically starts during adolescence and is lifelong. It is essential to exclude secondary causes, such as head trauma or hypersomnia owing to depression (Roth, 1976; Billiard and Dauvilliers, 2001). Some cases with prolonged sleep times have been reported to be familial, suggesting a genetic origin. Even less is known about idiopathic hypersomnia with normal sleep time. This condition is more variable and symptomatically defined. The cause of Kleine-Levin syndrome is unknown (Arnulf et al., 2005b).
Treatment
Treatment for these conditions is symptomatically based. Even in the case of narcolepsy in which the disorder is caused by hypocretin deficiency, current treatment does not aim at improving the defective neurotransmission (Mignot et al., 1993; Nishino and Mignot, 1997; Wisor et al., 2001). Behavioral measures, such as napping, support groups, and work arrangements are helpful but rarely sufficient. In most cases, pharmacological treatment is needed (Nishino and Mignot, 1997; Lammers and Overeem, 2003). However, as with other pharmaceuticals designed to treat sleep problems, large-scale clinical trails have not examined the efficacy and safety of drugs to treat narcolepsy in children and adolescents.
In narcolepsy with cataplexy, pharmacological treatment for daytime sleepiness involves modafinil or amphetamine-like stimulants, which likely act through increasing dopamine transmission. Cataplexy and abnormal REM sleep symptoms, sleep paralysis and hallucinations, are typically treated with tricyclic antidepressants or serotonin and norepinephrine reuptake inhibitors. Adrenergic reuptake inhibition is believed to be the primary mode of action. Sodium oxybate, or gamma hydroxybutyric acid, is also used at night to consolidate disturbed nocturnal sleep. This treatment is also effective on cataplexy and other symptoms.
The treatment of narcolepsy without cataplexy and idiopathic hypersomnia uses similar compounds, most notably modafinil and amphetamine-like stimulants (Billiard and Dauvilliers, 2001). Treatments, with the possible exception of lithium, of periodic hypersomnia and Kleine-Levin syndrome type are typically ineffective (Arnulf et al., 2005a).
PARASOMNIAS
Manifestations and Prevalence
Parasomnias are unpleasant or undesirable behaviors or experiences that occur during entry into sleep, during sleep, or during arousals from sleep (AASM, 2005). They are categorized as primary parasomnias, which predominantly occur during the sleep state, and secondary parasomnias, which are complications associated with disorders of organ systems that occur during sleep. Primary parasomnias can further be classified depending on which sleep state they originate in, REM sleep, NREM, or others that can occur during either state (Table 3-4).
Parasomnias typically manifest themselves during transition periods from one state of sleep to another, during which time the brain activity is reorganizing (Mahowald and Schenck, 2005). Activities associated with parasomnias are characterized by being potentially violent or injurious, disruptive to other household members, resulting in excessive daytime sleepiness, or associated with medical, psychiatric, or neurological conditions (Mahowald and Ettinger, 1990).
Disorders of Arousal, NREM
Disorders of arousal are the most common type of parasomnia, occurring in as much as 4 percent of the adult population (Ohayon et al., 1999) and up to 17 percent of children (Klackenberg, 1982). Typically the arousals occur during the first 60 to 90 minutes of sleep and do not cause full awakenings, but rather partial arousal from deep NREM sleep. Disorders of arousal manifest in a variety of ways, from barely audible mumbling, disoriented sleepwalking, to frantic bouts of shrieking and flailing of limbs (Wills and Garcia, 2002).
Confusional Arousals
Individuals who experience confusional arousals exhibit confused mental and behavioral activity following arousals from sleep. They are often disoriented in time and space, display slow speech, and blunted answers to questions (AASM, 2005). Episodes of resistive and even violent behavior can last several minutes to hours. Confusional arousals are more than three to four times more prevalent in children compared to individuals 15 years or older (around 3 percent) (Ohayon et al., 2000). They can be precipitated by forced arousals, particularly early in an individual’s sleep cycle.
TABLE 3-4 Selected Primary Sleep Parasomnias
Sleepwalking
Sleepwalking is characterized by a complex series of behaviors that culminate in walking around with an altered state of consciousness and impaired judgment (AASM, 2005). Individuals who are sleepwalking commonly perform routine and nonroutine behaviors at inappropriate times and have difficulty recalling episodic events. Like confusional arousals, the prevalence of sleepwalking is higher in children than adults (AASM, 2005).
There appears to be a genetic predisposition for sleepwalking. Children who have both parents affected by sleepwalking are 38 percent more likely to also be affected (Klackenberg, 1982; Hublin et al., 1997).
Sleep Terrors
Sleep terrors are characterized by arousal from SWS accompanied by a cry or piercing scream, in addition to autonomic nervous system and behavioral manifestations of intense fear (AASM, 2005). Individuals with sleep terrors are typically hard to arouse from sleep and, when they are awoken, are confused and disoriented. There does not appear to be a significant gender or age difference in prevalence or incidence of sleep terrors (AASM, 2005).
Disorders Associated with REM Sleep
Rapid Eye Movement Sleep Behavior Disorder
REM sleep behavior disorder is characterized by a complex set of behaviors that occur during REM sleep, including mild to harmful body movements associated with dreams and nightmares (AASM, 2005). Normally during REM sleep, muscles are temporarily paralyzed; however, in REM sleep behavior disorder this paralysis is absent, thus allowing individuals to “play out” their dreams. The overall prevalence in the general population is estimated to be less than half a percent, slightly higher in older persons (AASM, 2005), and affecting men more frequently than women.
REM sleep behavior disorder is frequently associated with neurological disorders and it has been suggested that it could be an early sign of neurodegeneration (Olson et al., 2000). At least 15 percent of individuals with Parkinson’s disease (Comella et al., 1998; Gagnon et al., 2002) and 44 percent of individuals with multiple system atrophy (Plazzi et al., 1997; 1998) also suffer from REM sleep behavior disorder. There are a number of effective pharmacological treatments, including a long-acting benzodiazepine (Schenck and Mahowald, 1990), clonazepam (Schenck et al., 1993), and dopamine agonists (Bamford, 1993; Fantini et al., 2003).
Nightmare Disorder
Nightmare disorder is characterized by recurrent disturbances of dreaming that are disturbing mental experiences that seem real and sometimes cause the individual to wake up. If awoken, individuals commonly have difficulty returning to sleep. Nightmares often occur during the second half of a normal period of sleep. Dream content involves a distressing theme, typically imminent physical danger. During nightmares, individuals experience increased heart and respiration rates (Fisher et al., 1970; Nielsen and Zadra, 2000).
Nightmares commonly affect children and adolescents and decrease in frequency and intensity as an individual grows older (AASM, 2005). Drugs and alcohol can trigger nightmares. Prevalence rates are also higher in individuals suffering from acute stress disorder and posttraumatic stress disorder.
SLEEP AND NEUROLOGICAL DISORDERS
Individuals suffering from dementia commonly experience sleep abnormalities. Although there are a variety of conditions associated with dementia—Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, Huntington’s disease, and Creutzfeldt-Jakob disease—there are some common patterns of sleep impairment associated with all dementias. Typically, sleep is more fragmented, leading to more awakenings and consequently less time asleep, and REM may be decreased (Petit et al., 2005). These sleep impairments usually worsen as the disease progresses.
Alzheimer’s Disease
Manifestations and Prevalence
Alzheimer’s disease is a neurodegenerative disorder characterized by memory loss and an intellectual decline that progresses with age and is caused by the degeneration of neurons in the brain. It is estimated that about 4 million individuals in the United States suffer from Alzheimer’s disease. Approximately one-quarter of these individuals have sleep disturbances (Tractenberg et al., 2005; Moran et al., 2005). Alzheimer’s disease causes an increase number of arousals and affects an individual’s sleep architecture. As a result of an increase in duration and number of awakenings, individuals spend an increased percentage of time in stage 1 sleep and a reduced percentage in stage 2 and SWS (Prinz et al., 1982a,b; Reynolds et al., 1985; Montplaisir et al., 1995).
Etiology and Risk Factors
There is limited information regarding the etiology of sleep disorders associated with Alzheimer’s disease and other conditions that cause dementia. Associations with sleep disturbance and other behavioral symptoms have been identified, including aggressiveness (Moran et al., 2005) and depression (Tractenberg et al., 2005). However, the pathophysiology of this association is not known. In addition to behavioral symptoms, OSA may also occur at a higher prevalence in individuals with Alzheimer’s disease than in the general population (Bliwise, 2002).
Treatment
Treatment options for demented individuals who suffer sleep disorders are typically the same as those received by individuals who do not have dementia. The approach is to address the sleep disorder based on its symptoms while managing and treating the underlying medical or psychiatric disorder (Petit et al., 2005). However, treating an individual’s sleep disorder is not effective in reducing dementia associated with Alzheimer’s disease.
Parkinson’s Disease
Manifestations and Prevalence
Sleep complaints and subsequent diminished quality of life are common in individuals who suffer from Parkinson’s disease. Parkinson’s disease is a neurological disorder that primarily affects the elderly—0.9 percent of people 65 to 69 years of age to upwards of 5 percent of people 80 to 84 years of age have Parkinson’s disease (de Rijk et al., 1997). It is characterized by trouble initiating walking and other movements, muscle tremor, a slow gait, and reduced facial expressions. Sleep disturbances associated with Parkinson’s disease include difficulty falling asleep, nocturnal akinesia, altered sleep architecture, abnormal motor activity, periodic limb movements, REM sleep behavior disorder (see above), and disturbed breathing. During the day, many Parkinson patients have excessive sleepiness.
Sleep disturbances typically increase with disease progression. Individuals suffer from increased sleep latency and frequent awakenings, spending as much as 30 to 40 percent of the night awake (Kales et al., 1971; Bergonzi et al., 1975). This causes reduced time spent in stages 3 and 4 and REM sleep and increased duration in stages 1 and 2 (Kales et al., 1971).
Etiology and Risk Factors
Sleep patterns are affected by abnormalities caused by neurodegeneration in regions of the brain that are involved in regulating the sleep-wake cycle. Dopaminergic neurons in the substantia nigra are dramatically reduced in number, as are noradrenerics neurons in the locus coeruleus (Jellinger, 1986) and cholinergic neurons in the pedunculopontine nucleus (Zweig et al., 1989). Braak and colleagues (2004) examined a large series of autopsy brains. They found that Lewy body degeneration begins in the lower brainstem and ascends to involve the substantia nigra only after several years, consistent with observations that REM alterations may precede the movement disorder by several years in many Parkinson’s disease patients. REM sleep behavioral disorder is often seen in patients with Parkin-
son’s disease and other parkinsonian syndromes, such as multiple systems atrophy and progressive supranuclear palsy. The ability to ameliorate the symptoms of REM sleep behavioral disorder with dopaminergic agonist drugs suggests that it may be an early sign of damage to the dopaminergic system (Trampus et al., 1991).
Treatment
Treating sleep disturbances associated with Parkinson’s disease is complicated owing to the different actions associated with dopaminergic medications. Medications used to treat this disorder often include dopamine precursors (levodopa/carbidopa) and dopamine agonists (pramipexole and ropinirole). When used in low doses, these medications can promote sleep, but high doses may cause increased nocturnal wakefulness, decreased SWS, and decreased sleep continuity (Leeman et al., 1987; Monti et al., 1988; Cantor and Stern, 2002). In contrast, excessive daytime sleepiness, including sleep attacks, has also been described in association with dopamine agonists (Paus et al., 2003); therefore, many patients with Parkinson’s disease require daytime stimulants such as modafinil or amphetamine to relieve excessive sleepiness.
Other classes of medication used to treat Parkinson’s disease include monoamine oxidase-B inhibitors (selegiline), presynaptic relating agents (amatadine, anticholingeric agents), and catechol-O-methyltransferase (COMT) inhibitors (hyoscyaine, benztropine). All may potentially affect sleep (Chrisp et al., 1991), particularly with regard to decreasing REM sleep, but the sleep effects of these medications remain to be well described (Kaakkola, 2000).
Epilepsy
Manifestations and Prevalence
Epilepsy refers to a group of various disorders characterized by abnormal electrical activity in the brain that manifests itself in individuals as a loss of or impaired consciousness and abnormal movements and behaviors. Sleep, sleep deprivation, and seizure activity are tightly intertwined. After stroke and Alzheimer’s disease, epilepsy is the third most common neurological disorder in the United States, with incidence between 1.5 to 3.1 percent (Shouse and Mahowald, 2005). It is estimated that sleep-related epilepsy may affect as many as 10 percent or more of epileptic individuals (AASM, 2005). Sixty percent of individuals who suffer partial complex localization related seizures—21.6 percent of the general epileptic population—exhibit convulsions only during sleep (Janz, 1962).
Disorders that cause seizures may affect an individual’s sleep cycle, leading to sleep deprivation. Similarly, sleep and sleep deprivation increase the incidence of seizure activity. Sleep-related epilepsy normally presents with at least two of the following features: arousals, abrupt awakenings from sleep, generalized tonic-clonic movements of the limbs, focal limb movement, facial twitching, urinary incontinence, apnea, tongue biting, and postictal confusion and lethargy (AASM, 2005). These features cause sleep fragmentation and daytime fatigue.
There are a number of common epileptic syndromes that manifest solely or predominately during the night, including nocturnal frontal lobe epilepsy, benign epilepsy of childhood with centrotemporal spikes, early-onset or late-onset childhood occipital epilepsy, juvenile myoclonic epilepsy, and continuous spike waves during non-REM sleep. Nocturnal frontal lobe epilepsy is characterized by severe sleep disruption, injuries caused by involuntary movements, and occasional daytime seizures. Juvenile myoclonic epilepsy is characterized by synchronous involuntary muscle contractions that often occur during awakening. Continuous spike waves during non-REM sleep epilepsy are commonly associated with neurocognitive impairment and sometimes with impairment of muscle activity and control.
Etiology and Risk Factors
Risk factors for sleep-related epilepsy include stress, sleep deprivation, other sleep disorders, and irregular sleep-wake rhythms. The etiologies for nocturnal seizures are not clearly understood. Genetic factors are likely important; however, as of yet no pathogenic markers have been associated with sleep-related epilepsy. There are specific patterns of rhythmic activity among neurons within specific regions of the brain—the hypothalamus and brainstem—that regulate sleep and arousal. Association of specific neuronal activity between these different regions is important for regulating sleep, while bursts of disassociated neuronal activity may contribute to nocturnal seizures (Tassinari et al., 1972; Velasco and Velasco, 1982; Applegate et al., 1986; Shouse et al., 1996).
Treatment
Treatments for seizures caused by sleep-related epileptic syndromes are typically similar to those of other seizure disorders (Dreifuss and Porter, 1997). Individuals with epilepsy are susceptible to nocturnal sleep disturbance and daytime sleepiness associated with commonly used medications. However, daytime hypersomnolence is not always treatable with antiepileptic drugs (Palm et al., 1992). In particular, phenobarbital, a mainstay of treatment for many years, causes daytime sedation in a dose depen-
dent manner (Brodie and Dichter, 1997). Daytime sedation is also observed with other antiepileptic agents including carbamazepine, alproate, phenytoin, and primidone. Some of the newer medication such as gabapentin, lamotrigine, bigabatrin, and zonisamide are often better tolerated (Salinsky et al., 1996). In addition to daytime sedation, these drugs also cause increased nocturnal sleep time. Vagal nerve stimulation, however, has been reported to improve daytime alertness (Rizzo et al., 2003), but it may also induce sleep apnea (Holmes et al., 2003).
Stroke
Manifestations and Prevalence
Stroke results in a sudden loss of consciousness, sensation, and voluntary movement caused by disruption of blood flow—and therefore oxygen supply—to the brain. Following a stroke an individual’s sleep architecture is often altered, causing a decrease in the total sleep time, REM sleep, and SWS (Broughton and Baron, 1978). Insomnia is a common complication of stroke that may result from medication, inactivity, stress, depression, and brain damage.
The annual incidence of stroke is 2 to 18 per 1000 individuals, and sleep-wake disturbances are found in at least 20 percent of stroke patients (Bassetti, 2005). In addition, over 70 percent of individuals who have suffered a mild stroke and are under 75 years of age suffer fatigue (Carlsson et al., 2003).
Sleep-Disordered Breathing May Be a Risk Factor
Risk factors for stroke include heart disease, hypertension, alcohol abuse, transient ischemic attacks, and, as described above, possibly sleep-disordered breathing (Diaz and Sempere, 2004). Studies investigating the association between sleep-disordered breathing and stroke found that 60 to 70 percent of individuals who have suffered a stroke exhibit sleep-disordered breathing with an apnea-hypopnea index of 10 or greater (Dyken et al., 1996; Bassetti et al., 1996). Sleep-disordered breathing has also been found in a high frequency of individuals with transient ischemic attacks (McArdle et al., 2003), hypertension (Morrell et al., 1999), myocardial infarction, and heart failure (Good et al., 1996; Shamsuzzaman et al., 2003).
Treatment
There are no specific therapies that relieve sleep-related symptoms caused by a stroke. Rather, treatments depend on the specific symptoms and are similar to the treatments of sleep disorders that arise indepen-
dent of a stroke. For example, CPAP is the treatment of choice for sleep disordered breathing, and insomnia and parasomnias are treated using similar temporary hypnotic drug therapies as typically used, zolpidem or benzo-diazepines. However, treatments for hypersomnia are not always as effective following a stroke (Bassetti, 2005).
SUDDEN INFANT DEATH SYNDROME
Manifestations and Prevalence
Sudden infant death syndrome (SIDS), the sudden and unexpected death of infants less than a year old during sleep, has no known cause (American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome, 2005; CDC, 2006). The syndrome is currently the third most common cause of infant death in the United States (CDC, 2006), responsible for approximately 3,000 infant deaths a year in this country (NICHD, 2006b). The majority of SIDS-related deaths occur in infants who are between 2 and 4 months old (NICHD, 2006a).
Etiology and Risk Factors
Although there are no known causes for SIDS, various hypotheses exist about the mechanisms underlying the syndrome. The leading theory is that developmental abnormalities in the infant’s cardiorespiratory system increase the child’s susceptibility to suffocation (Meny et al., 1994; Kinney et al., 1995; Verrier and Josephson, 2005). Infants who later die of SIDS have higher heart rates, narrower heart rate ranges, and problems with coordination of respiration, heart rate, and arterial blood pressure while sleeping (Kemp and Thach, 1991; Schechtman et al., 1995; Kinney et al., 1995; Verrier and Josephson, 2005). This lack of coordination in the cardiorespiratory system may be a result of defects in the region of the brain responsible for controlling breathing and arousal (Kinney et al., 1995; Panigraphy et al., 1997; AAP, 2000), possibly resulting in a baby being unable to wake up in response to troubled breathing.
Concordantly, risk factors attributed to SIDS typically relate to an infant’s ability to breathe easily while sleeping. The chief risk factor for SIDS is a prone sleeping position, otherwise known as stomach sleeping (Dwyer et al., 1991; Ponsonby et al., 1993; Irgens et al., 1995). More recently, side sleeping is thought to be attended by an intermediate level of risk (American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome, 2005; CDC, 2006). Other factors related to obstructed breathing space include bed sharing with adults or several family members, soft
sleep surfaces and/or loose bedding and overheating while sleeping (Hauck and Kemp, 1998; AAP, 2000).
Vulnerability to SIDS seems to depend on both gender and ethnicity. Male infants are more likely to die of SIDS than female babies (NICHD, 2006a); African American infants have twice the likelihood as Caucasian infants of dying from SIDS (Hauck et al., 2003; Daley, 2004; NICHD, 2006a), while Native American infants are three times as likely to be victims of this syndrome (AAP, 2000). SIDS has also been reported to occur at increased frequency in family members with OSA (Tishler et al., 1996), suggesting that there may be common genetic risk factors for both conditions.
Finally, general measures of poor health form the final category of risk factors. Smoking, drinking, or drug use by the mother during gestation are linked to an increased chance of SIDS-related deaths in infants, as is infant exposure to smoke (Schoendorf and Kiely, 1992; AAP, 2000; Iyasu et al., 2002). Infants born with low body weight, prematurely, or to mothers under the age of 20 are also at higher risk of SIDS (Malloy and Hoffman, 1995; AAP, 2000).
Prevention
Preventive measures have reduced the incidence of SIDS in the United States by more than 50 percent. A number of national intervention programs currently exist through various organizations. Of these, the most prominent intervention program is NIH’s “Back to Sleep” campaign (Chapter 5).
Additional preventive measures include no smoking, drinking, or drug use by the mother while pregnant; removal of loose bedding or other items that could suffocate an infant; and prevention of high temperatures in the baby’s sleeping environment (NICHD, 2006a).
SLEEP AND MOVEMENT DISORDERS
Restless Legs Syndrome
Manifestations and Prevalence
RLS is a neurological condition characterized by an irresistible urge to move the legs (it also may affect the arms, trunk, or head and neck). It is also associated with paresthesias—uncomfortable feelings—which individuals describe as creepy-crawly, jittery, itchy, or burning feelings. The symptoms are partially or completely relieved by movement. The urge to move and unpleasant sensations worsen during periods of rest or inactivity, espe-
cially in the evening and at night, causing most individuals difficulty falling asleep (Michaud et al., 2000). The discomfort associated with RLS also causes individuals to wake frequently during the night (Montplaisir et al., 1997). Individuals with RLS often experience periodic limb movements; however, periodic limb movement disorder (see below) is not always associated RLS (Michaud et al., 2000).
The prevalence of RLS has been reported to be at minimum 5 percent (Lavigne and Montplaisir, 1994; Rothdach et al., 2000; NSF, 2005a; Montplaisir et al., 2005; Phillips et al., 2006), which makes it one of the most common movement disorders and sleep disorders. This condition may be found in in adolescents and teenagers (Kryger et al., 2002a) and is more common in older adults and females (Rothdach et al., 2000; Allen and Earley, 2001; Nichols et al., 2002), affecting over 20 percent of pregnant women. RLS symptoms associated with pregnancy are caused by transient low levels of ferritin and folate; therefore, they typically disappear within 4 weeks after delivery (Lee et al., 2001).
RLS may also be associated with attention-deficit hyperactivity disorder (ADHD). In a cross-sectional survey of 866 children, ADHD symptoms were almost twice as likely to occur with symptoms of RLS as would be expected by chance alone (Chervin et al., 2002).
Etiology and Risk Factors
The exact cause of RLS is not completely understood. It likely results from altered dopamine and iron metabolism, and there is evidence for a genetic contribution. More than 50 percent of idiopathic cases are associated with a positive family history of RLS (Ekbom, 1945; Walters et al., 1996; Montplaisir et al., 1997; Winkelmann et al., 2002; Allen et al., 2003), and these cases segregate in an autosomal dominant fashion with high penetrance (90 to 100 percent) (Winkelmann et al., 2002). Susceptibility gene loci have been identified on chromosomes 12q (Desautels et al., 2001), 14q (Bonati et al., 2003), and 9p (Chen et al., 2004); however, no genetic markers or abnormalities have been identified.
RLS commonly occurs in individuals with iron deficiency, including end-stage renal disease, iron-deficiency anemia, pregnancy, and gastric surgery. Iron deficiency, for example caused by repeated blood donation, may also be associated with RLS (Silber et al., 2003; Ulfberg and Nystrom, 2004; Kryger et al., 2003). It is hypothesized that low levels of iron impair transmission of dopamine signals, which contributes to RLS. Iron levels are reduced in the substantia nigra (Allen et al., 2001; Connor et al., 2003), which is a region of the brain responsible for controlling voluntary movement through neurons that rely on dopamine to communicate with each other. The iron deficiency is consistent with abnormal regulation of the transferrin
receptor, which is responsible for transporting iron across cell membranes. Iron in turn is necessary for the synthesis of dopamine and the activity of the D2 dopamine receptor (Turjanski et al., 1999). The association between dopamine, iron deficiency, and RLS is further supported by observations that dopamine antagonists usually make RLS symptoms worse (Winkelmann et al., 2001), while dopaminergic agonists are used to treat RLS (Walters et al., 1988; Wetter et al., 1999; Stiasny et al., 2001).
Idiopathic RLS is not associated with an increased mortality rate; however, in secondary cases of RLS, such as in individuals treated with long-term hemodialysis for end-stage renal disease, RLS is associated with a greater mortality risk (Winkelman et al., 1996). In a survey of 894 dialysis patients that rated symptoms of RLS, severe symptoms were associated with an increased hazard ratio (OR = 1.39; 95% CI, 1.08–1.79) (Unruh et al., 2004).
Treatment
There are both behavioral and pharmacological treatments for RLS; however, there have been no clinical trials reporting the efficacy of non-pharmacological strategies to reduce RLS symptoms. Mild to moderate symptoms can sometimes be treated by lifestyle changes, including maintaining a normal sleeping pattern, taking supplements to manage iron deficiencies, and minimizing consumption of alcohol, caffeine, and tobacco (NINDS, 2005).
RLS is primarily treated using one of four classes of prescription medications: dopaminergic agents, benzodiazepines, opioids, or anticonvulsants (central nervous system depressants). Dopaminergic agents are the primary treatment option for individuals with RLS (Hening et al., 2004). Medications include the dopamine precursor levodopa (L-dopa). Although associated with some adverse effects, administration of L-dopa significantly reduces symptoms of RLS and periodic limb movements that occur throughout the night (Brodeur et al., 1988). However, dopaminergic agents can also have a stimulating effect that may exacerbate insomnia. Benzodiazepines are effective in improving sleep continuity and are therefore frequently prescribed in combination with dopaminergic agents. Opioids may be prescribed in patients with severe symptoms to help to induce relaxation and minimize pain (Walters et al., 1993, 2001). However, opioids may also exacerbate sleep apnea; therefore, they should be used cautiously in patients who snore (Montplaisir et al., 2005). Anticonvulsants are commonly prescribed as an alternative to dopaminergic agents, owing to their ability to minimize leg pain (Montplaisir et al., 2005). It is believed that anticonvulsants, such as carbamazepine and gabapentin, are less potent than dopaminergic agents; however, there have been no comparative studies performed. Furthermore,
there have been a limited number of studies that have examined the safety and efficacy of these treatments in children and adolescents.
Periodic Limb Movement Disorder
Periodic limb movement disorder is characterized by disruptions to sleep caused by periodic episodes of limb movements that occur during sleep, which cannot be explained by any other sleep disorder (AASM, 2005). Individuals with periodic limb movement disorder primarily complain of difficulty with sleep onset and sleep maintenance, insomnia, and/or hypersomnia. The periodic limb movements manifest themselves as rhythmic extensions of the big toe, dorsiflexions of the ankle, and occasional flexions of the knee and hip (Coleman, 1982). These are scored using the periodic limb movements index, which examines over the course of an hour the number of movements that are 0.5 to 5 seconds in duration, separated by an interval between 5 to 90 seconds, and in sequence of four or more an hour. An overnight index score of 5 or greater in children and 15 or greater in adults is considered pathogenic (AASM, 2005).
Periodic limb movements typically occur in the lower extremities and may result in autonomic arousal, cortical arousal, or an awakening. However, typically the individual is unaware of the movements. They are more frequent in the beginning of the night and cluster together. These events are associated with a fast heart rate, followed by a period of slow heart rate (Friedland et al., 1985). Periodic limb movements disorder is associated with above average rates of depression, memory impairment, attention deficits, oppositional behaviors, and fatigue (AASM, 2005). Similar to RLS, dopaminergic medications are helpful in alleviating the disorder’s symptoms.
Periodic limb movements are believed to be very common, especially in older persons, occurring in 34 percent of individuals over the age of 60 (AASM, 2005). However, the disorder—periodic limb movements associated with sleep disruption—is not as common. Periodic limb movements are very common in RLS, occurring in 80 to 90 percent of individuals. It is also observed in individuals with narcolepsy, REM sleep behavior disorder (Folstein et al., 1975), OSA (Montplaisir et al., 1996), and hypersomnia (Whitehouse et al., 1982). Children with ADHD have an increased prevalence of periodic limb movements (Picchietti et al., 1998), and children with periodic limb movement disorders are more likely to have ADHD (Picchietti et al., 1999; Ozminkowski et al., 2004). Sleep-disordered breathing may be a modulator that increases the association between periodic limb movements and ADHD (Chervin and Archbold, 2001).
SLEEP AND MEDICAL DISORDERS
A number of different medical disorders and diseases, from a common cold to cancer, frequently alter an individual’s sleep-wake cycle. These sleep problems often result from pain or infection associated with the primary condition. Although these are both known to cause problems with sleep-wake cycles, as will be shown below, very little is still known about the etiology.
Pain
Pain is described as an acute or chronic unpleasant sensory and emotional experience that varies from dull discomfort to unbearable agony that is associated with actual or potential tissue damage. It commonly causes sleep fragmentation and changes in an individual’s sleep architecture. The symptoms depend on the type and severity of the pain. They include daytime fatigue and sleepiness, poor sleep quality, delay in sleep onset, and decreased cognitive and motor performance (Table 3-5) (Bonnet and Arand, 2003).
Chronic pain affects at least 10 percent of the general adult population (Harstall, 2003), of whom 50 percent complain of poor sleep (Atkinson et al., 1988; Dao et al., 1994; Morin et al., 1998; Roizenblatt et al., 2001; Riley et al., 2001; Dauvilliers and Touchon, 2001; McCracken and Iverson, 2002; Perlis et al., 2005), and 44 percent complain of insomnia (Moldofsky, 2001). There are a number of clinical pain conditions that individuals report affect their sleep quality—RLS, irritable bowel, gastric ulcer, cancer, musculoskeletal disorders, dental and orofacial pain, spinal cord damage, burns, and other trauma (Lavigne et al., 2005).
Although progress has been made, there are still many unanswered questions about how pain affects regions of the brain responsible for regulating the sleep-wake cycle. Neurons that carry pain information to the brain do communicate with regions of the brain that are responsible for arousal—raphe magnus “off” cells (Foo and Mason, 2003). However, it is not known if hypocretin and other genes that regulate the circadian rhythms are affected by acute or chronic pain. Further, it is not known whether the hypothalamus, which is involved in sleep homeostasis, is affected by chronic pain (Kshatri et al., 1998; Mignot et al., 2002b). Because little is known about the interaction between pain and the circuitry in the brain that is responsible for regulating the sleep-wake cycle, much of the management of sleep problems focuses on managing and alleviating the pain or sleep quality.
TABLE 3-5 Selected Sleep-Related Symptoms and Findings in the Presence of Pain
|
Bedtime symptoms
|
|
Sleep time findings
|
|
Wake time symptoms
|
|
SOURCE: Lavigne et al. (2005). |
Infectious Disease
Infections caused by bacterial strains, viruses, and parasites may result in changes to sleep patterns. Although it is accepted that the activity of the immune system affects an individual’s sleep-wake cycle, very little is known about how these two systems interact. This is complicated by the unique effects that specific infections have on sleep patterns and the absence of a large body of clinical research.
Bacterial Infections and Sleep
Bacterial infections typically cause an increase in the total time spent in SWS and a decreased duration of REM sleep (Toth, 1999; Toth and Opp, 2002). Alterations of sleep patterns can be affected by the type of bacterial infection (Opp and Toth, 2003). For example, gram-negative bacteria induce enhanced sleep more rapidly than do gram-positive bacteria. Differences in the process and progression of the disease also affect the sleep-wake cycle.
Viral Infections and Sleep
Viral infections also have effects on the sleep-wake cycle. Individuals inoculated with rhinovirus or influenza virus report less sleep during the incubation period, while during the symptomatic period they slept longer (Smith, 1992). However, compared to healthy individuals there were no reported difference in sleep quality and number of awakenings.
The human immunodeficiency virus (HIV) also has been shown to alter sleep patterns. Individuals spend increased time in SWS during the second half of night (Darko et al., 1995) and suffer from frequent arousals and decreased time in REM sleep (Norman et al., 1990). As the infection progresses to AIDS, individuals develop increased sleep fragmentation, significant reductions in SWS, and disruption to the entire sleep architecture (Norman et al., 1990; Darko et al., 1995).
Cancer
Many patients with cancer also suffer pain or depression, which contributes to difficulty sleeping. These require treatment as in other patients with pain or depression as causes of insomnia. Excessive sleepiness may be caused by injury to the ascending arousal system due to brain metastases or by leptomeningeal carcinomatosis. These signs often alert physicians to the need to treat the underlying spread of cancer to the central nervous system. Other patients with cancer may develop antitumor antibodies that attack the brain. In particular, anti-Ma-2 antibodies tend to cause hypothalamic lesions and may precipitate daytime sleepiness and even cataplexy (Rosenfeld et al., 2001). Treatment of the underlying cancer may reverse the symptoms in some cases.
Sleeping Sickness
Fungal and parasitic infections also can alter the sleep-wake cycle. For example, sleeping sickness, or African trypanosomiasis, commonly occurs in individuals who have been infected with the Trypanosoma brucei (Tb) parasite. It is characterized by episodes of nocturnal insomnia and daytime sleep, but not hypersomnia (Lundkvist et al., 2004).
Sleeping sickness is found primarily in sub-Saharan African countries, where Tb is transmitted to humans as a result of bites received from tsetse flies (Lundkvist et al., 2004). The prevalence of this disorder is not known; however, over 60 million people live in areas where the Tb parasite is endemic.
Sleeping sickness is associated with altered sleep architecture. EEG recordings of individuals with sleeping sickness from Gambia demonstrate
periods of REM sleep that occur throughout the entire sleep-wake cycle, frequently without normal intermediate NREM periods (Buguet et al., 2001). Circadian fluctuations of hormones—cortisol, prolactin, and growth hormone—are also altered in individuals with sleeping sickness (Radomski et al., 1994). Therefore, it has been hypothesized that sleeping sickness may be a circadian rhythm disease that affects the neural pathways that interconnect the circadian-timing and sleep-regulating centers (Lundkvist et al., 2004).
Treatment Effects on Sleep
Numerous medical conditions are associated with a wide variety of sleep disorders including insomnia, hypersomnia, parasomnias, and sleep-related movement disorders. Although these disease-related sleep disorders have recently been receiving an increasing amount of attention, including addition to the latest International Classification of Sleep Disorders (AASM, 2005), the contribution that treatments for these medical conditions make to the development of sleep disturbances is less well appreciated. However, many medical therapies have iatrogenic effects on sleep-wake regulatory systems causing disturbed sleep, daytime sleepiness, and other related side effects.
Treatments for Cardiovascular Disease
Cardiovascular diseases, sometimes associated with sleep-related breathing disorders (Peters, 2005) (see above), are commonly treated with a wide range of medications including antihypertensives, hypolipidemics, and antiarrhythmics; each class of medication can adversely affect sleep and/or waking. For example, beta-antagonists, the mainstay of treatment for hypertension, are commonly associated with fatigue, insomnia, nightmares, and vivid dreams (McAinsh and Cruickshank, 1990). Sleep disturbances appear to be more severe with lipophilic drugs (e.g., propranolol) than with hydrophilic drugs (e.g., atenolol). However, even atenolol, one of the most hydrophilic beta-blockers, has been shown to increase total wake time (Van Den Heuvel et al., 1997). The mechanism underlying sleep disruption by beta-blocking agents may be their tendency to deplete melatonin, an important sleep-related hormone (Garrick et al., 1983; Dawson and Encel, 1993). Fatigue and somnolence have also been reported with other antihypertensive medications such as carvedilol, labetalol, clonidine, methyldopa, and reserpine (Paykel et al., 1982; Miyazaki et al., 2004). In contrast, angiotensin-converting enzyme inhibitors generally have very few effects on sleep (Reid, 1996). Hypolipidemic drugs, including atorvastatin and lovastatin, have
also been associated with reports of insomnia, but placebo-controlled clinical trials of lovastatin, simvastatin, and pravastatin did not appear to increase sleep disturbance (Bradford et al., 1991; Keech et al., 1996). Amiodarone, a widely use antiarrhythmic agent (Hilleman et al., 1998), can cause nocturnal sleep disturbance, and digoxin has been associated with both insomnia and daytime fatigue (Weisberg et al., 2002).
Treatments for Cancer
Patients with cancer receive multiple types of treatments designed at controlling the disease process including chemotherapy, biotherapy, radiotherapy, and medications. All can have important adverse effects on regulating the sleep-wake cycle. For example, sleep problems have been reported in patients undergoing chemotherapy (Broeckel et al., 1998; Berger and Higginbotham, 2000; Lee et al., 2004). However, objective measures of sleep in the patients and analyses of clinical correlates are very limited. Thus, the mechanisms underlying these sleep problems are poorly understood. Menopausal symptoms arising from chemotherapy and hormonal therapy, especially those of a vasomotor type (e.g., hot flashes, sweating), may be a contributing factor (Rombaux et al., 2000; Mourits et al., 2001; Carpenter et al., 2002). Nocturnal sleep disturbances and daytime sleepiness have also been reported in patients undergoing radiotherapy (Beszterczey and Lipowski, 1977; Miaskowski and Lee, 1999).
Cytokines (biotherapy), a diverse group of peptide molecules that regulate cell functions, are sometimes used as adjunct therapy (Dunlop and Campbell, 2000). Interferon, interleukin-2, and tumor necrosis factor are associated with a variety of side effects including daytime sleepiness, disturbed sleep, and depression (Capuron et al., 2000). Although very effective in reducing cancer-related pain, opioids often cause sleep disturbance and are associated with decreased REM and SWS (Cronin et al., 2001).
Treatments for Renal Disease
RLS, periodic limb movement disorder, sleep apnea, and excessive daytime sleepiness affect up to 70 percent of patients with end-stage renal disease receiving treatment with hemodialysis (Parker et al., 2000; Parker, 2003). Hemodialysis may alter biological systems controlling processes that regulate the sleep-wake cycle via several potential mechanisms. The rapid fluid, electrolyte, and acid/base changes that occur are often associated with central nervous system symptoms such as headache, restlessness, changes in arousal, and fatigue during or immediately after treatment. Several studies have reported an increase in cytokine production secondary to blood interactions with bioincompatible aspects of hemodialysis (such as blood expo-
sure to membranes, tubing, and cellular mechanical trauma) and backflow of endotoxins through the membrane (Panichi et al., 2000). Interleukin-1, tumor necrosis factor-α, and interleukin-6 are the major proinflammatory cytokines that may be involved (Pertosa et al., 2000). These substances have both somnogenic and pyrogenic properties and have been linked to a number of postdialytic symptoms (Konsman et al., 2002), including daytime sleepiness and sleep disturbances (Raison and Miller, 2001; Capuron et al., 2002). Dialysis-associated changes in melatonin levels and pattern of secretion and alterations in body temperature rhythm may also play a role in disrupting circadian systems (Vaziri et al., 1993, 1996; Parker et al., 2000; Parker, 2003).
Treatments for Rheumatologic and Immunologic Disorders
Numerous other classes of medications can alter sleep and waking. Corticosteroids are a class of medications that are used to treat a variety of medical conditions including rheumatologic and immunologic disorders, cancer, and asthma. Sleep disturbances, insomnia, daytime hyperactivity, and mild hypomania are common side effects (Wolkowitz et al., 1990); a significant decrease in REM sleep may also occur (Born et al., 1987). Theophylline, a respiratory stimulant and bronchodilator, is in the same class of medications as caffeine and can likewise disturb sleep—even in healthy subjects (Kaplan et al., 1993). Nonsteroidal anti-inflammatory agents may also affect sleep as they decrease the production of sleep-promoting prostaglandins, suppress normal surge of melatonin, and alter the daily rhythm of body temperature (Murphy et al., 1994, 1996). Pseudoephedrine and phenylpropanolamine, which have many of the same pharmacological properties of ephedrine, also cause sleep disruption—and many of these preparation are readily available over the counter (Lake et al., 1990; Bertrand et al., 1996).
Although the medications and treatments listed above are often necessary, it is essential for patients to be aware of potential side effects relating to the sleep-wake-related cycle. Unfortunately, patients often neglect to report such complaints as they think nothing can be done to alleviate the problems. However, numerous behavioral and pharmacological interventions are available to treat these iatrogenically induced problems with the sleep-wake cycle. In addition, administering treatment or medications at appropriate times of day in relationship to the sleep-wake schedule may potentially be beneficial and enhance clinical outcomes (Levi, 1994; Bliwise et al., 2001; Hermida and Smolensky, 2004). Research in this area is greatly needed.
|
BOX 3-3 Shift Work Disorder and Jet Lag Shift Work Disorder Shift work type circadian rhythm sleep disorder is characterized by complaints of insomnia or excessive sleepiness resulting from work hours that occur during the normal sleep period, including, night shifts, early morning shifts, and rotating shifts. Total sleep time is normally reduced by 1 to 4 hours and sleep quality is disturbed. During work shifts individuals can experience excessive sleepiness, reduced alertness, and reduced performance capacity. Individuals are also commonly more irritable, and the disorder may have negative social consequences. The condition is closely linked to work schedules; consequently, it abates in response to a conventional sleep schedule. Jet Lag Jet lag type is a temporary circadian rhythm sleep disorder that occurs when there is a transitory mismatch between the timing of the sleep-wake cycle caused by a change in time zone. Individuals with jet lag potentially experience disturbed sleep, decreased subjective alertness, general malaise, somatic symptoms such as gastrointestinal disturbance, and impaired daytime function. The severity and the duration of the symptoms are usually dependent on the number of time zones traveled and the direction of travel—eastern travel and travel through multiple time zones typically result in worse symptoms than western travel. |
CIRCADIAN RHYTHM SLEEP DISORDERS
Circadian rhythm sleep disorders arise from chronic alterations, disruptions, or misalignment of the circadian clock in relation to environmental cues and the terrestrial light-dark cycle. The 2005 update of the International Classification of Sleep Disorders designated nine different circadian disorders, including delayed sleep phase type, advanced sleep phase type, nonentrained sleep-wake type, irregular sleep-wake type, shift work type, and jet lag type (Box 3-3) (AASM, 2005). These disorders may be comorbid with other neurological or psychiatric disorders, making the diagnosis and treatment difficult (Reid and Zee, 2005). Diagnosis with a circadian rhythm disorder requires meeting the following three criteria:
-
Persistent or recurrent pattern of sleep disturbance due primarily to either an alteration of the circadian timekeeping system or a misalignment
-
between endogenous circadian rhythm and exogenous factors that affect timing and duration of sleep.
-
Circadian-related disruption leads to insomnia, excessive sleepiness, or both.
-
The sleep disturbance is associated with impairment of social, occupational, or other functions (AASM, 2005).
The following sections will describe two of the nine more common types of circadian rhythm sleep disorders, delayed sleep phase type and advanced sleep phase type.
Delayed Sleep Phase Syndrome
Manifestations and Prevalence
The sleep pattern of individuals suffering from delayed sleep phase syndrome (or delayed sleep phase type) is characterized by sleep onset and wake times that are typically delayed 3 to 6 hours relative to conventional sleep-wake times (Figure 3-7). An individual’s total sleep time is normal for his or her age (Weitzman et al., 1981), but individuals typically find it difficult to initiate sleep before 2:00 and 6:00 a.m., and prefer to wake up between 10:00 a.m. and 1:00 p.m. The impact of delayed sleep phase syndrome has not been fully investigated and is therefore limited. In a study that included 14 individuals it was reported that the syndrome may impair an individual’s job performance and may be associated with marital problems and financial difficulty (Alvarez et al., 1992). A second study investigated the impact of delayed sleep phase syndrome in 22 adolescents and found an association with increased daytime irritability, poor school performance, and mental disturbances (Regestein and Monk, 1995; AASM, 2005).
FIGURE 3-7 Representation of the temporal distribution of sleep.
SOURCE: Reid and Zee (2005).
The exact prevalence of delayed sleep phase syndrome in the general population is unknown. It is unclear what the prevalence of this disorder is; however, it may be more prevalent in adolescents and young adults (Weitzman et al., 1981; Pelayo et al., 1988; Regestein and Monk, 1995; AASM, 2005).
Etiology and Risk Factors
Night shift workers may be at higher risk for delayed sleep phase syndrome due to irregular circadian entrainment (Santhi et al., 2005). Similarly, individuals who live in extreme latitudes and are exposed to extended periods of light may also be at increased risk of suffering from delayed sleep phase syndrome (Lingjaerde et al., 1985; Pereira et al., 2005).
Biological, physiological, and genetic factors have been proposed to be responsible for causing delayed sleep phase syndrome. Behaviorally, late bedtimes and rise times delay an individual’s exposure to light, which may prevent entraining of the circadian clock. Furthermore, exposure to dim light in the late evening and at night, may also affect the circadian phase (Zeitzer et al., 2000; Gronfier et al., 2004).
Biological alterations to the endogenous circadian system also contribute to delayed sleep phase syndrome. Although levels of melatonin typically increase in the evening hours, individuals with this syndrome have a hypersensitivity to nighttime bright light exposure in the suppression of melatonin (Czeisler et al., 1981). It has also been hypothesized that the disorder may result from a circadian phase that has a reduced sensitivity to photic entrainment, or the free-running period of the circadian cycle is prolonged (Czeisler et al., 1981). Consistent with these hypotheses, polymorphisms in circadian genes influence the entraining and free-running period of the circadian cycle and may be associated with delayed sleep phase syndrome (Takahashi et al., 2000; Iwase et al., 2002; Hohjoh et al., 2003; Archer et al., 2003; Pereira et al., 2005). A recent study has also identified a candidate gene, human PER2, that results in familial advanced sleep phase syndrome (Xu et al., 2005).
Treatment
Treatment for delayed sleep phase syndrome requires resynchronizing to a more appropriate phase to the 24-hour light-dark cycle. In addition to a structured sleep-wake schedule and good sleep hygiene practices, potential therapies include resetting the circadian pacemaker with bright light, melatonin, or a combination of both. However, studies that have investigated the efficacy of bright light have provided mixed results (Pelayo et al., 1988; Rosenthal et al., 1990; Akata et al., 1993; Weyerbrock et al., 1996), partially owing to limitations in their study design and the numbers of par-
ticipants included in each study. Consequently, there are no standard criteria for its use. Similarly, there have been no large-scale controlled studies examining the efficacy of melatonin, and as of yet it has not been approved by the Food and Drug Administration for this indication (Reid and Zee, 2005).
Advanced Sleep Phase Syndrome
Manifestations and Prevalence
Advanced sleep phase syndrome (or advanced sleep phase type) is characterized by involuntary bedtimes and awake times that are more than 3 hours earlier than societal means (Figure 3-7) (Reid and Zee, 2005). As is the case with delayed sleep phase syndrome, the amount of sleep is not affected, unless evening activities result in later bedtimes. Therefore, the syndrome is primarily associated with impaired social and occupational activities.
The prevalence of advanced sleep phase syndrome is unknown; however, it has been estimated that as many as 1 percent of the middle-aged adults may suffer from it (Ando et al., 1995). One of the challenges in determining its prevalence is that affected individuals typically do not perceive it as a disorder and therefore do not seek medical treatment (Reid and Zee, 2005).
Etiology and Risk Factors
The causes of this syndrome are not known; however, as with delayed sleep phase type, biological and environmental factors likely contribute to the onset of advanced sleep phase type. Several familial cases of this syndrome have been reported (Jones et al., 1999; Ondze et al., 2001; Reid et al., 2001; Satoh et al., 2003), and these cases segregate in a dominant mode. Polymorphisms in circadian clock genes have been identified in a family with advanced sleep phase syndrome (Toh et al., 2001; Shiino et al., 2003). Changes in the activity of genes involved in circadian biology are consistent with observations that individuals with this syndrome have circadian rhythms that are less than 24 hours.
Treatment
Treatment options for individuals with advanced sleep phase syndrome are limited. Bright light therapy in the evening has been used successfully in a limited study to reduce awakenings (Campbell et al., 1993; Palmer et al., 2003). It is also hypothesized that administration of low levels of melatonin
in the early morning may also be used (Lewy et al., 1996), though there are no published reports verifying this option.
REFERENCES
AAP (American Academy of Pediatrics). 2000. Changing concepts of sudden infant death syndrome: Implications for infant sleeping environment and sleep position. American Academy of Pediatrics. Task Force on Infant Sleep Position and Sudden Infant Death Syndrome. Pediatrics 105(3 Pt 1):650–656.
AASM (American Academy of Sleep Medicine). 2005. The International Classification of Sleep Disorders. Westchester, IL: American Academy of Sleep Medicine.
Akata T, Sekiguchi S, Takahashi M, Miyamoto M, Higuchi T, Machiyama Y. 1993. Successful combined treatment with vitamin B12 and bright artificial light of one case with delayed sleep phase syndrome. Japanese Journal of Psychiatry and Neurology 47(2):439–440.
Al-Delaimy WK, Manson JE, Willett WC, Stampfer MJ, Hu FB. 2002. Snoring as a risk factor for type II diabetes mellitus: A prospective study. American Journal of Epidemiology 155(5):387–393.
Aldrich MS. 1996. The clinical spectrum of narcolepsy and idiopathic hypersomnia. Neurology 46(2):393–401.
Ali NJ, Pitson DJ, Stradling JR. 1993. Snoring, sleep disturbance, and behaviour in 4–5 year olds. Archives of Disease in Childhood 68(3):360–366.
Allen RP, Earley CJ. 2001. Restless legs syndrome: A review of clinical and pathophysiologic features. Journal of Clinical Neurophysiology 18(2):128–147.
Allen RP, Barker PB, Wehrl F, Song HK, Earley CJ. 2001. MRI measurement of brain iron in patients with restless legs syndrome. Neurology 56(2):263–265.
Allen RP, Picchietti D, Hening WA, Trenkwalder C. 2003. Restless legs syndrome: Diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Medicine 4(2):101–119.
Alvarez B, Dahlitz MJ, Vignau J, Parkes JD. 1992. The delayed sleep phase syndrome: Clinical and investigative findings in 14 subjects. Journal of Neurology, Neurosurgery, and Psychiatry 55(8):665–670.
American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome. 2005. The changing concept of sudden infant death syndrome: Diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics 116(5):1245–1255.
Amin RS, Carroll JL, Jeffries JL, Grone C, Bean JA, Chini B, Bokulic R, Daniels SR. 2004. Twenty-four-hour ambulatory blood pressure in children with sleep-disordered breathing. American Journal of Respiratory and Critical Care Medicine 169(8):950–956.
Amin RS, Kimball TR, Kalra M, Jeffries JL, Carroll JL, Bean JA, Witt SA, Glascock BJ, Daniels SR. 2005. Left ventricular function in children with sleep-disordered breathing. American Journal of Cardiology 95(6):801–804.
Ancoli-Israel S, Kripke DF, Klauber MR, Mason WJ, Fell R, Kaplan O. 1991. Sleep-disordered breathing in community-dwelling elderly. Sleep 14(6):486–495.
Ancoli-Israel S, Kripke DF, Klauber MR, Parker L, Stepnowsky C, Kullen A, Fell R. 1993. Natural history of sleep-disordered breathing in community dwelling elderly. Sleep 16(8 suppl):S25–S29.
Ancoli-Israel S, Klauber MR, Stepnowsky C, Estline E, Chinn A, Fell R. 1995. Sleep-disordered breathing in African-American elderly. American Journal of Respiratory Critical Care Medicine 152(6 Pt 1):1946–1949.
Ancoli-Israel S, Kripke DF, Klauber MR, Fell R, Stepnowsky C, Estline E, Khazeni N, Chinn A. 1996. Morbidity, mortality and sleep-disordered breathing in community dwelling elderly. Sleep 19(4):277–282.
Ando K, Kripke DF, Ancoli-Israel S. 1995. Estimated prevalence of delayed and advanced sleep phase syndromes. Sleep Research 24:509.
Andreas S, Schulz R, Werner GS, Kreuzer H. 1996. Prevalence of obstructive sleep apnoea in patients with coronary artery disease. Coronary Artery Disease 7(7):541–545.
Anic-Labat S, Guilleminault C, Kraemer HC, Meehan J, Arrigoni J, Mignot E. 1999. Validation of a cataplexy questionnaire in 983 sleep-disorders patients. Sleep 22(1): 77–87.
APA (American Psychiatric Association). 1994. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. 4th ed. Washington, DC: American Psychiatric Association.
Applegate CD, Burchfiel JL, Konkol RJ. 1986. Kindling antagonism: Effects of norepinephrine depletion on kindled seizure suppression after concurrent, alternate stimulation in rats. Experiments in Neurology 94(2):379–390.
Arand D, Bonnet M, Hurwitz T, Mitler M, Rosa R, Sangal RB. 2005. The clinical use of the MSLT and MWT. Sleep 28(1):123–144.
Archer SN, Robilliard DL, Skene DJ, Smits M, Williams A, Arendt J, von Schantz M. 2003. A length polymorphism in the circadian clock gene Per3 is linked to delayed sleep phase syndrome and extreme diurnal preference. Sleep 26(4):413–415.
Arnulf I, Mabrouk T, Mohamed K, Konofal E, Derenne JP, Couratier P. 2005a. Stages 1-2 non-rapid eye movement sleep behavior disorder associated with dementia: A new parasomnia? Movement Disorders 20(9):1223–1228.
Arnulf I, Zeitzer JM, File J, Farber N, Mignot E. 2005b. Kleine-Levin syndrome: A systematic review of 186 cases in the literature. Brain 128(Pt 12):2763–2776.
Atkinson JH, Slater MA, Grant I, Patterson TL, Garfin SR. 1988. Depressed mood in chronic low back pain: Relationship with stressful life events. Pain 35(1): 47–55.
Ayas NT, White DP, Manson JE, Stampfer MJ, Speizer FE, Malhotra A, Hu FB. 2003. A prospective study of sleep duration and coronary heart disease in women. Archives of Internal Medicine 163(2):205–209.
Babu AR, Herdegen J, Fogelfeld L, Shott S, Mazzone T. 2005. Type 2 diabetes, glycemic control, and continuous positive airway pressure in obstructive sleep apnea. Archives of Internal Medicine 165(4):447–452.
Baldwin DC Jr, Daugherty SR. 2004. Sleep deprivation and fatigue in residency training: Results of a national survey of first- and second-year residents. Sleep 27(2):217–223.
Bamford CR. 1993. Carbamazepine in REM sleep behavior disorder. Sleep 16(1):33–34.
Bassetti CL. 2005. Sleep and stroke. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 811–830.
Bassetti C, Aldrich MS. 1999. Sleep apnea in acute cerebrovascular diseases: Final report on 128 patients. Sleep 22(2):217–223.
Bassetti C, Aldrich MS, Chervin RD, Quint D. 1996. Sleep apnea in patients with transient ischemic attack and stroke: A prospective study of 59 patients. Neurology 47(5):1167– 1173.
Behlfelt K. 1990. Enlarged tonsils and the effect of tonsillectomy. Characteristics of the dentition and facial skeleton. Posture of the head, hyoid bone and tongue. Mode of breathing. Swedish Dental Journal: Supplement 72:1–35.
Benca RM. 2005a. Diagnosis and treatment of chronic insomnia: A review. Psychiatry Services 56(3):332–343.
Benca RM. 2005b. Mood disorder. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1311–1326.
Berger AM, Higginbotham P. 2000. Correlates of fatigue during and following adjuvant breast cancer chemotherapy: A pilot study. Oncology Nursing Forum 27(9):1443–1448.
Bergonzi P, Chiurulla C, Gambi D, Mennuni G, Pinto F. 1975. L-dopa plus dopa-decarboxylase inhibitor. Sleep organization in Parkinson’s syndrome before and after treatment. Acta Neurologica Belgica 75(1):5–10.
Bertrand B, Jamart J, Marchal JL, Arendt C. 1996. Cetirizine and pseudoephedrine retard alone and in combination in the treatment of perennial allergic rhinitis: A double-blind multicentre study. Rhinology 34(2):91–96.
Beszterczey A, Lipowski ZJ. 1977. Insomnia in cancer patients. Canadian Medical Association Journal 116(4):355.
Billiard M, Cadilhac J. 1988. Recurrent hypersomnia [in French]. Revue Neurologique 144(4):249–258.
Billiard M, Dauvilliers Y. 2001. Idiopathic hypersomnia. Sleep Medicine Reviews 5(5):349–358.
Billiard M, Dolenc L, Aldaz C, Ondze B, Besset A. 1994. Hypersomnia associated with mood disorders: A new perspective. Journal of Psychosomatic Research 38(suppl 1):41–47.
Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. 1998. Effects of age on sleep apnea in men: I. Prevalence and severity. American Journal of Respiratory and Critical Care Medicine 157(1):144–148.
Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Leiby BE, Vela-Bueno A, Kales A. 2000. Association of hypertension and sleep-disordered breathing. Archives of Internal Medicine 160(15):2289–2295.
Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A, Kales A. 2001. Prevalence of sleep-disordered breathing in women: Effects of gender. American Journal of Respiratory and Critical Care Medicine 163(3 Pt 1):608–613.
Bliwise DL. 2002. Sleep apnea, APOE4 and Alzheimer’s disease: 20 years and counting? Journal of Psychosomatic Research 53(1):539–546.
Bliwise DL, Bliwise NG, Partinen M, Pursley AM, Dement WC. 1988. Sleep apnea and mortality in an aged cohort. American Journal of Public Health 78(5):544–547.
Bliwise DL, Kutner NG, Zhang R, Parker KP. 2001. Survival by time of day of hemodialysis in an elderly cohort. Journal of the American Medical Association 286(21):2690–2694.
Bonati MT, Ferini-Strambi L, Aridon P, Oldani A, Zucconi M, Casari G. 2003. Autosomal dominant restless legs syndrome maps on chromosome 14q. Brain 126(6):1485–1492.
Bonnet MH, Arand DL. 2003. Clinical effects of sleep fragmentation versus sleep deprivation. Sleep Medicine Reviews 7(4):297–310.
Born J, Zwick A, Roth G, Fehm-Wolfsdorf G, Fehm HL. 1987. Differential effects of hydrocortisone, fluocortolone, and aldosterone on nocturnal sleep in humans. Acta Endocrinologica (Copenhagen) 116(1):129–137.
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. 2004. Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research 318(1):121–134.
Bradford RH, Shear CL, Chremos AN, Dujovne C, Downton M, Franklin FA, Gould AL, Hesney M, Higgins J, Hurley DP, et al. 1991. Expanded clinical evaluation of lovastatin (EXCEL) study results. I. Efficacy in modifying plasma lipoproteins and adverse event profile in 8245 patients with moderate hypercholesterolemia. Archives of Internal Medicine 151(1):43–49.
Bradley TD, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K, Belenkie I, Pfeifer M, Fleetham J, Hanly P, Smilovitch M, Tomlinson G, Floras JS. 2005. Continuous positive airway pressure for central sleep apnea and heart failure. New England Journal of Medicine 353(19):2025–2033.
Breslau N, Roth T, Rosenthal L, Andreski P. 1996. Sleep disturbance and psychiatric disorders: A longitudinal epidemiological study of young adults. Biological Psychiatry 39(6): 411–418.
Brodeur C, Montplaisir J, Godbout R, Marinier R. 1988. Treatment of restless legs syndrome and periodic movements during sleep with L-dopa: A double-blind, controlled study. Neurology 38(12):1845–1848.
Brodie MJ, Dichter MA. 1997. Established antiepileptic drugs. Seizure 6(3):159–174.
Broeckel JA, Jacobsen PB, Horton J, Balducci L, Lyman GH. 1998. Characteristics and correlates of fatigue after adjuvant chemotherapy for breast cancer. Journal of Clinical Oncology 16(5):1689–1696.
Brooks B, Cistulli PA, Borkman M, Ross G, McGhee S, Grunstein RR, Sullivan CE, Yue DK. 1994. Obstructive sleep apnea in obese noninsulin-dependent diabetic patients: Effect of continuous positive airway pressure treatment on insulin responsiveness. Journal of Clinical Endocrinology and Metabolism 79(6):1681–1685.
Broughton R, Baron R. 1978. Sleep patterns in the intensive care unit and on the ward after acute myocardial infarction. Electroencephalography and Clinical Neurophysiology 45(3):348–360.
Broughton RJ, Fleming JA, George CF, Hill JD, Kryger MH, Moldofsky H, Montplaisir JY, Morehouse RL, Moscovitch A, Murphy WF. 1997. Randomized, double-blind, placebo-controlled crossover trial of modafinil in the treatment of excessive daytime sleepiness in narcolepsy. Neurology 49(2):444–451.
Buguet A, Bourdon L, Bouteille B, Cespuglio R, Vincendeau P, Radomski MW, Dumas M. 2001. The duality of sleeping sickness: Focusing on sleep. Sleep Medicine Reviews 5(2):139–153.
Buxbaum SG, Elston RC, Tishler PV, Redline S. 2002. Genetics of the apnea hypopnea index in Caucasians and African Americans: I. Segregation analysis. Genetic Epidemiology 22(3):243–253.
Campbell SS, Dawson D, Anderson MW. 1993. Alleviation of sleep maintenance insomnia with timed exposure to bright light. Journal of American Geriatric Society 41(8):829–836.
Cantor CR, Stern MB. 2002. Dopamine agonists and sleep in Parkinson’s disease. Neurology 58(4 Suppl 1):S71–S78.
Caples SM, Gami AS, Somers VK. 2005. Obstructive sleep apnea. Annals of Internal Medicine 142(3):187–197.
Capuron L, Ravaud A, Dantzer R. 2000. Early depressive symptoms in cancer patients receiving interleukin 2 and/or interferon alfa-2b therapy. Journal of Clinical Oncology 18(10): 2143–2151.
Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH. 2002. Neurobehavioral effects of interferon-alpha in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26(5):643–652.
Carlsson GE, Moller A, Blomstrand C. 2003. Consequences of mild stroke in persons < 75 years—a 1-year follow-up. Cerebrovascular Disease 16(4):383–388.
Carpenter JS, Johnson D, Wagner L, Andrykowski M. 2002. Hot flashes and related outcomes in breast cancer survivors and matched comparison women. Oncological Nursing Forum 29(3): E16–E25.
Carskadon MA. 1990. Patterns of sleep and sleepiness in adolescents. Pediatrician 17(1):5–12.
Carskadon MA, Dement WC, Mitler MM, Roth T, Westbrook PR, Keenan S. 1986. Guidelines for the multiple sleep latency test (MSLT): A standard measure of sleepiness. Sleep 9(4):519–524.
Carskadon MA, Acebo C, Jenni OG. 2004. Regulation of adolescent sleep: Implications for behavior. Annals of the New York Academy of Sciences 1021:276–291.
CDC (Centers for Disease Control and Prevention). 2005. Percentage of adults who reported an average of 6 hours of sleep per 24-hour period, by sex and age group—United States, 1985 and 2004. Morbidity and Mortality Weekly Report 54(37):933.
CDC. 2006. Sudden Infant Death Syndrome (SIDS). [Online]. Available: http://www.cdc.gov/SIDS/index.htm [accessed January 17, 2006].
Chang PP, Ford DE, Mead LA, Cooper-Patrick L, Klag MJ. 1997. Insomnia in young men and subsequent depression. The Johns Hopkins precursors study. American Journal of Epidemiology 146(2):105–114.
Chen S, Ondo WG, Rao S, Li L, Chen Q, Wang Q. 2004. Genomewide linkage scan identifies a novel susceptibility locus for restless legs syndrome on chromosome 9p. American Journal of Human Genetics 74(5):876–885.
Chervin RD, Archbold KH. 2001. Hyperactivity and polysomnographic findings in children evaluated for sleep-disordered breathing. Sleep 24(3):313–320.
Chervin RD, Hedger AK, Dillon JE, Pituch KJ, Panahi P, Dahl RE, Guilleminault C. 2002. Associations between symptoms of inattention, hyperactivity, restless legs, and periodic leg movements. Sleep: Journal of Sleep Research and Sleep Medicine 25(2):213–218.
Chin K, Nakamura T, Takahashi K, Sumi K, Ogawa Y, Masuzaki H, Muro S, Hattori N, Matsumoto H, Niimi A, Chiba T, Nakao K, Mishima M, Ohi M, Nakamura T. 2003. Effects of obstructive sleep apnea syndrome on serum aminotransferase levels in obese patients. American Journal of Medicine 114(5):370–376.
Chrisp P, Mammen GJ, Sorkin EM. 1991. Selegiline. A review of its pharmacology, symptomatic benefits and protective potential in Parkinson’s disease. Drugs and Aging 1(3):228–248.
Cole MG, Dendukuri N. 2003. Risk factors for depression among elderly community subjects: A systematic review and meta-analysis. American Journal of Psychiatry 160(6):1147– 1156.
Coleman RM. 1982. Periodic movements in sleep (nocturnal myoclonus) and restless legs syndrome. In: Guilleminault C, ed. Sleep and Waking Disorders: Indications and Techniques. Menlo Park, CA: Addison-Wesley. Pp. 265–295.
Comella CL, Nardine TM, Diederich NJ, Stebbins GT. 1998. Sleep-related violence, injury, and REM sleep behavior disorder in Parkinson’s disease. Neurology 51(2):526–529.
Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Ondo WG, Earley CJ. 2003. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 61(3):304–309.
Cronin AJ, Keifer JC, Davies MF, King TS, Bixler EO. 2001. Postoperative sleep disturbance: Influences of opioids and pain in humans. Sleep 24(1):39–44.
Czeisler CA, Richardson GS, Zimmerman JC, Moore-Ede MC, Weitzman ED. 1981. Entrainment of human circadian rhythms by light-dark cycles: A reassessment. Photochemistry and Photobiology 34(2): 239–247.
D’Alessandro R, Magelli C, Gamberini G, Bacchelli S, Cristina E, Magnani B, Lugaresi E. 1990. Snoring every night as a risk factor for myocardial infarction: A case-control study. British Medical Journal 300(6739):1557–1558.
Daley KC. 2004. Update on sudden infant death syndrome. Current Opinion in Pediatrics 16(2):227–232.
Dao TT, Lavigne GJ, Charbonneau A, Feine JS, Lund JP. 1994. The efficacy of oral splints in the treatment of myofascial pain of the jaw muscles: A controlled clinical trial. Pain 56(1):85–94.
Darko DF, Mitler MM, Henriksen SJ. 1995. Lentiviral infection, immune response peptides and sleep. Advances in Neuroimmunology 5(1): 57–77.
Dauvilliers Y, Touchon J. 2001. Sleep in fibromyalgia: Review of clinical and polysomnographic data [in French]. Neurophysiologie Clinique 31(1):18–33.
Dauvilliers Y, Morin C, Cervena K, Carlander B, Touchon J, Besset A, Billiard M. 2005. Family studies in insomnia. Journal of Psychosomatic Research 58(3):271–278.
Dawson D, Encel N. 1993. Melatonin and sleep in humans. Journal of Pineal Research 15(1): 1–12.
de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, Manubens-Bertran JM, Alperovitch A, Rocca WA. 1997. Prevalence of parkinsonism and Parkinson’s disease in Europe: The EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. Journal of Neurology, Neurosurgery, and Psychiatry 62(1):10–15.
Desautels A, Turecki G, Montplaisir J, Sequeira A, Verner A, Rouleau GA. 2001. Identification of a major susceptibility locus for restless legs syndrome on chromosome 12q. American Journal of Human Genetics 69(6):1266–1270.
Diaz J, Sempere AP. 2004. Cerebral ischemia: New risk factors. Cerebrovascular Disease 17(suppl 1):43–50.
Dinges DF, Pack F, Williams K, Gillen KA, Powell JW, Ott GE, Aptowicz C, Pack AI. 1997. Cumulative sleepiness, mood disturbance, and psychomotor vigilance performance decrements during a week of sleep restricted to 4–5 hours per night. Sleep 20(4):267–277.
Dinges D, Ball E, Fredrickson P, Kiley J, Kryger MH, Richardson GS, Rogus S, Sheldon S, Wooten V, Zepf B. 1999. Recognizing problem sleepiness in your patients. American Family Physician 59(4):937–944.
Dinges D, Rogers N, Baynard. 2005. Chronic sleep deprivation. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/ Saunders. Pp. 67–76.
Doherty LS, Kiely JL, Swan V, McNicholas WT. 2005. Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest 127(6):2076–2084.
Drake CL, Roehrs T, Richardson G, Walsh JK, Roth T. 2004. Shift work sleep disorder: Prevalence and consequences beyond that of symptomatic day workers. Sleep 27(8):1453–1462.
Dreifuss FE, Porter RJ. 1997. Choice of antiepileptic drugs. In: Engel J, Pedley TA, eds. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven. Pp. 1233–1236.
Dunlop RJ, Campbell CW. 2000. Cytokines and advanced cancer. Journal of Pain and Symptom Management 20(3):214–232.
Duran J, Esnaola S, Rubio R, Iztueta A. 2001. Obstructive sleep apnea-hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. American Journal of Respiratory and Critical Care Medicine 163(3):685–689.
Dwyer T, Ponsonby AL, Newman NM, Gibbons LE. 1991. Prospective cohort study of prone sleeping position and sudden infant death syndrome. Lancet 337(8752):1244–1247.
Dyken ME, Somers VK, Yamada T, Ren ZY, Zimmerman MB. 1996. Investigating the relationship between stroke and obstructive sleep apnea. Stroke 27(3):401–407.
Eaker ED, Pinsky J, Castelli WP. 1992. Myocardial infarction and coronary death among women: Psychosocial predictors from a 20-year follow-up of women in the Framingham Study. American Journal of Epidemiology 135(8):854–864.
Edinger JD, Means MK. 2005. Overview of insomnia: Definitions, epidemiology, differential diagnosis, and assessment. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 702–713.
Ekbom KA. 1945. Restless legs. Acta Medica Scandinavia Supplement 158:1–123.
Fantini ML, Gagnon JF, Filipini D, Montplaisir J. 2003. The effects of pramipexole in REM sleep behavior disorder. Neurology 61(10):1418–1420.
Faulx MD, Larkin EK, Hoit BD, Aylor JE, Wright AT, Redline S. 2004. Sex influences endothelial function in sleep-disordered breathing. Sleep 27(6):1113–1120.
Fava M. 2004. Daytime sleepiness and insomnia as correlates of depression. Journal of Clinical Psychiatry 65(suppl 16):27–32.
Fawcett J, Scheftner WA, Fogg L, Clark DC, Young MA, Hedeker D, Gibbons R. 1990. Time-related predictors of suicide in major affective disorder. American Journal of Psychiatry 147(9):1189–1194.
Ferguson KA, Lowe AA. 2005. Oral appliances for sleep-disordered breathing. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1098–1108.
Ferguson KA, Ono T, Lowe AA, Ryan CF, Fleetham JA. 1995. The relationship between obesity and craniofacial structure in obstructive sleep apnea. Chest 108(2):375–381.
Fisher C, Byrne J, Edwards A, Kahn E. 1970. A psychophysiological study of nightmares. Journal of American Psychoanalysis Association 18(4):747–782.
Foley DJ, Monjan AA, Brown SL, Simonsick EM, Wallace RB, Blazer DG. 1995. Sleep complaints among elderly persons: An epidemiologic study of three communities. Sleep 18(6):425–432.
Foley DJ, Masaki K, White L, Larkin EK, Monjan A, Redline S. 2003. Sleep-disordered breathing and cognitive impairment in elderly Japanese-American men. Sleep 26(5):596–599.
Folstein MF, Folstein SE, McHugh PR. 1975. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research 12(3):189–198.
Foo H, Mason P. 2003. Brainstem modulation of pain during sleep and waking. Sleep Medicine Reviews 7(2):145–154.
Ford DE, Kamerow DB. 1989. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? Journal of the American Medical Association 262(11):1479–1484.
Fredriksen K, Rhodes J, Reddy R, Way N. 2004. Sleepless in Chicago: Tracking the effects of adolescent sleep loss during the middle school years. Child Development 75(1):84–95.
Friedland RP, Brun A, Budinger TF. 1985. Pathological and positron emission tomographic correlations in Alzheimer’s disease. Lancet 1(8422):228.
Gagnon JF, Bedard MA, Fantini ML, Petit D, Panisset M, Rompre S, Carrier J, Montplaisir J. 2002. REM sleep behavior disorder and REM sleep without atonia in Parkinson’s disease. Neurology 59(4):585–589.
Gami AS, Howard DE, Olson EJ, Somers VK. 2005. Day-night pattern of sudden death in obstructive sleep apnea. New England Journal of Medicine 352(12):1206–1214.
Garrick NA, Tamarkin L, Taylor PL, Markey SP, Murphy DL. 1983. Light and propranolol suppress the nocturnal elevation of serotonin in the cerebrospinal fluid of rhesus monkeys. Science 221(4609):474–476.
Giles DE, Kupfer DJ, Rush AJ, Roffwarg HP. 1998. Controlled comparison of electrophysiological sleep in families of probands with unipolar depression. American Journal of Psychiatry 155(2):192–199.
Gislason T, Benediktsdottir B. 1995. Snoring, apneic episodes, and nocturnal hypoxemia among children 6 months to 6 years old. An epidemiologic study of lower limit of prevalence. Chest 107(4):963–966.
Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. 1996. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 27(2):252–259.
Gottlieb DJ, Punjabi NM, Newman AB, Resnick HE, Redline S, Baldwin CM, Nieto FJ. 2005. Association of sleep time with diabetes mellitus and impaired glucose tolerance. Archives of Internal Medicine 165(8):863–867.
Gronfier C, Wright KP Jr, Kronauer RE, Jewett ME, Czeisler CA. 2004. Efficacy of a single sequence of intermittent bright light pulses for delaying circadian phase in humans. American Journal of Physiology—Endocrinology and Metabolism 287(1):174–181.
Grunstein R. 2005a. Continuous positive airway pressure treatment for obstructive sleep apnea-hypopnea syndrome. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1066– 1080.
Grunstein R. 2005b. Endocrine disorders. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1237– 1245.
Grunstein R, Wilcox I, Yang TS, Gould Y, Hedner J. 1993. Snoring and sleep apnoea in men: Association with central obesity and hypertension. International Journal of Obesity-Related Metabolic Disorders 17(9):533–540.
Guilleminault C, Connolly SJ, Winkle RA. 1983. Cardiac arrhythmia and conduction disturbances during sleep in 400 patients with sleep apnea syndrome. American Journal of Cardiology 52(5):490–494.
Guilleminault C, Partinen M, Hollman K, Powell N, Stoohs R. 1995. Familial aggregates in obstructive sleep apnea syndrome. Chest 107(6):1545–1551.
Gupta NK, Mueller WH, Chan W, Meininger JC. 2002. Is obesity associated with poor sleep quality in adolescents? American Journal of Human Biology 14(6):762–768.
Hajak G, Muller WE, Wittchen HU, Pittrow D, Kirch W. 2003. Abuse and dependence potential for the non-benzodiazepine hypnotics Zolpidem and Zopiclone: A review of case reports and epidemiological data. Addiction 98(10):1371–1378.
Halasz P, Ujszaszi J, Gadoros J. 1985. Are microarousals preceded by electroencephalographic slow wave synchronization precursors of confusional awakenings? Sleep 8(3):231–238.
Haponik EF. 1992. Sleep disturbances of older persons: Physicians’ attitudes. Sleep 15(2):168–172.
Harma M, Tenkanen L, Sjoblom T, Alikoski T, Heinsalmi P. 1998. Combined effects of shift work and life-style on the prevalence of insomnia, sleep deprivation and daytime sleepiness. Scandinavian Journal of Work, Environment and Health 24(4):300–307.
Harsch IA, Schahin SP, Radespiel-Troger M, Weintz O, Jahreiss H, Fuchs FS, Wiest GH, Hahn EG, Lohmann T, Konturek PC, Ficker JH. 2004. Continuous positive airway pressure treatment rapidly improves insulin sensitivity in patients with obstructive sleep apnea syndrome. American Journal of Respiratory and Critical Care Medicine 169(2):156–162.
Harstall C, Ospina M. 2003. How prevalent is chronic pain? Pain: Clinical Updates 11(2): 1–4.
Harvey AG. 2000. Sleep hygiene and sleep-onset insomnia. Journal of Nervous and Mental Disease 188(1):53–55.
Hasler G, Buysse DJ, Klaghofer R, Gamma A, Ajdacic V, Eich D, Rossler W, Angst J. 2004. The association between short sleep duration and obesity in young adults: A 13-year prospective study. Sleep 27(4):661–666.
Hasler G, Buysse DJ, Gamma A, Ajdacic V, Eich D, Rossler W, Angst J. 2005. Excessive daytime sleepiness in young adults: A 20-year prospective community study. Journal of Clinical Psychiatry 66(4):521–529.
Hauck FR, Kemp JS. 1998. Bedsharing promotes breastfeeding and AAP Task Force on Infant Positioning and SIDS. Pediatrics 102(3 Pt 1):662–664.
Hauck FR, Herman SM, Donovan M, Iyasu S, Merrick MC, Donoghue E, Kirschner RH, Willinger M. 2003. Sleep environment and the risk of sudden infant death syndrome in an urban population: The Chicago Infant Mortality Study. Pediatrics 111(5 Part 2): 1207– 1214.
Hauri PJ. 1997. Can we mix behavioral therapy with hypnotics when treating insomniacs? Sleep 20(12):1111–1118.
He J, Kryger MH, Zorick FJ, Conway W, Roth T. 1988. Mortality and apnea index in obstructive sleep apnea. Experience in 385 male patients. Chest 94(1):9–14.
Hening WA, Allen RP, Earley CJ, Picchietti DL, Silber MH, Restless Legs Syndrome Task Force of the Standards of Practice Committee of the American Academy of Sleep Medicine. 2004. An update on the dopaminergic treatment of restless legs syndrome and periodic limb movement disorder. Sleep 27(3):560–583.
Hermida RC, Smolensky MH. 2004. Chronotherapy of hypertension. Current Opinion in Nephrology and Hypertension 13(5):501–505.
Hilleman D, Miller MA, Parker R, Doering P, Pieper JA. 1998. Optimal management of amiodarone therapy: Efficacy and side effects. Pharmacotherapy 18(6 Pt 2):138S–145S.
Hohagen F, Rink K, Kappler C, Schramm E, Riemann D, Weyerer S, Berger M. 1993. Prevalence and treatment of insomnia in general practice. A longitudinal study. European Archives of Psychiatry and Clinical Neuroscience 242(6):329–336.
Hohjoh H, Takasu M, Shishikura K, Takahashi Y, Honda Y, Tokunaga K. 2003. Significant association of the arylalkylamine N-acetyltransferase (AA-NAT) gene with delayed sleep phase syndrome. Neurogenetics 4(3):151–153.
Holmes MD, Chang M, Kapur V. 2003. Sleep apnea and excessive daytime somnolence induced by vagal nerve stimulation. Neurology 61(8):1126–1129.
Hu FB, Willett WC, Manson JE, Colditz GA, Rimm EB, Speizer FE, Hennekens CH, Stampfer MJ. 2000. Snoring and risk of cardiovascular disease in women. Journal of the American College of Cardiologist 35(2):308–313.
Hublin C, Kaprio J, Partinen M, Heikkila K, Koskenvuo M. 1997. Prevalence and genetics of sleepwalking: A population-based twin study. Neurology 48(1):177–181.
Hung J, Whitford EG, Parsons RW, Hillman DR. 1990. Association of sleep apnoea with myocardial infarction in men. Lancet 336(8710):261–264.
Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. 2002. Obstructive sleep apnea is independently associated with insulin resistance. American Journal of Respiratory and Critical Care Medicine 165(5):670–676.
Irgens LM, Markestad T, Baste V, Schreuder P, Skjaerven R, Oyen N. 1995. Sleeping position and sudden infant death syndrome in Norway 1967-91. Archives of Disease in Childhood 72(6):478–482.
Iwase T, Kajimura N, Uchiyama M, Ebisawa T, Yoshimura K. 2002. Mutation screening of the human CLOCK gene in circadian rhythm sleep disorders. Psychiatry Research 109(2):121–128.
Iyasu S, Randall LL, Welty TK, Hsia J, Kinney HC, Mandell F, McClain M, Randall B, Habbe D, Wilson H, Willinger M. 2002. Risk factors for sudden infant death syndrome among northern plains Indians. Journal of the American Medical Association 288(21):2717– 2723.
Janz D. 1962. The grand mal epilepsies and the sleeping-waking cycle. Epilepsia 3:69–109.
Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA. 1998. Sleep apnea in 81 ambulatory male patients with stable heart failure: Types and their prevalences, consequences, and presentations. Circulation 97(21):2154–2159.
Jeans WD, Fernando DC, Maw AR, Leighton BC. 1981. A longitudinal study of the growth of the nasopharynx and its contents in normal children. British Journal of Radiology 54(638):117–121.
Jellinger K. 1986. Pathology of Parkinsonism. Fahn S, Marsden C, Goldstein M, Calne D, eds. Recent Developments in Parkinson’s Disease. New York: Raven Press. Pp. 33–66.
Jennum P, Hein HO, Suadicani P, Gyntelberg F. 1995. Risk of ischemic heart disease in self-reported snorers. A prospective study of 2,937 men aged 54 to 74 years: The Copenhagen male study. Chest 108(1):138–142.
Johnson JG, Cohen P, Kasen S, First MB, Brook JS. 2004. Association between television viewing and sleep problems during adolescence and early adulthood. Archives of Pediatrics and Adolescent Medicine 158(6):562–568.
Jones BE. 2005. Basic mechanisms of sleep-wake states. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 136–153.
Jones CR, Campbell SS, Zone SE, Cooper F, DeSano A, Murphy PJ, Jones B, Czajkowski L, Ptacek LJ. 1999. Familial advanced sleep-phase syndrome: A short-period circadian rhythm variant in humans. Nature Medicine 5(9): 1062–1065.
Juji T, Satake M, Honda Y, Doi Y. 1984. HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens 24(5):316–319.
Kaakkola S. 2000. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs 59(6):1233–1250.
Kales A, Ansel RD, Markham CH, Scharf MB, Tan TL. 1971. Sleep in patients with Parkinson’s disease and normal subjects prior to and following levodopa administration. Clinical Pharmacology and Therapeutics 12(2):397–406.
Kalra M, Inge T, Garcia V, Daniels S, Lawson L, Curti R, Cohen A, Amin R. 2005. Obstructive sleep apnea in extremely overweight adolescents undergoing bariatric surgery. Obesity Research 13(7):1175–1179.
Kanbayashi T, Inoue Y, Chiba S, Aizawa R, Saito Y, Tsukamoto H, Fujii Y, Nishino S, Shimizu T. 2002. CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. Journal of Sleep Research 11(1):91–93.
Kaplan J, Fredrickson PA, Renaux SA, O’Brien PC. 1993. Theophylline effect on sleep in normal subjects. Chest 103(1):193–195.
Kapsimalis F, Kryger MH. 2002. Gender and obstructive sleep apnea syndrome, part 1: Clinical features. Sleep 25(4):412–419.
Kapur VK, Redline S, Nieto F, Young TB, Newman AB, Henderson JA. 2002. The relationship between chronically disrupted sleep and healthcare use. Sleep 25(3):289–296.
Katz DA, McHorney CA. 1998. Clinical correlates of insomnia in patients with chronic illness. Archives of Internal Medicine 158(10):1099–1107.
Katz I, Stradling J, Slutsky AS, Zamel N, Hoffstein V. 1990. Do patients with obstructive sleep apnea have thick necks? American Review of Respiratory Disease 141(5 Pt 1):1228– 1231.
Keech AC, Armitage JM, Wallendszus KR, Lawson A, Hauer AJ, Parish SE, Collins R. 1996. Absence of effects of prolonged simvastatin therapy on nocturnal sleep in a large randomized placebo-controlled study. Oxford Cholesterol Study Group. British Journal of Clinical Pharmacology 42(4):483–490.
Kemp JS, Thach BT. 1991. Sudden death in infants sleeping on polystyrene-filled cushions. New England Journal of Medicine 324(26):1858–1864.
Kinney HC, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, White WF. 1995. Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 269(5229):1446–1450.
Klackenberg G. 1982. Somnambulism in childhood—prevalence, course and behavioral correlations. A prospective longitudinal study (6–16 years). Acta Paediatrica Scandinavia 71(3):495–499.
Kleitman N. 1987. Sleep and Wakefulness. Chicago: University of Chicago Press.
Klerman EB, Dijk DJ. 2005. Interindividual variation in sleep duration and its association with sleep debt in young adults. Sleep 28(10):1253–1259.
Konsman JP, Parnet P, Dantzer R. 2002. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends in Neuroscience 25(3):154–159.
Krahn LE. 2005. Psychiatric disorders associated with disturbed sleep. Seminars in Neurology 25(1):90–96.
Krahn LE, Pankratz VS, Oliver L, Boeve BF, Silber MH. 2002. Hypocretin (orexin) levels in cerebrospinal fluid of patients with narcolepsy: Relationship to cataplexy and HLA DQB1*0602 status. Sleep 25(7):733–736.
Kripke DF, Ancoli-Israel S, Klauber MR, Wingard DL, Mason WJ, Mullaney DJ. 1997. Prevalence of sleep-disordered breathing in ages 40-64 years: A population-based survey. Sleep 20(1):65–76.
Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR. 2002. Mortality associated with sleep duration and insomnia. Archives in General Psychiatry 59(2):131–136.
Kryger MH, Otake K, Foerster J. 2002. Low body stores of iron and restless legs syndrome: A correctable cause of insomnia in adolescents and teenagers. Sleep Medicine 3(2):127–132.
Kryger MH, Shepertycky M, Foerster J, Manfreda J. 2003. Sleep disorders in repeat blood donors. Sleep 26(5):625–626.
Kshatri AM, Baghdoyan HA, Lydic R. 1998. Cholinomimetics, but not morphine, increase antinociceptive behavior from pontine reticular regions regulating rapid-eye-movement sleep. Sleep 21(7):677–685.
Lake CR, Rosenberg DB, Gallant S, Zaloga G, Chernow B. 1990. Phenylpropanolamine increases plasma caffeine levels. Clinical Pharmacology and Therapeutics 47(6):675–685.
Lammers GJ, Overeem S. 2003. Pharmacological management of narcolepsy. Expert Opinions in Pharmacotherapy 4(10):1739–1746.
Larkin EK, Rosen CL, Kirchner HL, Storfer-Isser A, Emancipator JL, Johnson NL, Zambito AM, Tracy RP, Jenny NS, Redline S. 2005. Variation of C-reactive protein levels in adolescents: Association with sleep-disordered breathing and sleep duration. Circulation 111(15):1978–1984.
Lavie P, Lavie L, Herer P. 2005. All-cause mortality in males with sleep apnoea syndrome: Declining mortality rates with age. European Respiratory Journal 25(3):514–520.
Lavigne GJ, Montplaisir JY. 1994. Restless legs syndrome and sleep bruxism: Prevalence and association among Canadians. Sleep 17(8):739–743.
Lavigne GL, McMillan D, Zucconi M. 2005. Pain and sleep. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier Saunders. Pp. 1246–1255.
Lee KA, Zaffke ME, Baratte-Beebe K. 2001. Restless legs syndrome and sleep disturbance during pregnancy: The role of folate and iron. Journal of Women’s Health and Gender-based Medicine 10(4):335–341.
Lee KA, Landis C, Chasens ER, Dowling G, Merritt S, Parker KP, Redeker N, Richards KC, Rogers AE, Shaver JF, Umlauf MG, Weaver TE. 2004. Sleep and chronobiology: Recommendations for nursing education. Nursing Outlook 52(3):126–133.
Leeman AL, O’Neill CJ, Nicholson PW, Deshmukh AA, Denham MJ, Royston JP, Dobbs RJ, Dobbs SM. 1987. Parkinson’s disease in the elderly: Response to and optimal spacing of night time dosing with levodopa. British Journal of Clinical Pharmacology 24(5):637–643.
Levi F. 1994. Chronotherapy of cancer: Biological basis and clinical application [in French]. Pathologie-Biologie (Paris) 42(4):338–341.
Lewy AJ, Ahmed S, Sack RL. 1996. Phase shifting the human circadian clock using melatonin. Behavioural Brain Research 73(1–2):131–134.
Li KK, Kushida C, Powell NB, Riley RW, Guilleminault C. 2000. Obstructive sleep apnea syndrome: A comparison between Far-East Asian and white men. Laryngoscope 110(10 Pt 1):1689–1693.
Lindberg E, Janson C, Svardsudd K, Gislason T, Hetta J, Boman G. 1998. Increased mortality among sleepy snorers: A prospective population-based study. Thorax 53(8):631–637.
Lingjaerde O, Bratlid T, Hansen T. 1985. Insomnia during the “dark period” in northern Norway. An explorative, controlled trial with light treatment. Acta Psychiatrica Scandinavia 71(5):506–512.
Liu X. 2004. Sleep and adolescent suicidal behavior. Sleep 27(7):1351–1358.
Liu X, Uchiyama M, Kim K, Okawa M, Shibui K, Kudo Y, Doi Y, Minowa M, Ogihara R. 2000. Sleep loss and daytime sleepiness in the general adult population of Japan. Psychiatry Research 93(1):1–11.
Liu Y, Tanaka H, Fukuoka Heart Study Group. 2002. Overtime work, insufficient sleep, and risk of non-fatal acute myocardial infarction in Japanese men. Occupational and Environmental Medicine 59(7):447–451.
Livingston G, Blizard B, Mann A. 1993. Does sleep disturbance predict depression in elderly people? A study in inner London. British Journal of General Practice 43(376): 445–448.
Locard E, Mamelle N, Billette A, Miginiac M, Munoz F, Rey S. 1992. Risk factors of obesity in a five-year-old population: Parental versus environmental factors. International Journal of Obesity and Related Metabolic Disorders 16(10):721–729.
Lundkvist GB, Kristensson K, Bentivoglio M. 2004. Why trypanosomes cause sleeping sickness. Physiology (Bethesda) 19(4):198–206.
Mahowald MW, Ettinger MG. 1990. Things that go bump in the night: The parasomnias revisited. Journal of Clinical Neurophysiology 7(1):119–143.
Mahowald MW, Schenck CH. 2005. Insights from studying human sleep disorders. Nature 437(7063):1279–1285.
Malloy MH, Hoffman HJ. 1995. Prematurity, sudden infant death syndrome, and age of death. Pediatrics 96(3 Pt 1):464–471.
Marcus CL, Greene MG, Carroll JL. 1998. Blood pressure in children with obstructive sleep apnea. American Journal of Respiratory and Critical Care Medicine 157(4 Pt 1):1098–1103.
Marcus CL, Chapman D, Ward SD, McColley SA, Herrerias CT, Stillwell PC, Howenstine M, Light MJ, McColley SA, Schaeffer DA, Wagener JS, Laskosz LN. 2002. Clinical practice guideline: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 109(4):704–712.
Marin JM, Carrizo SJ, Vicente E, Agusti AG. 2005. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: An observational study. Lancet 365(9464):1046–1053.
Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR. 1992. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: Results of a 25-year follow-up study. Lancet 340(8825):925–929.
McAinsh J, Cruickshank JM. 1990. Beta-blockers and central nervous system side effects. Pharmacology and Therapeutics 46(2):163–197.
McArdle N, Riha RL, Vennelle M, Coleman EL, Dennis MS, Warlow CP, Douglas NJ. 2003. Sleep-disordered breathing as a risk factor for cerebrovascular disease: A case-control study in patients with transient ischemic attacks. Stroke 34(12):2916–2921.
McClusky HY, Milby JB, Switzer PK, Williams V, Wooten V. 1991. Efficacy of behavioral versus triazolam treatment in persistent sleep-onset insomnia. American Journal of Psychiatry 148(1):121–126.
McCracken LM, Iverson GL. 2002. Disrupted sleep patterns and daily functioning in patients with chronic pain. Pain Research and Management: The Journal of the Canadian Pain Society 7(2):75–79.
Meier-Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF, Mullington JM. 2004. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. Journal of the American College of Cardiologists 43(4):678–683.
Mellinger GD, Balter MB, Uhlenhuth EH. 1985. Insomnia and its treatment: Prevalence and correlates. Archives of General Psychiatry 42(3):225–232.
Meny RG, Carroll JL, Carbone MT, Kelly DH. 1994. Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics 93(1):44–49.
Miaskowski C, Lee KA. 1999. Pain, fatigue, and sleep disturbances in oncology outpatients receiving radiation therapy for bone metastasis: A pilot study. Journal of Pain and Symptom Management 17(5):320–332.
Michaud M, Chabli A, Lavigne G, Montplaisir J. 2000. Arm restlessness in patients with restless legs syndrome. Movement Disorders 15(2):289–293.
Mignot E. 1998. Genetic and familial aspects of narcolepsy. Neurology 50(2 suppl 1):S16–S22.
Mignot E. 2001. A hundred years of narcolepsy research. Archives of Italian Biology 139(3): 207–220.
Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. 1993. Canine cataplexy is preferentially controlled by adrenergic mechanisms: Evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology (Berlin) 113(1):76–82.
Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, Vankova J, Black J, Harsh J, Bassetti C, Schrader H, Nishino S. 2002a. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Archives of Neurology 59(10):1553–1562.
Mignot E, Taheri S, Nishino S. 2002b. Sleeping with the hypothalamus: Emerging therapeutic targets for sleep disorders. Nature Neuroscience 5(suppl):1071–1075.
Mitler MM, O’Malley MB. 2005. Wake-promoting medications: Efficacy and adverse effects. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 484–498.
Miyazaki S, Uchida S, Mukai J, Nishihara K. 2004. Clonidine effects on all-night human sleep: Opposite action of low- and medium-dose clonidine on human NREM-REM sleep proportion. Psychiatry and Clinical Neuroscience 58(2):138–144.
Moldofsky H. 2001. Sleep and pain. Sleep Medicine Reviews 5(5):385–396.
Mondini S, Guilleminault C. 1985. Abnormal breathing patterns during sleep in diabetes. Annals of Neurology 17(4):391–395.
Monk TH. 2005. Shift work: Basic principles. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 673–679.
Monti JM, Hawkins M, Jantos H, D’Angelo L, Fernandez M. 1988. Biphasic effects of dopamine D-2 receptor agonists on sleep and wakefulness in the rat. Psychopharmacology (Berlin) 95(3):395–400.
Montplaisir J, Petit D, Lorrain D, Gauthier S, Nielsen T. 1995. Sleep in Alzheimer’s disease: Further considerations on the role of brainstem and forebrain cholinergic populations in sleep-wake mechanisms. Sleep 18(3):145–148.
Montplaisir J, Petit D, McNamara D, Gauthier S. 1996. Comparisons between SPECT and quantitative EEG measures of cortical impairment in mild to moderate Alzheimer’s disease. European Neurology 36(4):197–200.
Montplaisir J, Boucher S, Poirier G, Lavigne G, Lapierre O, Lesperance P. 1997. Clinical, polysomnographic, and genetic characteristics of restless legs syndrome: A study of 133 patients diagnosed with new standard criteria. Movement Disorders 12(1):61–65.
Montplaisir J, Allen RP, Walters AD, Lerini-Strambi L. 2005. Restless legs syndrome and periodic limb movements during sleep. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 839–852.
Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. 1996a. Sleep-disordered breathing in men with coronary artery disease. Chest 109(3):659–663.
Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. 1996b. Sleep-disordered breathing in women: Occurrence and association with coronary artery disease. American Journal of Medicine 101(3):251–256.
Moran M, Lynch CA, Walsh C, Coen R, Coakley D, Lawlor BA. 2005. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Medicine 6(4):347–352.
Morin CM. 2005. Psychological and behavioral treatments for primary insomnia. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 726–737.
Morin CM, Gibson D, Wade J. 1998. Self-reported sleep and mood disturbance in chronic pain patients. Clinical Journal of Pain 14(4):311–314.
Morin CM, Mimeault V, Gagne A. 1999. Nonpharmacological treatment of late-life insomnia. Journal of Psychosomatic Research 46(2):103–116.
Morrell MJ, Heywood P, Moosavi SH, Guz A, Stevens J. 1999. Unilateral focal lesions in the rostrolateral medulla influence chemosensitivity and breathing measured during wakefulness, sleep, and exercise. Journal of Neurology, Neurosurgery, and Psychiatry 67(5):637–645.
Morrison DN, McGee R, Stanton WR. 1992. Sleep problems in adolescence. Journal of the American Academy of Child and Adolescent Psychiatry 31(1):94–99.
Morton S, Rosen C, Larkin E, Tishler P, Aylor J, Redline S. 2001. Predictors of sleep-disordered breathing in children with a history of tonsillectomy and/or adenoidectomy. Sleep 24(7):823–829.
Moser NJ, Phillips BA, Berry DT, Harbison L. 1994. What is hypopnea, anyway? Chest 105(2):426–428.
Mourits MJ, De Vries EG, Willemse PH, Ten Hoor KA, Hollema H, Van der Zee AG. 2001. Tamoxifen treatment and gynecologic side effects: A review. Obstetrics and Gynecology 97(5 Pt 2):855–866.
Murdey ID, Cameron N, Biddle SJ, Marshall SJ, Gorely T. 2005. Short-term changes in sedentary behaviour during adolescence: Project STIL (Sedentary Teenagers and Inactive Lifestyles). Annals of Human Biology 32(3):283–296.
Murphy PJ, Badia P, Myers BL, Boecker MR, Wright KP Jr. 1994. Nonsteroidal anti-inflammatory drugs affect normal sleep patterns in humans. Physiology and Behavior 55(6): 1063–1066.
Murphy PJ, Myers BL, Badia P. 1996. Nonsteroidal anti-inflammatory drugs alter body temperature and suppress melatonin in humans. Physiology and Behavior 59(1):133–139.
Namen AM, Wymer A, Case D, Haponik EF. 1999. Performance of sleep histories in an ambulatory medicine clinic: Impact of simple chart reminders. Chest 116(6):1558–1563.
Namen AM, Landry SH, Case LD, McCall WV, Dunagan DP, Haponik EF. 2001. Sleep histories are seldom documented on a general medical service. Southern Medical Journal 94(9):874–879.
Narkiewicz K, Somers VK. 2003. Sympathetic nerve activity in obstructive sleep apnoea. Acta Physiologica Scandinavia 177(3):385–390.
Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK. 1999. Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 100(23):2332–2335.
Netzer NC, Hoegel JJ, Loube D, Netzer CM, Hay B, Alvarez-Sala R, Strohl KP, Sleep in Primary Care International Study Group. 2003. Prevalence of symptoms and risk of sleep apnea in primary care. Chest 124(4):1406–1414.
Newman AB, Spiekerman CF, Enright P, Lefkowitz D, Manolio T, Reynolds CF, Robbins J. 2000. Daytime sleepiness predicts mortality and cardiovascular disease in older adults. The Cardiovascular Health Study Research Group. Journal of the American Geriatric Society 48(2):115–123.
NHLBI (National Heart, Lung, and Blood Institute). 2003. National Sleep Disorders Research Plan, 2003. Bethesda, MD: National Institutes of Health.
NICHD (National Institute of Child Health and Human Development). 2006a. Safe Sleep for Your Baby: Ten Ways to Reduce the Risk of Sudden Infant Death Syndrome (SIDS). [Online]. Available: http://www.nichd.nih.gov/sids/reduce_infant_risk.htm [accessed January 17, 2006].
NICHD. 2006b. SIDS Facts. [Online]. Available: www.nichd.nih.gov/sids/PART_II.pdf [accessed March 13, 2006].
Nichols DA, Allen RP, Grauke JH, Brown JB, Rice ML, Hyde PR, Dement WC, Kushida CA. 2002. Restless legs syndrome symptoms in primary care: A prevalence study. Archives of Internal Medicine 163(18):2323–2329.
Nielsen TA, Zadra A. 2000. Dreaming disorders. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 3rd ed. Philadelphia: Elsevier Saunders. Pp. 753–772.
Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG. 2000. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. Journal of the American Medical Association 283(14):1829–1836.
Nieto FJ, Herrington DM, Redline S, Benjamin EJ, Robbins JA. 2004. Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. American Journal of Respiratory and Critical Care Medicine 169(3):354–360.
NIH (National Institutes of Health). 2005. NIH State-of-the-Science Conference Statement on Manifestations and Management of Chronic Insomnia in Adults [Online]. Available: http://consensus.nih.gov/2005/2005InsomniaSOS026html.htm [accessed March 6, 2006].
NINDS (National Institute of Neurological Disorders and Stroke). 2005. Restless Legs Syndrome Fact Sheet. [Online]. Available: http://www.ninds.nih.gov/disorders/restless_legs/detail_restless_legs.htm [accessed June 13, 2005].
Nishino S, Mignot E. 1997. Pharmacological aspects of human and canine narcolepsy. Progress in Neurobiology 52(1):27–78.
Nofzinger EA, Buysse DJ, Germain A, Carter CS, Luna B, Price JC, Meltzer CC, Miewald JM, Reynolds CF, and Kupfer DJ. 2004a. Increased activation of anterior paralimbic and executive cortex from waking to REM sleep in depression. Archives of General Psychiatry 61(7):695–702.
Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, and Kupfer DJ. 2004b. Functional neuroimaging evidence for hyperarousal in insomnia. American Journal of Psychiatry 161(11):2126–2128.
Nofzinger EA, Buysse DJ, Germain A, Price JC, Meltzer CC, Miewald JM, Kupfer DJ. 2005. Alterations in regional cerebral glucose metabolism across waking and non-rapid eye movement sleep in depression. Archives of General Psychiatry 62(4):387–396.
Norman SE, Chediak AD, Kiel M, Cohn MA. 1990. Sleep disturbances in HIV-infected homosexual men. AIDS 4(8):775–781.
Nowell PD, Buysse DJ, Reynolds CF III, Hauri PJ, Roth T, Stepanski EJ, Thorpy MJ, Bixler E, Kales A, Manfredi RL, Vgontzas AN, Stapf DM, Houck PR, Kupfer DJ. 1997. Clinical factors contributing to the differential diagnosis of primary insomnia and insomnia related to mental disorders. American Journal of Psychiatry 154(10):1412–1416.
NSF (National Sleep Foundation). 2000a. 2000 Omnibus Sleep in America Poll. [Online]. Available: www.sleepfoundation.org/publications/2001poll.html [accessed May 25, 2005].
NSF. 2005b. 2005 Sleep in America Poll. [Online]. Available: http://www.sleepfoundation.org/_content/hottopics/2005_summary_of_findings.pdf [accessed June 7, 2005].
NSF. 2005c. Shift work: Coping. [Online]. Available: http://www.sleepfoundation.org/sleeptionary/index.php?id=20&subsection=coping [accessed June 7, 2005].
Ohayon MM. 2002. Epidemiology of insomnia: What we know and what we still need to learn. Sleep Medicine Reviews 6(2):97–111.
Ohayon MM, Roth T. 2003. Place of chronic insomnia in the course of depressive and anxiety disorders. Journal of Psychiatric Research 37(1):9–15.
Ohayon MM, Caulet M, Guilleminault C. 1997. How a general population perceives its sleep and how this relates to the complaint of insomnia. Sleep 20(9):715–723.
Ohayon MM, Guilleminault C, Priest RG. 1999. Night terrors, sleepwalking, and confusional arousals in the general population: Their frequency and relationship to other sleep and mental disorders. Journal of Clinical Psychiatry 60(4):268–277.
Ohayon MM, Priest RG, Zulley J, Smirne S. 2000. The place of confusional arousals in sleep and mental disorders: Findings in a general population sample of 13,057 subjects. Journal of Nervous Mental Disease 188(6):340–348.
Olson EJ, Boeve BF, Silber MH. 2000. Rapid eye movement sleep behaviour disorder: Demographic, clinical and laboratory findings in 93 cases. Brain 123 (Pt 2):331–339.
Ondze B, Espa F, Ming LC, Chakkar B, Besset A, Billiard M. 2001. Advanced sleep phase syndrome [in French]. Reviews of Neurology 157(11 Pt 2):S130–S134.
Opp MR, Toth LA. 2003. Neural-immune interactions in the regulation of sleep. Frontiers of Bioscience 8:d768–d779.
Overeem S, Scammell TE, Lammers GJ. 2002. Hypocretin/orexin and sleep: Implications for the pathophysiology and diagnosis of narcolepsy. Current Opinion in Neurology 15(6): 739–745.
Ozminkowski R, Wang S, Trautman H, Orsini L. 2004. Estimating the cost burden of insomnia for health plans. Journal of Managed Care Pharmacy 10(5):467.
Palm L, Anderson H, Elmqvist D, Blennow G. 1992. Daytime sleep tendency before and after discontinuation of antiepileptic drugs in preadolescent children with epilepsy. Epilepsia 33(4):687–691.
Palmer CR, Kripke DF, Savage HC Jr, Cindrich LA, Loving RT, Elliott JA. 2003. Efficacy of enhanced evening light for advanced sleep phase syndrome. Behavioral Sleep Medicine 1(4):213–226.
Palmer LJ, Buxbaum SG, Larkin E, Patel SR, Elston RC, Tishler PV, Redline S. 2003. A whole-genome scan for obstructive sleep apnea and obesity. American Journal of Human Genetics 72(2):340–350.
Palmer LJ, Buxbaum SG, Larkin EK, Patel SR, Elston RC, Tishler PV, Redline S. 2004. Whole genome scan for obstructive sleep apnea and obesity in African-American families. American Journal of Respiratory and Critical Care Medicine 169(12):1314–1321.
Panichi V, Migliori M, De Pietro S, Taccola D, Andreini B, Metelli MR, Giovannini L, Palla R. 2000. The link of biocompatibility to cytokine production. Kidney International Supplement 76(suppl):S96–S103.
Panigraphy A, Filiano JJ, Sleep LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, White WF, Kinney HC. 1997. Decreased kainate receptor binding in the arcuate nucleus of the sudden infant death syndrome. Journal of Neuropathology and Experimental Neurology 56(11):1253–1261.
Parker KP. 2003. Sleep disturbances in dialysis patients. Sleep Medicine Reviews 7(2): 131–143.
Parker KP, Bliwise DL, Rye DB. 2000. Hemodialysis disrupts basic sleep regulatory mechanisms: Building hypotheses. Nursing Research 49(6):327–332.
Parra O, Arboix A, Bechich S, Garcia-Eroles L, Montserrat JM, Lopez JA, Ballester E, Guerra JM, Sopena JJ. 2000. Time course of sleep-related breathing disorders in first-ever stroke or transient ischemic attack. American Journal of Respiratory and Critical Care Medicine 161(2):375–380.
Partinen M, Hublin C. 2005. Epidemiology of sleep disorders. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/ Saunders. Pp. 626–647.
Patel SR, White DP, Malhotra A, Stanchina ML, Ayas NT. 2003. Continuous positive airway pressure therapy for treating sleepiness in a diverse population with obstructive sleep apnea: Results of a meta-analysis. Archives of Internal Medicine 163(5): 565–571.
Patel SR, Ayas NT, Malhotra MR, White DP, Schernhammer ES, Speizer FE, Stampfer MJ, Hu FB. 2004. A prospective study of sleep duration and mortality risk in women. Sleep 27(3):440–444.
Paus S, Brecht HM, Koster J, Seeger G, Klockgether T, Wullner U. 2003. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson’s disease. Movement Disorders 18(6):659–667.
Paykel ES, Fleminger R, Watson JP. 1982. Psychiatric side effects of antihypertensive drugs other than reserpine. Journal of Clinical Psychopharmacology 2(1):14–39.
Pelayo R, Thorpy MJ, Govinsky P. 1988. Prevalence of delayed sleep phase syndrome among adolescents. Sleep Research 17:392.
Peppard PE, Young T, Palta M, Skatrud J. 2000. Prospective study of the association between sleep-disordered breathing and hypertension. New England Journal of Medicine 342(19): 1378–1384.
Pereira DS, Tufik S, Louzada FM, Benedito-Silva AA, Lopez AR, Lemos NA, Korczak AL, D’Almeida V, Pedrazzoli M. 2005. Association of the length polymorphism in the human Per3 gene with the delayed sleep-phase syndrome: Does latitude have an influence upon it? Sleep 28(1):29–32.
Perlis ML, Smith MT, Pigeon WR. 2005. Etiology and pathophysiology of insomnia. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 714–725.
Pertosa G, Grandaliano G, Gesualdo L, Schena FP. 2000. Clinical relevance of cytokine production in hemodialysis. Kidney International Supplement 76:S104–S111.
Peters RW. 2005. Obstructive sleep apnea and cardiovascular disease. Chest 127(1):1–3.
Petit D, Montplaisir J, Boeve B. 2005. Alzheimer’s disease and other dementias. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 853–862.
Peyron C, Faraco J, Rogers W, Ripley B, Overeem S. 2000. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nature Medicine 6(9):991–997.
Phillips BG, Hisel TM, Kato M, Pesek CA, Dyken ME, Narkiewicz K, Somers VK. 1999. Recent weight gain in patients with newly diagnosed obstructive sleep apnea. Journal of Hypertension 17(9):1297–1300.
Phillips BG, Kato M, Narkiewicz K, Choe I, Somers VK. 2000. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. American Journal of Physiology—Heart and Circulatory Physiology 279(1): H234–H237.
Phillips B, Hening W, Britz P, Mannino D. 2006. Prevalence and correlates of restless legs syndrome: 2 Results from the 2005 National Sleep Foundation poll. Chest 129(1): 76–80.
Picchietti DL, England SJ, Walters AS, Willis K, Verrico T. 1998. Periodic limb movement disorder and restless legs syndrome in children with attention-deficit hyperactivity disorder. Journal of Child Neurology 13(12): 588–594.
Picchietti DL, Underwood DJ, Farris WA, Walters AS, Shah MM, Dahl RE, Trubnick LJ, Bertocci MA, Wagner M, Hening WA. 1999. Further studies on periodic limb movement disorder and restless legs syndrome in children with attention-deficit hyperactivity disorder. Movement Disorders 14(6):1000–1007.
Pilcher JJ, Huffcutt AI. 1996. Effects of sleep deprivation on performance: A meta-analysis. Sleep 19(4):318–326.
Pillar G, Lavie P. 1995. Assessment of the role of inheritance in sleep apnea syndrome. American Journal of Respiratory and Critical Care Medicine 151(3 Pt 1): 688–691.
Plazzi G, Corsini R, Provini F, Pierangeli G, Martinelli P, Montagna P, Lugaresi E, Cortelli P. 1997. REM sleep behavior disorders in multiple system atrophy. Neurology 48(4):1094– 1097.
Plazzi G, Cortelli P, Montagna P, De Monte A, Corsini R, Contin M, Provini F, Pierangeli G, Lugaresi E. 1998. REM sleep behaviour disorder differentiates pure autonomic failure from multiple system atrophy with autonomic failure. Journal of Neurology, Neurosurgery and Psychiatry 64(5):683–685.
Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Wang YG. 1993. Factors potentiating the risk of sudden infant death syndrome associated with the prone position. New England Journal of Medicine 329(6):377–382.
Powell NB, Riley RW, Guilleminault C. 2005. Surgical management of sleep-disordered breathing. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1081–1097.
Prinz PN, Peskind ER, Vitaliano PP, Raskind MA, Eisdorfer C, Zemcuznikov N, Gerber CJ. 1982a. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. Journal of the American Geriatrics Society 30(2):86–93.
Prinz PN, Vitaliano PP, Vitiello MV, Bokan J, Raskind M, Peskind E, Gerber C. 1982b. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiology of Aging 3(4):361–370.
Punjabi NM, Beamer BA. 2005. Sleep apnea and metabolic dysfunction. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1034–1042.
Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. 2002. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. American Journal of Respiratory and Critical Care Medicine 165(5):677–682.
Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE, Sleep Heart Health Study Investigators. 2004. Sleep-disordered breathing, glucose intolerance, and insulin resistance: The Sleep Heart Health Study. American Journal of Epidemiology 160(6):521–530.
Qureshi AI, Giles WH, Croft JB, Bliwise DL. 1997. Habitual sleep patterns and risk for stroke and coronary heart disease: A 10-year follow-up from NHANES I. Neurology 48(4):904–911.
Radomski MW, Buguet A, Bogui P, Doua F, Lonsdorfer A, Tapie P, Dumas M. 1994. Disruptions in the secretion of cortisol, prolactin, and certain cytokines in human African trypanosomiasis patients. Bulletin de la Societe de Pathologie Exotique (Paris) 87(5): 376–379.
Raison CL, Miller AH. 2001. The neuroimmunology of stress and depression. Seminars in Clinical Neuropsychiatry 6(4):277–294.
Rao U, Dahl RE, Ryan ND, Birmaher B, Williamson DE, Giles DE, Rao R, Kaufman J, Nelson B. 1996. The relationship between longitudinal clinical course and sleep and cortisol changes in adolescent depression. Biological Psychiatry 40(6):474–484.
Redline S, Tishler PV, Tosteson TD, Williamson J, Kump K, Browner I, Ferrette V, Krejci P. 1995. The familial aggregation of obstructive sleep apnea. American Journal of Respiratory and Critical Care Medicine 151(3 Pt 1):682–687.
Redline S, Tishler PV, Hans MG, Tosteson TD, Strohl KP, Spry K. 1997. Racial differences in sleep-disordered breathing in African-Americans and Caucasians. American Journal of Respiratory and Critical Care Medicine 155(1):186–192.
Redline S, Tishler PV, Schluchter M, Aylor J, Clark K, Graham G. 1999. Risk factors for sleep-disordered breathing in children: Associations with obesity, race, and respiratory problems. American Journal of Respiratory and Critical Care Medicine 159(5):1527–1532.
Redline S, Kapur VK, Sanders MH, Quan SF, Gottlieb DJ, Rapoport DM, Bonekat WH, Smith PL, Kiley JP, Iber C. 2000. Effects of varying approaches for identifying respiratory disturbances on sleep apnea assessment. American Journal of Respiratory and Critical Care Medicine 161(2 Pt 1):369–374.
Regestein QR, Monk TH. 1995. Delayed sleep phase syndrome: A review of its clinical aspects. American Journal of Psychiatry 152(4):602–608.
Reid JL. 1996. New therapeutic agents for hypertension. British Journal of Clinical Pharmacology 42(1):37–41.
Reid KJ, Zee PC. 2005. Circadian disorders of the sleep-wake cycle. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/ Saunders. Pp. 691–701.
Reid KJ, Chang AM, Dubocovich ML, Turek FW, Takahashi JS, Zee PC. 2001. Familial advanced sleep phase syndrome. Archives of Neurology 58(7):1089–1094.
Reynolds CF III, Kupfer DJ, Taska LS, Hoch CC, Spiker DG, Sewitch DE, Zimmer B, Marin RS, Nelson JP, Martin D, Morycz R. 1985. EEG sleep in elderly depressed, demented, and healthy subjects. Biological Psychiatry 20(4):431–442.
Reynolds CF III, Frank E, Houck PR, Mazumdar S, Dew MA, Cornes C, Buysse DJ, Begley A, Kupfer DJ. 1997. Which elderly patients with remitted depression remain well with continued interpersonal psychotherapy after discontinuation of antidepressant medication? American Journal of Psychiatry 154(7):958–962.
Riemann D, Voderholzer U. 2003. Primary insomnia: A risk factor to develop depression? Journal of Affective Disorders 76(1-3):255–259.
Riley JL III, Benson MB, Gremillion HA, Myers CD, Robinson ME, Smith CL Jr, Waxenberg LB. 2001. Sleep disturbance in orofacial pain patients: Pain-related or emotional distress? Cranio 19(2):106–113.
Rizzo P, Beelke M, De Carli F, Canovaro P, Nobili L, Robert A, Tanganelli P, Regesta G, Ferrillo F. 2003. Chronic vagus nerve stimulation improves alertness and reduces rapid eye movement sleep in patients affected by refractory epilepsy. Sleep 26(5):607–611.
Robinson GV, Pepperell JC, Segal HC, Davies RJ, Stradling JR. 2004a. Circulating cardiovascular risk factors in obstructive sleep apnoea: Data from randomised controlled trials. Thorax 59(9):777–782.
Robinson GV, Stradling JR, Davies RJ. 2004b. Sleep 6: Obstructive sleep apnoea/hypopnoea syndrome and hypertension. Thorax 59(12):1089–1094.
Roizenblatt S, Moldofsky H, Benedito-Silva AA, Tufik S. 2001. Alpha sleep characteristics in fibromyalgia. Arthritis and Rheumatism 44(1):222–230.
Rombaux P, Hamoir M, Plouin-Gaudon I, Liistro G, Aubert G, Rodenstein D. 2000. Obstructive sleep apnea syndrome after reconstructive laryngectomy for glottic carcinoma. European Archives of Otorhinolaryngology 257(9):502–506.
Rosen CL, D’Andrea L, Haddad GG. 1992. Adult criteria for obstructive sleep apnea do not identify children with serious obstruction. American Review of Respiratory Diseases 146(5 Pt 1):1231–1234.
Rosen CL, Larkin EK, Kirchner HL, Emancipator JL, Bivins SF, Surovec SA, Martin RJ, Redline S. 2003. Prevalence and risk factors for sleep-disordered breathing in 8- to 11-year-old children: Association with race and prematurity. Journal of Pediatrics 142(4): 383–389.
Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. 2001. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Annals of Neurology 50(3):339–348.
Rosenthal NE, Joseph-Vanderpool JR, Levendosky AA, Johnston SH, Allen R, Kelly KA, Souetre E, Schultz PM, Starz KE. 1990. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep 13(4):354–361.
Roth B. 1976. Narcolepsy and hypersomnia: Review and classification of 642 personally observed cases. Schweizer Archiv fur Neurologie, Neurochirurgie und Psychiatrie 119(1): 31–41.
Roth T, Ancoli-Israel S. 1999. Daytime consequences and correlates of insomnia in the United States: Results of the 1991 National Sleep Foundation Survey. II. Sleep 22(suppl 2):S354–S358.
Rothdach AJ, Trenkwalder C, Haberstock J, Keil U, Berger K. 2000. Prevalence and risk factors of RLS in an elderly population: The MEMO study. Memory and morbidity in Augsburg elderly. Neurology 54(5):1064–1068.
Ryan ND, Puig-Antich J, Ambrosini P, Rabinovich H, Robinson D, Nelson B, Iyengar S, Twomey J. 1987. The clinical picture of major depression in children and adolescents. Archives of General Psychiatry 44(10):854–861.
Salinsky MC, Oken BS, Binder LM. 1996. Assessment of drowsiness in epilepsy patients receiving chronic antiepileptic drug therapy. Epilepsia 37(2):181–187.
Santhi N, Duffy JF, Horowitz TS, Czeisler CA. 2005. Scheduling of sleep/darkness affects the circadian phase of night shift workers. Neuroscience Letters 384(3):316–320.
Satoh K, Mishima K, Inoue Y, Ebisawa T, Shimizu T. 2003. Two pedigrees of familial advanced sleep phase syndrome in Japan. Sleep 26(4):416–417.
Scammell TE. 2003. The neurobiology, diagnosis, and treatment of narcolepsy. Annals of Neurology 53(2):154–166.
Schechtman VL, Harper RK, Harper RM. 1995. Aberrant temporal patterning of slow-wave sleep in siblings of SIDS victims. Electroencephalography and Clinical Neurophysiology 94(2):95–102.
Schenck C, Mahowald M. 1990. A polysomnographic neurologic, psychiatric and clinical outcome report on 70 consecutive cases with REM sleep behavior disorder (RBD): sustained clonazepam efficacy in 89.5% of 57 treated patients. Cleveland Clinic Journal of Medicine 57(10):10–24.
Schenck CH, Hurwitz TD, Mahowald MW. 1993. Symposium: Normal and abnormal REM sleep regulation: REM sleep behaviour disorder: An update on a series of 96 patients and a review of the world literature. Journal of Sleep Research 2(4):224–231.
Schmidt-Nowara WW, Coultas DB, Wiggins C, Skipper BE, Samet JM. 1990. Snoring in a Hispanic-American population. Risk factors and association with hypertension and other morbidity. Archives of Internal Medicine 150(3):597–601.
Schoendorf KC, Kiely JL. 1992. Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics 90(6):905–908.
Schwartz SW, Cornoni-Huntley J, Cole SR, Hays JC, Blazer DG, Schocken DD. 1998. Are sleep complaints an independent risk factor for myocardial infarction? Annals of Epidemiology 8(6):384–392.
Sekine M, Yamagami T, Handa K, Saito T, Nanri S, Kawaminami K, Tokui N, Yoshida K, Kagamimori S. 2002. A dose-response relationship between short sleeping hours and childhood obesity: Results of the Toyama birth cohort study. Child: Care, Health and Development 28(2):163–170.
Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier Nieto F, O’Connor GT, Boland LL, Schwartz JE, Samet JM. 2001. Sleep-disordered breathing and cardiovascular disease: Cross-sectional results of the Sleep Heart Health Study. American Journal of Respiratory and Critical Care Medicine 163(1):19–25.
Shahar E, Redline S, Young T, Boland LL, Baldwin CM, Nieto FJ, O’Connor GT, Rapoport DM, Robbins JA. 2003. Hormone replacement therapy and sleep-disordered breathing. American Journal of Respiratory and Critical Care Medicine 167(9):1186–1192.
Shamsuzzaman AS, Gersh BJ, Somers VK. 2003. Obstructive sleep apnea: Implications for cardiac and vascular disease. Journal of the American Medical Association 290(14):1906– 1914.
Shepertycky MR, Banno K, Kryger MH. 2005. Differences between men and women in the clinical presentation of patients diagnosed with obstructive sleep apnea syndrome. Sleep 28(3):309–314.
Shiino Y, Nakajima S, Ozeki Y, Isono T, Yamada N. 2003. Mutation screening of the human period 2 gene in bipolar disorder. Neuroscience Letters 338(1):82–84.
Shouse MN, Mahowald M. 2005. Epilepsy, sleep, and sleep disorders. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 863–878.
Shouse MN, da Silva AM, Sammaritano M. 1996. Circadian rhythm, sleep, and epilepsy. Journal of Clinical Neurophysiology 13(1):32–50.
Silber MH, Richardson JW. 2003. Multiple blood donations associated with iron deficiency in patients with restless legs syndrome. Mayo Clinic Proceedings 78(1):52–54.
Simon GE, VonKorff M. 1997. Prevalence, burden, and treatment of insomnia in primary care. American Journal of Psychiatry 154(10):1417–1423.
Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. 1999. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. American Journal of Respiratory and Critical Care Medicine 160(4):1101–1106.
Singh M, Drake C, Roehrs T, Koshorek G, Roth T. 2005. The prevalence of SOREMPs in the general population. Sleep 28(abstract suppl):A221.
Smith A. 1992. Sleep, colds, and performance. In: Broughton RJ, Ogilvie R, eds. Sleep Arousal and Performance. Boston: Birkhouser.
Smith MT, Perlis ML, Park A, Smith MS, Pennington J, Giles DE, Buysse DJ. 2002. Comparative meta-analysis of pharmacotherapy and behavior therapy for persistent insomnia. American Journal of Psychiatry 159(1):5–11.
Somers VK, Mark AL, Abboud FM 1988. Sympathetic activation by hypoxia and hypercapnia—implications for sleep apnea. Clinical and Experimental Hypertension: Part A, Theory and Practice 10(suppl 1):413–422.
Somers VK, Dyken ME, Mark AL, Abboud FM. 1992. Parasympathetic hyperresponsiveness and bradyarrhythmias during apnoea in hypertension. Clinical Autonomic Research 2(3):171–176.
Somers VK, Dyken ME, Clary MP, Abboud FM. 1995. Sympathetic neural mechanisms in obstructive sleep apnea. Journal of Clinical Investigation 96(4):1897–1904.
Spiegel K, Leproult R, Van Cauter E. 1999. Impact of sleep debt on metabolic and endocrine function. Lancet 354(9188):1435–1439.
Spiegel K, Tasali E, Penev P, Van Cauter E. 2004. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Annals of Internal Medicine 141(11):846–850.
Stiasny K, Wetter TC, Winkelmann J, Brandenburg U, Penzel T, Rubin M, Hundemer HP, Oertel WH, Trenkwalder C. 2001. Long-term effects of pergolide in the treatment of restless legs syndrome. Neurology 56(10):1399–1402.
Strine TW, Chapman DP. 2005. Associations of frequent sleep insufficiency with health-related quality of life and health behaviors. Sleep Medicine 6(1):23–27.
Strohl KP, Redline S. 1996. Recognition of obstructive sleep apnea. American Journal of Respiratory and Critical Care Medicine 154(2 Pt 1):279–289.
Strollo PJ, Atwood CW Jr, Sanders MH. 2005. Medical therapy for obstructive sleep apnea-hypopnea syndrome. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1053–1065.
Sulit LG, Storfer-Isser A, Rosen CL, Kirchner HL, Redline S. 2005. Associations of obesity, sleep-disordered breathing, and wheezing in children. American Journal of Respiratory and Critical Care Medicine 171(6):659–664.
Szymczak JT, Jasinska M, Pawlak E, Zwierzykowska M. 1993. Annual and weekly changes in the sleep-wake rhythm of school children. Sleep 16(5):433–435.
Taasan VC, Block AJ, Boysen PG, Wynne JW. 1981. Alcohol increases sleep apnea and oxygen desaturation in asymptomatic men. American Journal of Medicine 71(2):240–245.
Taheri S, Lin L, Austin D, Young T, Mignot E. 2004. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. Public Library of Science Medicine 1(3):210–217.
Takahashi Y, Hohjoh H, Matsuura K. 2000. Predisposing factors in delayed sleep phase syndrome. Psychiatry and Clinical Neuroscience 54(3):356–358.
Tamakoshi A, Ohno Y, JACC Study Group. 2004. Self-reported sleep duration as a predictor of all-cause mortality: Results from the JACC study, Japan. Sleep 27(1):51–54.
Tassinari CA, Mancia D, Bernardina BD, Gastaut H. 1972. Pavor nocturnus of non-epileptic nature in epileptic children. Electroencephalography and Clinical Neurophysiology 33(6):603–607.
Terzano MG, Parrino L, Spaggiari MC. 1988. The cyclic alternating pattern sequences in the dynamic organization of sleep. Electroencephalography and Clinical Neurophysiology 69(5):437–447.
Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. 2000. Reduced number of hypocretin neurons in human narcolepsy. Neuron 27(3):469–474.
Thorpy MJ. 2005. Classification of sleep disorders. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 615–625.
Tishler PV, Redline S, Ferrette V, Hans MG, Altose MD. 1996. The association of sudden unexpected infant death with obstructive sleep apnea. American Journal of Respiratory and Critical Care Medicine 153(6 Pt 1):1857–1863.
Tochikubo O, Ikeda A, Miyajima E, Ishii M. 1996. Effects of insufficient sleep on blood pressure monitored by a new multibiomedical recorder. Hypertension 27(6):1318– 1324.
Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. 2001. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291(5506):1040–1043.
Toth LA. 1999. Microbial modulation of sleep. In: Lydic R, Baghdoyan HA, eds. Handbook of Behavioral State Control: Cellular and Molecular Mechanisms. Boca Raton, FL: CRC Press.
Toth LA, Opp MR. 2002. Infection and sleep. In: Lee CT, Sateia M, Carskadon M. Sleep Medicine. Philadelphia, PA: Hanley and Belfus.
Tractenberg RE, Singer CM, Kaye JA. 2005. Symptoms of sleep disturbance in persons with Alzheimer’s disease and normal elderly. Journal of Sleep Research 14(2):177–185.
Trampus M, Ferri N, Monopoli A, Ongini E. 1991. The dopamine D1 receptor is involved in the regulation of REM sleep in the rat. European Journal of Pharmacology 194(2–3):189–194.
Tune GS. 1968. Sleep and wakefulness in normal human adults. British Medical Journal 2(600):269–271.
Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. 2005. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308(5724):1043–1045.
Turjanski N, Lees AJ, Brooks DJ. 1999. Striatal dopaminergic function in restless legs syndrome: 18F-dopa and 11C-raclopride PET studies. Neurology 52(5):932–937.
Ulfberg J, Nystrom B. 2004. Restless legs syndrome in blood donors. Sleep Medicine 5(2):115–118.
United States Census Bureau. 1990. Time Leaving Home to Go to Work for the United States: 1990 Census. [Online] Available: http://www.census.gov/population/socdemo/journey/usdeptim.txt [accessed March 7, 2006].
Unruh ML, Levey AS, D’Ambrosio C, Fink NE, Powe NR, Meyer KB. 2004. Restless legs symptoms among incident dialysis patients: Association with lower quality of life and shorter survival. American Journal of Kidney Diseases 43(5):900–909.
Van Den Heuvel CJ, Reid KJ, Dawson D. 1997. Effect of atenolol on nocturnal sleep and temperature in young men: Reversal by pharmacological doses of melatonin. Physiology and Behavior 61(6):795–802.
Vaziri ND, Oveisi F, Wierszbiezki M, Shaw V, Sporty LD. 1993. Serum melatonin and 6-sulfatoxymelatonin in end-stage renal disease: Effect of hemodialysis. Artificial Organs 17(9):764–769.
Vaziri ND, Oveisi F, Reyes GA, Zhou XJ. 1996. Dysregulation of melatonin metabolism in chronic renal insufficiency: Role of erythropoietin-deficiency anemia. Kidney International 50(2):653–656.
Veasey S, Rosen R, Barzansky B, Rosen I, Owens J. 2002. Sleep loss and fatigue in residency training: A reappraisal. Journal of the American Medical Association 288(9):1116–1124.
Velasco M, Velasco F. 1982. Brain stem regulation of cortical and motor excitability: Effects on experimental and focal motor seizures. In: Sterman MB, Shouse MN, Passouant P, eds. Sleep and Epilepsy. New York: Academic Press. Pp. 53–61.
Verrier RL, Josephson ME. 2005. Cardiac arrhythmogenesis during sleep: Mechanisms, diagnosis, and therapy. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1171–1191.
Vgontzas AN, Kales A. 1999. Sleep and its disorders. Annual Review of Medicine 50(1):387–400.
Vgontzas AN, Tan TL, Bixler EO, Martin LF, Shubert D, Kales A. 1994. Sleep apnea and sleep disruption in obese patients. Archives of Internal Medicine 154(15):1705–1711.
Vgontzas AN, Bixler EO, Lin HM, Prolo P, Mastorakos G, Vela-Bueno A, Kales A, Chrousos GP. 2001. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: Clinical implications. Journal of Clinical Endocrinology and Metabolism 86(8): 3787–3794.
Vincent NK, Hameed H. 2003. Relation between adherence and outcome in the group treatment of insomnia. Behavioral Sleep Medicine 1(3):125–139.
Vioque J, Torres A, Quiles J. 2000. Time spent watching television, sleep duration and obesity in adults living in Valencia, Spain. International Journal of Obesity and Related Metabolic Disorders 24(12):1683–1688.
von Kries R, Toschke AM, Wurmser H, Sauerwald T, Koletzko B. 2002. Reduced risk for overweight and obesity in 5- and 6-y-old children by duration of sleep—a cross-sectional study. International Journal of Obesity and Related Metabolic Disorders: Journal of the International Association for the Study of Obesity 26(5):710–716.
Walsh JK, Dement WC, Dinges DF. 2005. Sleep medicine, public policy, and public health. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 648–656.
Walters AS, Hening WA, Kavey N, Chokroverty S, Gidro-Frank S. 1988. A double-blind randomized crossover trial of bromocriptine and placebo in restless legs syndrome. Annals of Neurology 24(3):455–458.
Walters AS, Wagner ML, Hening WA, Grasing K, Mills R, Chokroverty S, Kavey N. 1993. Successful treatment of the idiopathic restless legs syndrome in a randomized double-blind trial of oxycodone versus placebo. Sleep 16(4):327–332.
Walters AS, Hickey K, Maltzman J, Verrico T, Joseph D, Hening W, Wilson V, Chokroverty S. 1996. A questionnaire study of 138 patients with restless legs syndrome: The “night-walkers” survey. Neurology 46(1):92–95.
Walters AS, Winkelmann J, Trenkwalder C, Fry JM, Kataria V, Wagner M, Sharma R, Hening W, Li L. 2001. Long-term follow-up on restless legs syndrome patients treated with opioids. Movement Disorders 16(6):1105–1109.
Weisberg RB, Bruce SE, Machan JT, Kessler RC, Culpepper L, Keller MB. 2002. Non-psychiatric illness among primary care patients with trauma histories and posttraumatic stress disorder. Psychiatry Services 53(7): 848–854.
Weissman MM, Greenwald S, Nino-Murcia G, Dement WC. 1997. The morbidity of insomnia uncomplicated by psychiatric disorders. General Hospital Psychiatry 19(4):245–250.
Weitzman ED, Czeisler CA, Coleman RM, Spielman AJ, Zimmerman JC, Dement W, Richardson G, Pollak CP. 1981. Delayed sleep phase syndrome. A chronobiological disorder with sleep-onset insomnia. Archives of General Psychiatry 38(7):737–746.
Wesensten NJ, Belenky G, Kautz MA, Thorne DR, Reichardt RM, Balkin TJ. 2002. Maintaining alertness and performance during sleep deprivation: Modafinil versus caffeine. Psychopharmacology (Berlin) 159(3):238–247.
Wetter TC, Stiasny K, Winkelmann J, Buhlinger A, Brandenburg U, Penzel T, Medori R, Rubin M, Oertel WH, Trenkwalder C. 1999. A randomized controlled study of pergolide in patients with restless legs syndrome. Neurology 52(5):944–950.
Weyerbrock A, Timmer J, Hohagen F, Berger M, Bauer J. 1996. Effects of light and chronotherapy on human circadian rhythms in delayed sleep phase syndrome: Cytokines, cortisol, growth hormone, and the sleep-wake cycle. Biological Psychiatry 40(8):794–797.
White DP. 2005. Central sleep apnea. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 969–982.
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. 1982. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 215(4537):-1237–1239.
Wills L, Garcia J. 2002. Parasomnias: Epidemiology and management. CNS Drugs 16(12):803–810.
Winkelman JW, Chertow GM, Lazarus JM. 1996. Restless legs syndrome in end–stage renal disease. American Journal of Kidney Disease 28(3):372–378.
Winkelmann J, Schadrack J, Wetter TC, Zieglgansberger W, Trenkwalder C. 2001. Opioid and dopamine antagonist drug challenges in untreated restless legs syndrome patients. Sleep Medicine 2(1):57–61.
Winkelmann J, Muller-Myhsok B, Wittchen HU, Hock B, Prager M, Pfister H, Strohle A, Eisensehr I, Dichgans M, Gasser T, Trenkwalder C. 2002. Complex segregation analysis of restless legs syndrome provides evidence for an autosomal dominant mode of inheritance in early age at onset families. Annals of Neurology 52(3):297–302.
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. 2001. Dopaminergic role in stimulant-induced wakefulness. Journal of Neuroscience 21(5):1787–1794.
Wolfson AR, Carskadon MA. 1998. Sleep schedules and daytime functioning in adolescents. Child Development 69(4):875–887.
Wolkowitz OM, Rubinow D, Doran AR, Breier A, Berrettini WH, Kling MA, Pickar D. 1990. Prednisone effects on neurochemistry and behavior. Preliminary findings. Archives of General Psychiatry 47(10):963–968.
Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH. 2005. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434(7033):640–644.
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. 2005. Obstructive sleep apnea as a risk factor for stroke and death. New England Journal of Medicine 353(19):2034–2041.
Young T, Javaheri S. 2005. Systemic and pulmonary hypertension in obstructive sleep apnea. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier/Saunders. Pp. 1192–1202.
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. 1993. The occurrence of sleep-disordered breathing among middle-aged adults. New England Journal of Medicine 328(17):1230–1235.
Young T, Blustein J, Finn L, Palta M. 1997a. Sleep-disordered breathing and motor vehicle accidents in a population-based sample of employed adults. Sleep 20(8):608–613.
Young T, Evans L, Finn L, Palta M. 1997b. Estimation of the clinically diagnosed proportion of sleep apnea syndrome in middle-aged men and women. Sleep 20(9):705–706.
Young T, Peppard PE, Gottlieb DJ. 2002a. Epidemiology of obstructive sleep apnea: A population health perspective. American Journal of Respiratory and Critical Care Medicine 165(9):1217–1239.
Young T, Shahar E, Nieto FJ, Redline S, Newman AB, Gottlieb DJ, Walsleben JA, Finn L, Enright P, Samet JM, Sleep Heart Health Study Research Group. 2002b. Predictors of sleep-disordered breathing in community-dwelling adults: The Sleep Heart Health Study. Archives of Internal Medicine 162(8):893–900.
Young T, Rabago D, Zgierska A, Austin D, Laurel F. 2003. Objective and subjective sleep quality in premenopausal, perimenopausal, and postmenopausal women in the Wisconsin Sleep Cohort Study. Sleep 26(6):667–672.
Zadra AL, Nielsen TA, Donderi DC. 1998. Prevalence of auditory, olfactory, and gustatory experiences in home dreams. Perceptual and Motor Skills 87(3 Pt 1):819–826.
Zeitzer JM, Dijk DJ, Kronauer R, Brown E, Czeisler C. 2000. Sensitivity of the human circadian pacemaker to nocturnal light: Melatonin phase resetting and suppression. Journal of Physiology 526 (Pt 3):695–702.
Zucconi M, Oldani A, Ferini-Strambi L, Smirne S. 1995. Arousal fluctuations in non-rapid eye movement parasomnias: The role of cyclic alternating pattern as a measure of sleep instability. Journal of Clinical Neurophysiology 12(2):147–154.
Zweig RM, Jankel WR, Hedreen JC, Mayeux R, Price DL. 1989. The pedunculopontine nucleus in Parkinson’s disease. Annals of Neurology 26(1):41–44.


















































































