
3
Kidney Toxicity and Cancer
This chapter reviews information on the effects of trichloroethylene on the kidney, with emphasis on information generated since the U.S. Environmental Protection Agency released its draft health risk assessment on this chemical (EPA 2001b). The review focuses on scientific issues raised during the review process that have relevance in carrying out a human health risk assessment. Studies published before the draft risk assessment are sometimes discussed to provide the context of current knowledge. Noncancer and cancer toxicity are addressed separately; toxic effects of trichloroethylene on the nephron tubule have been proposed to have a role in cancer development, functioning as a promoter. That role is considered later in this chapter.
ROLE OF METABOLISM IN RENAL EFFECTS
Trichloroethylene nephrotoxicity, like that of several haloalkenes, is associated with a multistep metabolic pathway that includes hepatic or renal glutathione S-conjugate formation, enzymatic hydrolysis of the glutathione S-conjugates to cysteine S-conjugates, and renal uptake of cysteine S-conjugates. It is generally accepted that the cysteine S-conjugate S-(1,2-dichlorovinyl)-L-cysteine is the penultimate nephrotoxicant. S-(1,2-Dichlorovinyl)-L-cysteine can undergo bioactivation by renal cysteine S-conjugate β-lyase to reactive species (Figure 3-1), whose reaction with cellular proteins is associated with cell damage and death (Dekant et al. 1987, 1991; Pähler et al. 1999). A second pathway of haloalkene S-conjugates’ bioactivation and toxification involving sulfoxidation of haloalkene cysteine and mercapturic acid conjugates has been identified (Sausen and
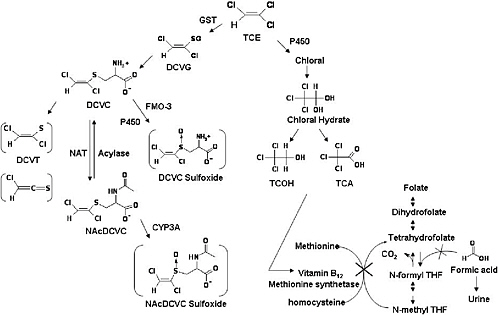
FIGURE 3-1 Composite figure of metabolic pathways relevant to renal toxicity demonstrated in mammalian tissue (see text for references). Abbreviations: DCVC, S-(1,2-dichlorovinyl)-L-cysteine; DCVG, S-(1,2-dichlorovinyl)glutathione; DCVT, 1,2-dichlorovinylthiol; GST, glutathions S-transferase; NAcDCVC, N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine; NAT, N-acetyl transferase; TCA, trichloroacetic acid; TCOH, trichloroethanol; THF, tetrahydrofolate.
Elfarra 1991; Park et al. 1992; Lash et al. 1994; Werner et al. 1995a,b, 1996; Birner et al. 1998). Sulfoxidation of haloalkyl cysteine S-conjugates can constitute a toxification independent of β-lyase-mediated bioactivation (Lash et al. 1994; Werner et al. 1995a,b, 1996; Birner et al. 1998). Lash et al. (2000a,b) extensively reviewed biotransformation and bioactivation of trichloroethylene. Since then, there have been additional investigations of the renal metabolism and effects of trichloroethylene, some with a focus on sulfoxidation, as well as the sulfoxidation and toxicity of other haloalkyl nephrotoxicants (see below).
The sulfoxidation and toxicity of trichloroethylene S-conjugates (involving hepatic or kidney microsomal sulfoxidation of cysteine and mercapturic acid conjugates) have been clearly established (Sausen and Elfarra 1991; Lash et al. 1994; Werner et al. 1996; Krause et al. 2003; Lash et al. 2003). The first report of enzymatic trichloroethylene S-conjugate sulfoxidation was by Ripp et al. (1997), who demonstrated rabbit liver microsomal sulfoxidation of S-(1,2-dichlorovinyl)-L-cysteine. Sulfoxidation was cata-
lyzed mainly by flavin monooxygenase, rather than by cytochrome P-450 (CYP450), and was specific for rabbit flavin monooxygenase-3 (Ripp et al. 1997). S-(1,2-Dichlorovinyl)-L-cysteine sulfoxidation was also catalyzed by human flavin monooxygenase-3 but not by other isoforms of flavin monooxygenase (Krause et al. 2003). Human liver microsomes also catalyzed S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation (Krause et al. 2003). Sulfoxidation was not detected with human kidney microsomes, although only one kidney sample was evaluated (Krause et al. 2003). The lack of metabolism was attributed to the low and variable concentrations of flavin monooxygenase-3 expression in kidney, which ranged from trace amounts to 1.3 pg/mg protein, compared with liver (Krause et al. 2003). S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide, whether formed in the liver and translocated to the kidney or potentially formed renally in situ, was considered to play a possible role in trichloroethylene nephrotoxicity (Krause et al. 2003). The mercapturic acid conjugates of dichlorovinyl cysteine, N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(2,2-dichlorovinyl)-L-cysteine, also undergo sulfoxidation, as shown for rat liver microsomes (Werner et al. 1996). Unlike S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation, S-(1,2-dichlorovinyl)-L-cysteine mercapturate sulfoxidation was catalyzed mainly if not exclusively by CYP450, and a role for flavin monooxygenase was excluded. Specifically, rat liver microsomal N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation was catalyzed predominantly by CYP3A isoforms (Werner et al. 1996).
Haloalkyl S-conjugates undergo sulfoxidation primarily in the liver. S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide was quantified after S-(1,2-dichlorovinyl)-L-cysteine incubation with microsomes from human liver but was not detected in microsomes from human kidney (Krause et al. 2003). Sulfoxidation of both S- and N-acetyl cysteine conjugates of cis- and trans-1,3-dichloropropene was detected in pig liver but not in rat kidney microsomes (Park et al. 1992). Nevertheless, mercapturate sulfoxidation by human kidney microsomes has been observed, albeit at rates much slower than for liver microsomes (Altuntas et al. 2004). Whether microsomes from human liver or kidney catalyze the sulfoxidation of N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine, and the relative activities, remains unknown.
In general, S-conjugate sulfoxidation might be mediated by CYP or by flavin monooxygenase. For example, sulfoxidation of S-allyl-L-cysteine and S-benzyl-L-cysteine and, at a lower rate, S-(1,2-dichlorovinyl)-L-cysteine and S-(1,2,2-trichlorovinyl)-L-cysteine, was catalyzed by flavin monooxygenases (Ripp et al. 1997; Krause et al. 2003). In contrast, sulfoxidation of N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine, N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine, N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine, N-acetyl-S-(2,2-dichlorovinyl)-L-cysteine, N-acetyl-S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine, and N-acetyl-S-(1-fluoro-
2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine was catalyzed mainly by CYP450 (Werner et al. 1995a,b, 1996; Altuntas et al. 2004). The relative contribution of CYP450 and flavin monooxygenase toward cysteine S-conjugate S-oxidation depends on the conjugate structure. Generally, nucleophilic sulfur atoms are oxidized preferentially by flavin monooxygenase, whereas nonnucleophilic sulfur atoms are oxidized preferentially by CYP450 (Ripp et al. 1997; Damani and Houdi 1988). Thus, cysteine conjugates with more nucleophilic sulfur atoms (S-allyl-L-cysteine, S-benzyl-L-cysteine) were much better microsomal flavin monooxygenase substrates in human kidney and liver and in rabbit liver than were those with less nucleophilic sulfur atoms (the various haloalkyl cysteine and mercapturic acid conjugates) (Ripp et al. 1997; Krause et al. 2003). This is likely because the sulfur atoms of allyl and benzyl compounds are more nucleophilic than that of vinyl compounds and because flavin monooxygenases tend to oxidize strong nucleophiles (Damani and Houdi 1988). Lipophilicity might also affect haloalkene S-conjugate sulfoxidation by flavin monooxygenase. S-Benzyl-L-cysteine is relatively lipophilic, with a nucleophilic sulfur atom, and has been shown to be a selective substrate for flavin monooxygenase (Sausen et al. 1993). N-Acetyl-S-(1-fluoro-2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine has a vinylic sulfur atom as well as strong electron-withdrawing fluorine atoms, which make the sulfur atom much less nucleophilic than those of S-allyl-L-cysteine, S-benzyl-L-cysteine, S-(1,2-dichlorovinyl)-L-cysteine, and S-(1,2,2-trichlorovinyl)-L-cysteine. N-Acetyl-S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine and N-acetyl-S-(1-fluoro-2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine are less lipophilic than S-benzyl-L-cysteine, rendering them theoretically less susceptible to flavin monooxygenase sulfoxidation, potentially also explaining the lack of flavin monooxygenase activity toward their sulfoxidation.
Rat liver microsomal N-acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-2,2-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation was catalyzed predominantly, if not exclusively, by CYP3A isoforms (Werner et al. 1996). This conclusion was based on induction of sulfoxidation by phenobarbital and dexamethasone, inhibition by troleandomycin, and correlation with CYP3A activity. Indeed, CYP3A has been shown to be the predominant CYP isoform catalyzing the rat or human liver microsomal sulfoxidation of all haloalkyl mercapturic acid conjugates studied to date, including N-acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine, N-acetyl-2,2-S-(1,2-dichlorovinyl)-L-cysteine, N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine, N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine, N-acetyl-S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine, and N-acetyl-S-(1-fluoro-2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine (Werner et al. 1995a,b, 1996; Altuntas et al. 2004), which has been confirmed with cDNA-expressed CYP450s (Werner et al. 1995b; Altuntas et al. 2004). The role of CYP3A
in sulfoxidation, together with the polymorphic expression of CYP3A5 in humans, raises the possibility of pharmacogenetic differences in sulfoxidation and hence toxicity in persons exposed to trichloroethylene. Indeed, sulfoxidation of N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine, N-acetyl-S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine, and N-acetyl-S-(1-fluoro-2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine was also catalyzed by expressed CYP3A5 as well as by CYP3A4 (Werner et al. 1995b; Altuntas et al. 2004). These studies showing CYP3A-catalyzed mercapturate sulfoxidation were performed in vitro. The first evidence for the role of CYP3A in any S-conjugate sulfoxidation in rats in vivo was recently obtained with a related haloalkene (Sheffels et al. 2004).
Sulfoxidation of trichloroethylene S-conjugates can constitute a toxification pathway independent of β-lyase-mediated bioactivation (Sausen and Elfarra 1991; Lash et al. 1994; Werner et al. 1995a,b, 1996; Birner et al. 1998). Sulfoxides of trichloroethylene S-conjugates are stable but can react readily with nonprotein thiols. Thus, S-(1,2-dichlorovinyl)-L-cysteine sulfoxide and N-acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide react spontaneously with glutathione as an electrophile and Michael acceptor (Sausen and Elfarra 1991; Ripp et al. 1997; Rosner and Dekant 1999). N-Acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide reactivity is greater than that of other mercapturate sulfoxides, including those of N-acetyl-2,2-S-(1,2-dichlorovinyl)-L-cysteine, S-(1,2,2-trichlorovinyl)-L-cysteine sulfoxide, and N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine, which react only slowly or require bioactivation by glutathione S-transferase for conjugate formation (Ripp et al. 1997; Rosner et al. 1998; Rosner and Dekant 1999).
Toxicity of the S-conjugate sulfoxides of trichloroethylene, and other haloalkenes, has been evaluated in vitro and in vivo. Qualitatively, S-(1,2-dichlorovinyl)-L-cysteine sulfoxide replicated the rat renal tubular cell injury also caused by S-(1,2-dichlorovinyl)-L-cysteine (Lash et al. 1994). Quantitatively, S-(1,2-dichlorovinyl)-L-cysteine sulfoxide was significantly more nephrotoxic than S-(1,2-dichlorovinyl)-L-cysteine to isolated rat distal, but not proximal, tubular cells in vitro (Lash et al. 1994). Like S-(1,2-dichlorovinyl)-L-cysteine sulfoxide, the sulfoxide of the mercapturate N-acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine was significantly more cytotoxic than equivalent concentrations of 1,2-S-(1,2-dichlorovinyl)-L-cysteine in rat renal proximal tubular cells (Werner et al. 1996). Greater cytotoxicity in rat renal tubular cells of N-acetyl-2,2-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide, N-acetyl-S-(1,2,2-trichlorovinyl)-L-cysteine sulfoxide, and N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine compared with their corresponding mercapturic acids was also observed (Birner et al. 1995; Werner et al. 1996). In rats in vivo, S-(1,2-dichlorovinyl)-L-cysteine sulfoxide caused the same type of renal proximal tubular cell histologic changes
as trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine (Lash et al. 1994). S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide, however, was significantly more nephrotoxic than S-(1,2-dichlorovinyl)-L-cysteine at equivalent doses (Lash et al. 1994). A recent investigation evaluated the effects of S-(1,2-dichlorovinyl)-L-cysteine sulfoxide on human renal proximal tubular cells (Lash et al. 2003). S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide caused obvious morphologic abnormalities and cellular necrosis at concentrations as low as 10 µM. S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide also caused apoptosis. Apoptosis occurred rapidly and at low toxic concentrations, whereas necrosis occurred at later incubation times and at higher sulfoxide concentrations. Compared with S-(1,2-dichlorovinyl)-L-cysteine, S-(1,2-dichlorovinyl)-L-cysteine sulfoxide caused greater and more rapid depletion of both ATP and cellular glutathione than S-(1,2-dichlorovinyl)-L-cysteine. Less apoptosis was observed with S-(1,2-dichlorovinyl)-L-cysteine sulfoxide than with S-(1,2-dichlorovinyl)-L-cysteine, which was attributed to the more rapid depletion of ATP. These results suggested a role for both S-(1,2-dichlorovinyl)-L-cysteine and S-(1,2-dichlorovinyl)-L-cysteine sulfoxide in human renal tubular cell toxicity.
Other haloalkyl mercapturate sulfoxides demonstrate similar characteristics. N-Acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine sulfoxide was significantly nephrotoxic in rats in vivo (Birner et al. 1998). More recently, the effects of the cysteine-S-, mercapturic acid, and corresponding sulfoxide conjugates of the nephrotoxicant fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl ether were compared in human proximal tubular cells (Altuntas et al. 2003). Both S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine sulfoxide and (Z)-N-acetyl-S-(1-fluoro-2-fluoromethoxy-2-[trifluoromethyl]vinyl)-L-cysteine sulfoxide caused greater cytotoxicity than the corresponding equimolar cysteine conjugates.
Toxicity of trichloroethylene S-conjugate sulfoxides occurs via a mechanism independent of β-lyase. Whereas the β-lyase inhibitor aminooxyacetic acid partially protected against S-(1,2-dichlorovinyl)-L-cysteine renal toxicity in vitro and in vivo, it failed to protect against S-(1,2-dichlorovinyl)-L-cysteine sulfoxide toxicity in both settings (Lash et al. 1994). Similarly, N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide and N-acetyl-S-(2,2-dichlorovinyl)-L-cysteine sulfoxide toxicities also were not blocked by aminooxyacetic acid (Werner et al. 1996). The α-methyl analog of N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine sulfoxide, which is not a substrate for renal β-lyase, also caused renal tubular necrosis in rats in vivo (Birner et al. 1998). S-(1,2-Dichlorovinyl)-L-cysteine sulfoxidation has been proposed as a mechanism to explain the observation that the D- and L- isomers of S-(1,2-dichlorovinyl)-L-cysteine are nearly equally nephrotoxic in rats, yet only the L-isomer is a substrate for β-lyase (Sausen and Elfarra 1991). Thus, both β-lyase-dependent metabolism of cysteine S-conjugates,
and CYP450- or flavin monooxygenase-dependent sulfoxidation of cysteine S-conjugates or their mercapturates, can contribute to the bioactivation and renal toxicity of trichloroethylene and other haloalkenes.
Several questions remain unaddressed, the answers to which might have important implications for human trichloroethylene biotransformation, toxification, and individual susceptibility. Sulfoxides are more potent nephrotoxicants than their parent S-conjugates. Whereas rat liver microsomes catalyze S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation, and human liver microsomes catalyze S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation, whether human liver microsomes form N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxides remains unknown. The enzymes responsible for human liver (and kidney, if extant) S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation, and particularly the role of CYP3A4 and CYP3A5, remain unknown. Interindividual variability in human S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation remains unknown. CYP3A5 is polymorphic for high expression in Caucasian (30%); Japanese (30%); Chinese (40%); and African American, Southeast Asian, Pacific Islander, and Southwestern American Indian (50%) populations (Hustert et al. 2001; Kuehl et al. 2001; see OMIM 2006a). Assuming that, like rat CYP3A, human CYP3A catalyzes these reactions, together with human CYP3A5 polymorphic expression, suggests that the potential exists for pharmacogenetic differences in sulfoxidation and hence susceptibility to toxicity. This remains unknown, as does the ability of human kidney (which constitutively expresses CYP3A as the major CYP isoform) to catalyze N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation.
More fundamentally, the existence of S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxidation in vivo has not been documented either in rats or in humans. Myriad investigations of in vivo trichloroethylene disposition in rodents and humans after controlled as well as occupational exposure (Birner et al. 1993; Bernauer et al. 1996; Bruning et al. 1998; Bloemen et al. 2001) were evaluated, including one with 10 metabolites of S-(1,2-dichlorovinyl)-L-cysteine (Bloemen et al. 2001); none evaluated the potential existence of trichloroethylene S-conjugates sulfoxides in urine. Similarly, little is known about sulfoxidation in vivo for any nephrotoxic haloalkene. Only two reports have evaluated sulfoxidation in vivo. In rats, N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-L-cysteine sulfoxide was qualitatively identified in urine after administration of hexachlorobutadiene but was not quantified (Birner et al. 1995). N-Acetyl-S-(1,1-difluoro-2-fluoromethoxy-2-[trifluoromethyl]ethyl)-L-cysteine sulfoxide was identified and quantified in the urine of rats given fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl ether (Sheffels et al. 2004). Although sulfoxidation was apparently a quantitatively small fraction of
the overall metabolism, it appeared to be a toxicologically significant route of biotransformation of fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl ether and its S-conjugates. S-(1,2-Dichlorovinyl)-L-cysteine sulfoxide and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide were shown to be formed by rodent liver microsomes (Werner et al. 1996; Ripp et al. 1997) and to be highly reactive renal tubular cell nephrotoxicants in rats in vitro and in vivo (Sausen and Elfarra 1991; Lash et al. 1994, 2003; Rosner and Dekant 1999) and were proposed as important determinants of trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine nephrotoxicity (Krause et al. 2003; Lash et al. 2003); yet no published studies have evaluated S-conjugate sulfoxidation from trichloroethylene or S-(1,2-dichlorovinyl)-L-cysteine, either in rats or in humans, or the toxicologic significance of the conjugates. Such studies may be complicated, however, by the reactivity of S-(1,2-dichlorovinyl)-L-cysteine sulfoxide and N-acetyl-1,2-S-(1,2-dichlorovinyl)-L-cysteine sulfoxide (Sausen and Elfarra 1991; Ripp et al. 1997; Rosner and Dekant 1999), which might therefore not be excreted unchanged. For example, when rats were administered S-(1,2-dichlorovinyl)-L-cysteine sulfoxide, the glutathione conjugate of this sulfoxide was excreted in bile (Sausen and Elfarra 1991; Rosner and Dekant 1999). Therefore, measurement of the glutathione conjugate of S-(1,2-dichlorovinyl)-L-cysteine sulfoxide or its metabolites may provide a method to assess S-(1,2-dichlorovinyl)-L-cysteine sulfoxide formation after trichloroethylene or S-(1,2-dichlorovinyl)-L-cysteine exposure in vivo.
NONCANCER TOXICITY
Animal Studies
Tubular Toxicity
Trichloroethylene has been shown to cause toxicity to renal tubules in bioassay studies, and mechanisms of this toxicity have been pursued in experimental studies. Lash et al. (2000b) reviewed mechanistic studies and those will not be recapitulated here. The committee directed its efforts to studies since that review.
Trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine are toxic to primary cultures of rat proximal and distal tubular cells (Cummings et al. 2000). Glutathione-related enzyme activities were well maintained in the cells, whereas CYP activities were not. The response to S-(1,2-dichlorovinyl)-L-cysteine was greater than the response to trichloroethylene; however, the proximal and distal tubule cells had similar responses even though the proximal tubule is the target in vivo. The authors attributed this to the fact that the proximal tubule is exposed before the distal tubule in vivo and to
possible differences in uptake transporters. They did not address the extent to which transporters were maintained in the cultured cells.
The same group also assessed the toxicity of trichloroethylene and its metabolites S-(1,2-dichlorovinyl)-L-cysteine and S-(1,2-dichlorovinyl)glutathione using in vitro techniques (Lash et al. 2001b). Their goal was to determine whether in vitro techniques are valid indicators of species-, sex-, and tissue-related differences in sensitivity. Experiments using isolated cells were performed only with tissues from Fischer 344 rats, and lactate dehydrogenase release was used as the measure of cellular toxicity. The effects were greater in males. S-(1,2-Dichlorovinyl)-L-cysteine and trichloroethylene had similar effects, but S-(1,2-dichlorovinyl)glutathione exhibited increased efficacy compared with trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine. Mitochondrial toxicity was assessed in both Fischer 344 rats and B6C3F1 mice. Renal mitochondria from male rats and mice responded similarly; a greater effect was seen in female mice. Thus, although these studies show S-(1,2-dichlorovinyl)-L-cysteine to be more toxic than trichloroethylene and S-(1,2-dichlorovinyl)glutathione, the magnitude of the effect was not much different and species differences are not consistent with the effects observed in long-term bioassays. This suggests that in vitro data be used with caution in risk assessment, being mindful that in vitro experiments avoid in vivo pharmacokinetic and metabolic processes.
In LLC-PK1 cells, S-(1,2-dichlorovinyl)-L-cysteine causes loss of mitochondrial membrane potential, mitochondrial swelling, release of cytochrome c, caspase activation, and apoptosis (Chen et al. 2001). Thus, S-(1,2-dichlorovinyl)-L-cysteine is toxic to mitochondria, resulting in either apoptosis or necrosis. S-(1,2-Dichlorovinyl)-L-cysteine-induced apoptosis also has been reported in primary cultures of human proximal tubule cells (Lash et al. 2001a).
Korrapati et al. (2005) builds upon a series of investigations of hetero-(by HgCl2) and homo-(by S-(1,2-dichlorovinyl)-L-cysteine, 15 mg/kg) protection against a lethal dose of S-(1,2-dichlorovinyl)-L-cysteine (75 mg/kg), in which priming, or preconditioning, was said to augment and sustain cell division and tissue repair, hence protecting against the subsequent lethal S-(1,2-dichlorovinyl)-L-cysteine dose (Vaidya et al. 2003a,b,c). Korrapati et al. (2005) showed that a lethal dose of S-(1,2-dichlorovinyl)-L-cysteine downregulates phosphorylation of endogenous retinoblastoma protein (pRb), which is considered critical in renal proximal tubular and mesangial cells for the passage of cells from G1 to S-phase, thereby leading to a block of renal tubule repair. Priming, in contrast, upregulated P-pRb which was sustained even after the administration of a lethal dose of S-(1,2-dichlorovinyl)-L-cysteine, thereby stimulating S-phase DNA synthesis, which was concluded to result in tissue repair and recovery from acute renal failure
and death. While these studies are indeed fascinating, they inform more on the mechanism of autoprotection rather than on the mechanism of initial injury caused by S-(1,2-dichlorovinyl)-L-cysteine. In addition, the priming injury (not innocuous, as it caused 25-50% necrosis and elevated blood urea nitrogen) may have influenced the toxicokinetics of the second S-(1,2-dichlorovinyl)-L-cysteine injection. This remains unknown.
Mensing et al. (2002) reported on the nephrotoxicity of trichloroethylene in male Long Evans rats after 6 months of inhalation exposure (500 ppm). Results were expressed relative to urine creatinine to account for individual differences in urine volume that can affect the concentration of urine constituents. Urinary excretion of albumin was not affected (although the high end of the range was about twice that of the control group) and high-molecular-weight proteins showed an upward trend but were not significantly increased (creatinine at 36 mg/g [4-81 mg/g] versus 41 mg/g [not detected-215 mg/g]). Increased excretion of low-molecular-weight proteins and N-acetylglucosaminidase was noted. The increase in N-acetylglucosaminidase was small (8.4 units [U]/g [5.7-8.9 U/g] versus 9.7 U/g [not detected-12.4 U/g); the increase in low-molecular-weight proteins was 332 U/g (176-659 U/g) versus 637 U/g (293-1,910 U/g). The histopathology description does not mention tubular damage, whereas interstitial infections and glomerulonephritis are described for the treated group.
Proteinuria has long been recognized as a sign of kidney damage, and it is a reliable predictor of ultimate outcome; more recently, it has been recognized that an elevated filtered load of protein is damaging to tubules (Verhave et al. 2004; Zandi-Nejad et al. 2004). Proteinuria can be characterized as glomerular, tubular, or mixed, based on the causal defect. Proteins less than about 40 kDa or 30 Å are readily filtered at the glomerulus, and are reabsorbed in the proximal tubule. Proteins larger than 100 kDa or 55 Å are not filtered. Albumin is considered an intermediate-sized protein that is normally filtered sparingly, largely because of its negative charge being repulsed by a fixed negative charge in the glomerular barrier. The glomerular pattern is excretion of high-molecular-weight proteins, such as IgG, and indicates increased permeability or decreased selectivity of the glomerular barrier. Damage to the proximal tubule impairs reabsorption of low-molecular-weight proteins; thus, a tubular pattern is one that has increased excretion of albumin and low-molecular-weight proteins, such as α1-microglobulin. N-Acetylglucosaminidase is a lysosomal protein released by tubules during processing of filtered protein. Increased amounts of N-acetylglucosaminidase are expected when the tubules are presented with elevated amounts of protein, and thus it is an indicator of protein load. Elevated urinary N-acetylglucosaminidase is not an index of cell death, as is release of alkaline phosphatase from cultured cells or release of trans-
aminase enzymes from liver cells into the plasma. However, elevation of N-acetylglucosaminidase in urine is a sign of proteinuria, which is a sign of kidney malfunction (Zandi-Nejad et al. 2004).
While Mensing et al. (2002) did not report tubular toxicity, the urinary protein profile is consistent with impairment of tubule reabsorption of filtered protein and perhaps increased glomerular permeability to proteins.
Role of Formic Acid in Trichloroethylene Nephrotoxicity
Some investigators (Green et al. 1998, 2003; Dow and Green 2000) have proposed that the mode of trichloroethylene nephrotoxicity is related to formic acid. They demonstrated that exposure to either trichloroethanol or trichloroacetic acid causes increased formation and urinary excretion of formic acid (Green et al. 1998). The formic acid does not come from trichloroethylene (Figure 3-1). Rather, trichloroethylene (or a metabolite) causes a functional depletion of vitamin B12, which is required for the methionine salvage pathway of folate metabolism. Vitamin B12 depletion results in folate depletion. Folate is a cofactor in one-carbon metabolism and depletion of folate allows formic acid to accumulate, and then to be excreted in the urine (Dow and Green 2000).
The effects of trichloroethanol-induced formic acid accumulation were determined in a 1-year chronic toxicity study in male Fischer 344 rats (Green et al. 2003). Trichloroethanol was administered in drinking water to achieve a urine formic acid concentration similar to that found in rats exposed by inhalation to trichloroethylene at 500 parts per million (ppm). The pathology of formic acid (induced by trichloroethanol administration) is initially increased tubular basophilia and hyaline drop accumulation (12-16 weeks) followed by tubular degeneration at 40 weeks (“increased cellular eosinophilia, tubular vacuolation and intratubular cast formation”) and an increased amount of pigmentation in the S2 portion of the proximal tubules and hyaline droplet accumulation. At 52 weeks, hyaline droplet and tubular degeneration were not found, but increased tubular pigmentation was observed. It was also noted that foci of “atypical” tubular hyperplasia occurred in two of the trichloroethanol-treated rats. The authors stated these changes were consistent with the nephrotoxicity seen in the 2-year cancer bioassays.
Results from the National Toxicology Program’s 2-year cancer bioassays of trichloroethylene administered by gavage to rats and mice are provided in Tables 3-1 and 3-2. Nonneoplastic kidney lesions were found in all animals dosed for 2 years, including mice that did not develop kidney cancer (NTP 1988). In rats, both studies noted cytomegaly and karyomegaly of tubular cells in the area of the corticomedullary border (specified as pars recta by NTP [1990], which is situated in the corticomedullary region).
TABLE 3-1 Summary of Renal Toxicity and Tumor Findings in Gavage Studies of Trichloroethylene by NTP (1990)
|
Sex |
Dose (mg/kg)a |
Cytomegaly and Karyomegaly |
Tumor Incidence (overall, survival) |
|
|
Incidence |
Severityb |
|||
|
13-wk study, F344/N rats |
||||
|
Male |
0, 125, 250, 500, 100 |
Tissues not evaluated |
— |
None reported in this study |
|
|
2,000 |
8/9 |
Minimal/mild |
|
|
Female |
0, 62.5, 125, 250, 500 |
Tissues not evaluated |
— |
|
|
|
1,000 |
5/10 |
Equivocal/minimal |
|
|
13-wk study, B6C3F1mice |
||||
|
Male |
0, 375, 750, 1,500 |
Tissues not evaluated |
— |
None reported in this study |
|
|
3,000 |
7/10c |
Mild/moderate |
|
|
|
6,000 |
—d |
— |
|
|
Female |
0, 375, 750, 1,500 |
Tissues not evaluated |
— |
|
|
|
3,000 |
9/10 |
Mild/moderate |
|
|
|
6,000 |
1/10 |
Mild/moderate |
|
|
103-wk study, F344/N rats |
||||
|
Male |
0 |
0% |
0 |
0/48; 0/33 |
|
|
500 |
98% |
2.8 |
0/49; 0/20 |
|
|
1,000 |
98% |
3.1 |
3/49; 3/16e |
|
Female |
0 |
0% |
0 |
0/50 |
|
|
500 |
100% |
1.9 |
0/49 |
|
|
1,000 |
100% |
2.7 |
1/48 |
|
103-wk study, B6C3F1mice |
||||
|
Male |
0 |
0% |
0 |
1/49 |
|
|
1,000 |
90% |
1.5 |
1/50 |
|
Female |
0 |
0% |
0 |
None |
|
|
1,000 |
98% |
1.8 |
None |
|
aCorn oil vehicle. bNumerical scores reflect the average grade of the lesion in each group (1, slight; 2, moderate; 3, well marked; and 4, severe). cObserved in four mice that died after 7-13 wk and in three that survived the study. dAll mice died during the first week. eP = 0.028. |
||||
Cytomegaly and karyomegaly were seen early in the bioassays and there were signs of these changes in the 13-week study (NTP 1988) (which were noted on reexamination of the slides after changes were seen in the 2-year bioassay); cytomegaly was noted at 26 weeks in (NTP 1990). Kidneys with more extensive damage had similar changes in cortical area. Both reports noted additional lesions: dilation of tubules and loss of tubular cells lining the basement membrane (“stripped appearance” [NTP 1988] or flattening
TABLE 3-2 Summary of Toxicity and Tumor Findings in Gavage Studies of Trichloroethylene by NTP (1988)
|
Sex |
Dose (mg/kg)a |
Cytomegaly |
Toxic Nephropathy |
Adenoma |
Adenocarcinoma |
|
2-yr study, ACI rats |
|||||
|
Male |
0 |
0/50 |
0/50 |
0/50 |
0/50 |
|
|
500 |
40/49 (82%) |
18/49 (37%) |
0/49 |
1/49 |
|
|
1,000 |
48/49 (98%) |
18/49 (37%) |
0/49 |
0/49 |
|
Female |
0 |
0/48 |
0/48 |
0/48 |
0/48 |
|
|
500 |
43/47 (91%) |
21/47 (45%) |
2/47 |
1/47 |
|
|
1,000 |
42/43 (98%) |
19/43 (44%) |
0/43 |
1/43 |
|
2-yr study, August rats |
|||||
|
Male |
0 |
0/50 |
0/50 |
0/50 |
0/50 |
|
|
500 |
46/50 (92%) |
10/50 (20%) |
1/50 |
1/50 |
|
|
1,000 |
46/49 (94%) |
31/49 (63%) |
1/49 |
0/49 |
|
Female |
0 |
0/49 |
0/49 |
1/49 |
0/49 |
|
|
500 |
46/48 (96%) |
8/48 (17%) |
2/48 |
2/48 |
|
|
1,000 |
50/50 (100%) |
29/50 (58%) |
0/50 |
0/50 |
|
2-yr study, Marshall rats |
|||||
|
Male |
0 |
0/49 |
0/49 |
0/49 |
0/49 |
|
|
500 |
48/50 (96%) |
18/50 (36%) |
1/50 |
0/50 |
|
|
1,000 |
47/47 (100%) |
23/47 (49%) |
0/47 |
1/47 |
|
Female |
0 |
0/50 |
0/50 |
1/50 |
0/50 |
|
|
500 |
46/48 (96%) |
30/48 (63%) |
1/48 |
1/48 |
|
|
1,000 |
43/44 (98%) |
30/44 (68%) |
0/44 |
1/44 |
|
2-yr study, Osborne-Mendel rats |
|||||
|
Male |
0 |
0/50 |
0/50 |
0/50 |
0/50 |
|
|
500 |
48/50 (96%) |
39/50 (78%) |
6/50 |
0/50 |
|
|
1,000 |
49/50 (98%) |
35/50 (70%) |
1/50 |
1/50 |
|
Female |
0 |
0/50 |
0/50 |
0/50 |
0/50 |
|
|
500 |
48/50 (96%) |
30/50 (60%) |
0/50 |
0/50 |
|
|
1,000 |
49/49 (100%) |
39/49 (80%) |
1/49 |
0/49 |
of these cells [NTP 1990]). This toxic nephropathy was infrequent before 52 weeks but then increased in severity with longer exposure. Only NTP (1990) commented on intratubular material and noted that the tubules were empty or “contained wisps of eosinophilic material.”
Maltoni et al. (1988) reported cancer bioassays after inhalation exposure of Sprague-Dawley rats and Swiss and B6C3F1 mice to trichloroethylene (see Table 3-3). No renal effects were reported for mice, but renal adenocarcinomas were found in male rats at the high dose (600 ppm) at 2 years. Male rats also experienced cytokaryomegaly or megalonucleocytosis
TABLE 3-3 Summary of Toxicity and Tumor Findings in Inhalation Studies of Trichloroethylene by Maltoni et al. (1988)
|
Sex |
Concentration (ppm) |
Megalonucleocytosis |
Renal Adenocarcinoma |
|
2-yr study, Sprague-Dawley rats |
|||
|
Male |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
300 |
16.9% |
— |
|
|
600 |
77.7% |
3.1% |
|
Female |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
300 |
— |
— |
|
|
600 |
— |
0.7% |
|
78-wk study, Swiss mice |
|||
|
Male |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
600 |
— |
— |
|
Female |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
600 |
— |
— |
|
78-wk study, B6C3F1mice |
|||
|
Male |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
600 |
— |
— |
|
Female |
0 |
— |
— |
|
|
100 |
— |
— |
|
|
600 |
— |
— |
(77% of the high-dose group and 17% of the medium-dose group (300 ppm). There was no indication of pathology at earlier times.
The lesions due to formic acid (induced by trichloroethanol administration) and trichloroethylene exposure differ in the nature and the time course of the lesions. They are similar in that the same region of the kidney is affected. However, that region of the kidney is most often affected by nephrotoxic chemicals and by hypoxia and ischemia. Green et al. (2003) did not observe flattening or loss of tubular epithelial cells nor did they report tubular dilation. Hyaline droplets and tubular degeneration were found at 40 weeks, but not at 52 weeks, which is when tubular degeneration (albeit with different characteristics) was noted in the cancer bioassays. Toxic nephropathy was infrequent before 52 weeks, but then increased in severity with longer exposure (NTP 1990). Intratubular cast formation was noted as part of the tubular degeneration following exposure to formic acid (produced by trichloroethanol exposure), but with trichloroethylene exposure
tubules were described as empty or containing “wisps” of material. Because dosing with trichloroethanol was selected to achieve the concentrations observed after exposure to daily inhalation of trichloroethylene at 500 ppm, similar to that used in the Maltoni et al. (1988) study, it is noteworthy that the histopathologic descriptions of the Maltoni et al. study differ from those of the Green et al. (2003) study.
Dow and Green (2000) noted that trichloroacetic acid also induced formic acid accumulation in rats. If formic acid is the actual trichloroethylene nephrotoxicant, then trichloroacetic acid would be expected to cause similar pathology. Mather et al. (1990) reported an increase of kidney-weight to body-weight ratio in rats after 90 days of exposure to trichloroacetic acid in drinking water at 5,000 ppm but reported no histopathologic changes in the kidney. DeAngelo et al. (1997) reported no effects of trichloroacetic acid on kidney weight or histopathology in rats in a 2-year cancer bioassay. The amount of formic acid produced after administration of trichloroethanol or trichloroacetic acid in drinking water was similar at 2-4 weeks (about 20 mg/day for each compound) and was the same at the two doses used (1 and 5 g/L for trichloroacetic acid and 0.5 and 1.0 g/L for trichloroethanol). The studies with trichloroethanol were carried out for a longer time and excretion of formic acid at the high dose increased to about 60 mg/day. However, because the dose-response relationship was lost, folate was added to the regime of the low-dose animals, and this decreased their formic acid excretion. The formic acid exposure in the groups exposed to trichloroacetic acid at 5,000 ppm in the Mather et al. (1990) and DeAngelo et al. (1997) studies would be similar to that of the group treated at 1 g/L in the Green et al. (2003) study.
In summary, on the basis of dissimilarities between the pathologic responses of the kidney to formic acid and trichloroethylene and the lack of nephrotoxicity from trichloroacetic acid, which also results in formic acid production, it is difficult to accept formic acid formation as a mechanism or mode of action for trichloroethylene.
α2µ-Globulin Accumulation
There is no evidence that trichloroethylene induces α2µ-globulin accumulation (Goldsworthy et al. 1988), and the histopathologic effects of trichloroethylene are not consistent with that histopathology (EPA 1991). Trichloroethanol was recently reported to cause hyaline droplet accumulation and an increase in α2µ-globulin accumulation that was insufficient to account for the hyaline droplet nephropathy (Green et al. 2003). Similar to tubular damage, the hyaline droplet accumulation was seen at 40 weeks but not at 52 weeks.
Peroxisome Proliferation
The role of peroxisome proliferation as a mode of action of trichloroethylene was considered in the review by Lash et al. (2000b). They concluded that the published literature does not support peroxisome proliferation as a mode of action for renal carcinogenesis from trichloroethylene or its metabolites.
Human Studies
Tubular Toxicity
Trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine have been shown to be toxic to fresh human proximal tubule cells (Cummings and Lash 2000) and S-(1,2-dichlorovinyl)-L-cysteine is toxic to cultures of human proximal tubule cells (Lash et al. 2001a). S-(1,2-Dichlorovinyl)-L-cysteine produced necrosis, apoptosis, and an increase in the percentage of cells in S phase, an indication of cell proliferation. The authors noted that effects were observed in the 10 to 100 µM range, judged to be occupationally relevant because the concentration of S-(1,2-dichlorovinyl)glutathione in the blood is 45 µM after 4 hours of exposure at 100 ppm.
Biological monitoring of persons who previously experienced “high” exposures to trichloroethylene (100-500 ppm) in the workplace has been performed. These studies have used generalized proteinuria and urinary excretion of specific tubular proteins as an index of tubular toxicity. Brüning et al. (1999a) reported results supporting nephrotoxicity in kidney cancer patients. They compared the highly trichloroethylene-exposed group with nonexposed renal cancer patients and with healthy, unexposed controls. Both renal cancer groups were about 2.5 years postnephrectomy. In the renal cancer groups, 95% (39 of 41) of trichloroethylene-exposed patients had elevated proteinuria, 85% had tubular proteinuria, 7% had a combined pattern, and 2% had glomerular proteinuria. In comparison, only 44% (22 of 50) of the nonexposed renal cancer patients had tubular proteinuria and 2% had a combined pattern (54% had no proteinuria). The trichloroethyleneexposed patients had elevated excretion of α1-microglobin compared with the nonexposed renal cell cancer patients. The authors concluded that their results support an initiation-promotion model, in which the repeated toxicity, evidenced by increased incidence of proteinuria, serves as the promoter for the genotoxic1 metabolites produced via the glutathione pathway.
Bolt et al. (2004) measured α1-microglobulin excretion in patients from
the case-control study by Brüning et al. (2003) (details provided later in this chapter). Some subjects in this study were highly exposed; of the 134 with renal cell cancer, 19 reported past exposures that led to narcotic effects and 18 of the 401 controls, experienced similar effects (odds ratio [OR] = 3.71, 95% confidence interval [CI] 1.80-7.54). The study of Bolt et al. was based on urine samples obtained from 74% of the patients and 75% of the controls. They found that α1-microglobulin excretion increased in exposed renal cancer patients compared with nonexposed patients. Of the exposed cancer patients, 15% had normal α1-microglobulin excretion, whereas 52% of the nonexposed patients did. On the high end, 55% of the exposed patients had α1-microglobulin excretion greater than 11 mg/L, compared with 29% of the nonexposed cases. The results of this case-control study agree with their previous study (Brüning et al. 1999a).
Proteinuria was also observed in exposed male workers who were not cancer patients (Brüning et al. 1999b). Severe tubular proteinuria was seen in 35% of exposed workers but in none of the nonexposed workers; slight tubular proteinuria was seen in 20% of exposed workers and in 2% of nonexposed workers. α1-Microglobulin excretion was significantly increased in the exposed group compared with controls.
Green et al. (2004) measured biomarkers of the proposed formate mode of action and exposure in a group of workers currently exposed to trichloroethylene. They found that urinary excretion of albumin, total N-acetylglucosaminidase and formate were increased in the exposed group compared with the unexposed group. As discussed above under Animal Studies, Tubular Toxicity, elevation of N-acetylglucosaminidase in urine is a sign of proteinuria, and proteinuria is both a sign and a cause of kidney malfunction (Zandi-Nejad et al. 2004). The exposed workers excreted an average of 9.7 (standard deviation [SD] = 11.6) mg of albumin per g of creatinine, significantly different from the nonexposed group value of 5.5 (SD = 4.3). For a urine sample, 10-17 mg of albumin per g of creatinine is considered to be suspected albuminuria in males (15-25 in females) (De Jong and Brenner 2004). Thus, the results presented provide evidence for kidney damage at current occupational exposure conditions. Nevertheless, Green et al. (2004) state that N-acetylglucosaminidase does not indicate nephropathy, or damage, but rather is an indicator of functional change in the kidney.
Green et al. (2004) performed further analyses to examine the exposure-response relationship. Trichloroethylene exposure was estimated by applying the German occupational exposure limit (maximale arbeitsplatz konzentration, MAK) standard to urine trichloroacetic acid and assuming that the linear relationship holds for exposures above 100 ppm. Neither N-acetylglucosaminidase nor albumin concentration correlated to trichloroacetic acid and, therefore, to estimated exposure; they concluded that
increased urinary albumin or N-acetylglucosaminidase was not related to trichloroethylene exposure.
This conclusion is predicated on the assumption that trichloroacetic acid in urine reliably estimates trichloroethylene exposure and that the relationship of urine trichloroacetic acid to trichloroethylene is linear up to 250 ppm (their highest estimate). The published literature indicates that urinary trichloroacetic acid concentration is not a reliable predictor of trichloroethylene exposure. This results from: the variability within the data, the linearity of the relationship, and genetic variations within the populations. These are discussed in the paragraphs below.
Studies reporting urinary excretion of trichloroethylene metabolites show considerable variability between individuals. Ikeda et al. (1972) measured exposure in workshops and trichloroethylene metabolites in urine from workers. They presented results as the mean of five measurements of exposure for each workshop and the mean and standard deviation of urinary trichloroethylene metabolite measurements from workers at that workshop. They did not include goodness-of-fit characterization. The variation around the data is considerable; a urine trichloroacetic acid concentration of 100 mg/L could be obtained after an exposure ranging from 20 to 60 ppm trichloroethylene and a concentration of 200 mg/L could be obtained after an exposure of 50 to 200 ppm. The MAK values Green et al. (2004) used to estimate exposure fall within the range of results of Ikeda et al. (1972). Inoue et al. (1989) used personal diffusive samplers to measure time-weighted exposures during the shift of individual workers and compared them with various metabolites (trichloroacetic acid, trichloroethanol, and its metabolite glucuronide). They reported a correlation coefficient (r) of 0.457 for the males and females combined; the r2 would be 0.209, indicating that about 21% of the variation of trichloroacetic acid excretion among subjects is due to trichloroethylene exposure.
The second concern is the exposure range for which metabolite excretion is linear. Ikeda et al. (1972) noted that the relationship between trichloroethylene exposure and urinary trichloroacetic acid was nonlinear at trichloroethylene concentrations above 50 ppm, reaching a plateau at 100 ppm, thus indicating saturation of trichloroacetic acid formation. Inoue et al. (1989) did not observe saturation of metabolism, and suggested it was because most of the workers were exposed to concentrations below 50 ppm. The slope of the exposure-response relationship was much flatter (0.31 for males) than that of Ikeda et al. (2.74). Fisher et al. (1998) exposed human volunteers to trichloroethylene in support of developing a physiologically based pharmacokinetic model for trichloroethylene. They reported cumulative trichloroacetic acid excretion in urine over time, not concentrations in urine. They used two exposures, 50 and 100 ppm, and two each of the males and females were exposed to both concentrations (additional subjects
were exposed to only one concentration). One male and one female did not have a higher cumulative urinary excretion of trichloroacetic acid at 100 ppm. These results are consistent with a saturation of metabolism above 50 ppm for some subjects.
Genetic differences in study populations might contribute to the differences observed. The Ikeda study (1972) appears to involve Japanese workers. The subjects of the Inoue et al. (1989) paper are from China. Green et al. (2004) did not state the nationality of the study subjects, but several of the authors are located in Singapore and China, and genetic polymorphisms are known to occur for CYP2E1 within Asian populations (Hayashi et al. 1991;OMIM 2006b). Inoue et al. (1986) reported that Japanese men have higher rates of a CYP2E1-mediated reaction (toluene metabolism to hippuric acid) than Chinese men and Japanese women; they suggested the higher rates may be related to higher alcohol consumption by Japanese males. CYP3A5 is polymorphic for high expression in Caucasian (30%); Japanese (30%); Chinese (40%); and African American, Southeast Asian, Pacific Islander, and Southwestern American Indian (50%) populations (Hustert et al. 2001; Kuehl et al. 2001; OMIM 2006a).
Green et al. (2004) concluded that increased protein excretion was not related to the extent of trichloroethylene exposure as assessed by urinary trichloroacetic acid concentration. Because the relationship of urinary trichloroacetic acid concentration to ambient trichloroethylene concentration is highly variable and nonlinear, the committee does not consider urinary concentrations of trichloroacetic acid to be sufficiently reliable to use as a quantitative measure of exposure. Therefore, analyses based on urinary trichloroacetic acid measurements should not be used to conclude that trichloroethylene does not cause nephrotoxicity. Rather, weight of evidence indicates that proteinuria is occurring at current occupational exposures and that kidney damage is occurring.
Although generalized proteinuria and urinary excretion of specific tubular proteins have been used to evaluate renal tubular cell toxicity in animals and humans exposed to trichloroethylene, it should be noted that while proteinuria does result from tubular toxicity, it is not specific for trichloroethylene or tubular nephrotoxins in general (D’Amico and Bazzi 2003; Han and Bonventre 2004; Lane 2004). Proteinuria can, for example, result from nonxenobiotic tubular injury and from glomerular disease, and is also associated with diabetes, cardiovascular disease, and inflammation. Considerable effort has been directed toward identifying urinary biomarkers that detect early and subclinical acute renal tubular injury, but this remains an unattained ideal. Development and validation of a biomarker for nephrotoxicity from trichloroethylene or, more likely, haloalkenes in general, remains an area for future investigation.
In summary, recent studies show that humans exposed to trichloroethyl-
ene have tubular proteinuria and, thus, have experienced toxic insult similar to that observed in rats.
Formate
Green et al. (2004) measured formate in urine and used it as a mode-of-action marker. They did not establish a dose-formate relationship—the study was not adequate to establish a dose-response relationship. Formate did correlate with trichloroacetic acid formation and with methylmalonic acid and glutathione S-transferase in urine, all of which were considered to be mode-of-action markers.
Formic acid nephrotoxicity has been reported in humans following deliberate poisonings. Hematuria is noted within a few hours, followed by acute renal failure (Rajan et al. 1985).
KIDNEY CANCER
Hazard Identification from Epidemiology Studies
The committee was charged with evaluating the strengths and limitations of the body of epidemiologic evidence on trichloroethylene and kidney cancer. The guidelines for evaluating epidemiologic studies developed in Chapter 2 are used in this assessment. To identify the relevant studies, the committee used all studies listed in previous assessments by Wartenberg et al. (2000), Kelsh et al. (2005), and the Institute of Medicine (IOM 2003). In addition, the committee reviewed materials (published and unpublished) submitted during the course of its study. Although the committee is unsure that this represents all the epidemiologic literature on kidney cancer and trichloroethylene, it suffices to illustrate the essential methodological issues.
As discussed in Chapter 2, it is important to include detailed tables and figures that summarize the main design characteristics of the epidemiologic studies in any risk assessment. Many formats can be used for that purpose. In this report, the committee used the format the Institute of Medicine (IOM 2003) developed for its Gulf War study regarding the chronic health effects from exposure to organic solvents and insecticides. Tables provide the essential design characteristics of the cohort and case-control studies and their principal findings, including all newly identified studies since publication of the IOM report.
Cohort Studies
The committee focused its evaluation on occupational cohort studies conducted in a variety of industries in which workers were exposed to tri-
chloroethylene, including aircraft and aerospace workers (Garabrant et al. 1988; Costa et al. 1989; Blair et al. 1998; Morgan et al. 1998; Boice et al. 1999), biologically monitored workers in national programs of Scandinavia (Axelson et al. 1994; Antilla et al. 1995; Hansen et al. 2001), rubber workers (Wilcosky et al. 1984), cardboard and paperboard workers (Sinks et al. 1992; Henschler et al. 1995), uranium-processing workers (Ritz 1999), electronics workers (Greenland et al. 1994; Chang et al. 2003), and workers in other industries. The design characteristics of 18 cohort studies are presented in Table 3-4 and selected results from the studies are provided in Table 3-5. The committee has attempted to compile a complete list of studies that provide insights into the association between exposure to trichloroethylene and kidney cancer. Estimates of relative risk for all cancer sites were not provided in all papers. For example, Shindell and Ulrich (1985) only reported on major disease categories because of the small size of the cohort.
Subjects in the studies were mostly men and their age range when they entered the cohorts was typical of working populations. Studies of dry-cleaning workers were not considered because it appears unlikely that substantial numbers of them were exposed frequently to sufficient amounts of trichloroethylene (Stewart and Dosemeci 2005). In particular, trichloroethylene was not used extensively in the dry-cleaning industry between the 1930s and 1960s and it was used rarely in subsequent decades, although it was used in spot stain removal throughout the century.
In the following sections, the committee identifies the critical issues to consider in evaluating cohort studies of kidney cancer.
Follow-Up and Misclassification of Incidence
Rate ratios (and power) estimated in cohort studies of kidney cancer could be underestimated if the follow-up period was not long enough to account for latency (say, mean latent periods on the order of 15-25 years). In mortality studies, further attenuations of power would occur because some incident cases could have been lost through misclassification of the cause of death. Nondifferential misclassification of the outcome in cohort studies (independent of exposure and reference groups) will lead to attenuation of the rate ratios, although the magnitude is difficult to predict. Thus, the rate ratios estimated in the mortality cohort studies of kidney cancer (e.g., Shindell and Ulrich 1985; Garabrant et al. 1988; Sinks et al. 1992; Axelson et al. 1994; Greenland et al. 1994; Blair et al. 1998; Morgan et al. 1998; Boice et al. 1999; Ritz 1999) are likely underestimated to some extent. The magnitude of this bias can be calculated theoretically if the sensitivity and specificity of attributing the cause of death as kidney cancer are known or estimated. In addition, methods for correcting the estimated relative risks
TABLE 3-4 Selected Cohort Studies That Present Associations Between Cancer and Exposure to Trichloroethylene
|
Reference |
Description |
Study Group (No. of Subjects) |
Comparison Group |
Exposure Assessment and Other Relevant Exposures |
Analysis and Adjustment for Potential Confounders |
|
Aircraft and Aerospace Workers |
|||||
|
Garabrant et al. 1988 |
Mortality experience (1958-1982) of aircraft manufacturing workers (at least 1 day) at an aircraft manufacturing facility in San Diego County, California (with at least 4 yr of cumulative company employment). |
14,067 total 11,898 men 2,169 women |
U.S. general population |
Employment determined through company work records and interviews; about 37% of jobs had exposure to TCE, based on 70 subjects. |
SMR Age, sex, race, calendar year, duration of employment, year of death |
|
Costa et al. 1989 |
Mortality experience (1955-1981) of workers in Turin, Italy, involved in manufacturing aircraft and aerospace components. Subjects were those working at the plant in 1954 and newly employed until 1981. |
8,626 total 7,676 men 950 women |
Italian general population |
Employment determined through company work records. No exposure assessment carried out. Hazardous exposures included cutting fluids, rubber plastic paint dyes, organic solvents, paints, welding fumes, epoxy resins and hardeners, asbestos, manmade mineral fibers, and ionizing radiation (testing of components). No specific mention of TCE. |
SMR Age, sex, race, calendar year |
|
Reference |
Description |
Study Group (No. of Subjects) |
Comparison Group |
Exposure Assessment and Other Relevant Exposures |
Analysis and Adjustment for Potential Confounders |
|
Spirtas et al. 1991; Blair et al. 1998 |
Incidence and mortality experience (1952-1990) of aircraft maintenance workers (at least 1 yr in 1952-1956) at Hill Air Force Base in Utah. |
14,457 total 10,730 men 3,727 women |
Utah white population |
Industrial hygienist assessment from interviews, surveys, hygiene files, position descriptions Exposure to TCE from dipping large parts and cleaning small electrical components with squeeze bottles. Exposure scored by department or job; rank- ordered index of intensity. Other exposures: Stoddard solvent, isopropyl alcohol, 1,1,1-trichloroethane, acetone, toluene, methyl ethyl ketone, methylene chloride. Degreasing: 1950-1960, TCE replaced Stoddard solvent and carbon tetrachloride. Early 1960, concentration of TCE about 400 ppm during the usual 15 min of degreasing. This was reduced to about 200 ppm in late 1960s. After 1978: TCE replaced by 1,1,1-trichloroethane 1968: cold state solvent: TCE replaced by 1,1,1-trichloroethane. |
SMR, RR (Poisson) Age, sex, calendar period |
|
Morgan et al. 1998 |
Mortality experience (1950-1993) of aerospace workers (at least 6 months) at Hughes Aircraft plant in Arizona. |
20,508 total (4,733 exposed) 13,742 men 6,766 women |
U.S. general population |
Exposure matrices generated by employees and industrial hygienists. TCE used in vapor degreasing (>50 ppm) 1952-1977. Coded as none (0), low (1), medium (4), high (9). |
SMR, Cox proportional hazards model Age, sex |
|
Boice et al. 1999 |
Mortality experience (1960-1996) of aircraft manufacturing workers (at least 1 yr) at Lockheed Martin facility in California. |
77,965 total 62,477 men 15,488 women |
California general population of white workers |
Abstracted from walk-through surveys, hygiene files, job descriptions. TCE used until 1966 and Perc after 1996. 70% of workers using TCE or perchloroethylene also exposed to chromates. Jobs with exposure to TCE or perchloroethylene: process equipment operator, electroplater, metal bond assembler, heat transfer, sheet metal forming. Other exposures: chromate, Perc, mixed solvents. |
SMR, RR (Poisson) Age, sex, race, dates of first and last employment |
|
Reference |
Description |
Study Group (No. of Subjects) |
Comparison Group |
Exposure Assessment and Other Relevant Exposures |
Analysis and Adjustment for Potential Confounders |
|
Zhao et al. 2005 |
Mortality and cancer incidence experience of 6,107 male workers at the Santa Susana Field Laboratory (Boeing) in California. Workers were employed between1950 and 1992 with follow-up until the end of 2001. |
6,107 men |
Internal comparison |
Job-exposure matrix developed through assessment of the workplace. Exposure scores for TCE, benzene, polycyclic aromatic hydrocarbons, mineral oil, and hydrazine. Exposure score = intensity of exposure (none, low, medium, high) × number of years exposed summed over all jobs. |
Cox model adjusted for pay type, time since hire, and age. Additional adjustments for other exposures, including hydrazine. |
|
Other Cohort Studies |
|||||
|
Axelson et al. 1978, 1994 |
Mortality experience (1955-1986) of Swedish workers occupationally exposed during the 1950s and 1960s. Cohort included workers in manufacturing plants as well as users. |
1,670 total 1,421 men 249 women |
Swedish general population |
Biological monitoring for U-TCA |
|
|
Wilcoskyet al. 1984 |
Cases, age 40-84 years, selected retrospectively from a cohort of active and retired male rubber workers in a plant in Akron, Ohio, in 1964-1973; an age-stratified, 20% random sample from the original cohort served as the control group. |
NA |
1,336 (20% of 6,678) |
Linkage of worker histories to plant solvent-use records; work in process area with known solvent use equates to exposure. Other exposures: TCE, Perc, toluene, xylenes, naphthas, ethanol, acetone, phenol. |
Race-specific ORs Age |
|
Shindell and Ulrich 1985 |
Mortality experience of white and nonwhite men and women, >3 months employment, 1957-1983, in a manufacturing plant in Illinois that used TCE as a degreasing agent. |
2,140 white men 76 nonwhite men 430 women |
External comparison with U.S. general population |
None |
SMR Age, sex, calendar year |
|
Seldén and Ahlborg 1991 |
Mortality and incidence experience among male Swedish Armed Forces personnel possibly exposed to military aircraft fuel (MC77, MC25) during 1972 to 1974. Follow-up from 1975 to 1984 (mortality) and from 1975 to 1983 (cancer incidence). Exposure assessments from 1930 to 1983. Percent of eligible cohort included in study, 2,176/2,181 = 99.8%. |
2,176 men 1,865 from Air Force |
Swedish general population |
Estimates of probability of exposure to different types of military aircraft fuel attributed by a senior aircraft technician from subjects’ personnel files. Fuels considered: MC55, MC75, MC77, MC25. If exposure to TCE, then it would be part of the complex mixture of the aircraft fuels. |
SMR, SIR Age, sex, time period |
|
Sinks et al. 1992 |
Mortality experience of paperboard printing workers, >1 day employment, 1957-1988. Based on a “cluster” reported by a physician. |
1,765 white men 63 black men 219 white women 3 black women |
External comparison with U.S. general population; internal comparison |
None |
SMR and SIR Age, sex, calendar year; nested case-control study on renal cancer |
|
Reference |
Description |
Study Group (No. of Subjects) |
Comparison Group |
Exposure Assessment and Other Relevant Exposures |
Analysis and Adjustment for Potential Confounders |
|
Greenland et al. 1994 |
Mortality experience of white men in a transformer manufacturing plant. Subjects were employed before 1985, died in the period 1969-1984, as reported by the company pension plan, and had a job history, were between ages 21 and 90 at death, ended work after 1946. |
512 cases of different sites of cancer. 1,202 controls with other causes of death. Controls were excluded if they died of diseases of the blood and blood-forming organs (ICD8 280-289), mental disorders (290-315), and diseases of the digestive system (580-629). 21 cases and 68 controls did not meet the eligibility criteria. |
Internal comparison |
Job titles rated for exposure by industrial hygienist and created a job-exposure matrix for specific solvents, including TCE (coded as unexposed/exposed). |
Logistic analyses adjusting for age, year of death, and other covariates that altered the exposure estimate by >20% |
|
Anttila et al. 1995; Tola et al. 1980 |
Incidence experience (1967-1992) of workers biologically monitored for occupational exposure to halogenated solvents (1965-1982) at the Finnish Institute of Occupational Health. |
3,974 total 2,050 men 1,924 women |
Finnish general population |
Biological monitoring for U-TCA and blood metabolites of Perc and trichloroethane. Other exposures and biomonitoring: 1,1,1-trichloroethane, Perc |
SIR Age, sex, time period |
|
Henschler et al. 1995 |
Mortality and incidence experience of cardboard manufacturing plant, Germany, >1 yr, 1956-1975, follow-up until 1992. |
183 exposed men; 169 participated in study. Control cohort of 190 unexposed men, matched on age and physical job activity to exposed cohort. Unclear how the cohorts were constituted and followed. Appears to be based on a cluster of 5 cases. Case finding may not have been adequate. |
Internal comparison uses Danish rates to calculate expected. External comparison with Danish and German cancer registry incidence rates. Cause of death from hospital records; cause of death for external rates based on death certificates. |
TCE exposure, used for cleaning between 1956 and 1975. In 1976, other solvents used in small quantities. Walk-through I survey. High exposures incardboard-machine area. Lower but continuous exposures in locksmith and electrical workshops. |
Internal comparison using SIRS, adjusted for age. External: SIR calculations. Body weight, height, blood pressure, intake of diuretics, smoking, and alcohol assessed. |
|
Reference |
Description |
Study Group (No. of Subjects) |
Comparison Group |
Exposure Assessment and Other Relevant Exposures |
Analysis and Adjustment for Potential Confounders |
|
Ritz 1999 |
Mortality experience (1951-1989) of male uranium-processing plant workers (at least 3 yr, with first hire in 1951-1972) in Ohio. |
3,814 men |
(1) External comparison with U.S. general population. (2) Internal comparison among workers monitored for exposure. |
Exposure matrices generated by employees and industrial hygienists for TCE, cutting fluids, kerosene. Workers classified as not exposed or as low (1), medium (2), high (3). |
SMR, RR (conditional logistic regression) Age, calendar year, time sincefirst hired, pay type, internal and external radiation dose |
|
Hansen et al. 2001 |
Incidence experience (1968-1996) in Danish workers (1947-1989) occupationally exposed. |
803 total 658 men 145 women |
Danish general population. Nonrandom sample; loss of subjects because they could not be linked. |
Biological monitoring for U-TCA. |
SIR Age, sex, calendar year, period of first employment, employment duration |
|
Raaschou-Nielsen et al. 2003 |
Incidence experience of workers at 347 small plants in Denmark using TCE, follow-up 1968-1997. |
40,049 men and women |
External comparison with Danish cancer registry |
Duration of employment, year of first employment positively correlated, and number of employees negatively correlated with exposure to TCE. |
External SIR calculations, adjusted for sex, age, calendar year |
|
Chang et al. 2003 |
Mortality experience of workers at an electronics manufacturing plant in Taiwan employed between 1978 and 1997 and followed from 1985 to 1997. Subjects at the plant were identified by Bureau of Labor Insurance files, United Labor Association, and labor-insurance hospitalization data. |
86,868 total 16,133 men 70,735 women |
Person-years calculated from 1985 onward; external comparison with Taiwanese national mortality |
Exposures to organic solvents at the plant were due to TCE and perchloroethylene. The primary index of exposure was duration of employment at the plant. Because no company records were available, duration was based on insurance records: from inception to termination of labor insurance coverage. Durations were likely underestimated as dates of commencement (n = 6,508; 7.5%) and termination (n = 6.0%) of insurance coverage were incomplete. |
External SMR calculations, adjusted for sex, age, calendar year |
|
ABBREVIATIONS: ICD, International Classification of Disease; NA, not available; OR, odds ratio; Perc, tetrachloroethylene; RR, relative risk; SIR, standardized incidence ratio; SMR, standardized mortality ratio; TCE, trichloroethylene; U-TCA, urinary trichloroacetic acid. SOURCE: Adapted from IOM 2003. |
|||||
TABLE 3-5 Selected Results from Cohort Studies of Kidney Cancer and Occupational Exposure to Trichloroethylene
|
Reference |
Study Population |
Exposed Cases |
Estimated RR (95% CI) |
Approximate Statistical Power to Detect a RR = 2 |
|
Incidence |
|
|
|
|
|
Sinks et al. 1992 |
Cardboard manufacturers, USA SIR |
6 |
3.7 (1.4-8.1) |
27.9 |
|
Hansen et al. 2001 |
Danish workers occupationally exposed to TCE (U-TCA monitoring) |
|
||
|
|
Men, ever exposed |
3 |
0.9 (0.2-2.6) |
44.9 |
|
|
Women, ever exposed |
1 |
2.4 (0.03-14) |
13.5 |
|
Blair et al. 1998 |
Male aircraft maintenance workers in Utah |
|
|
|
|
|
Total |
15 |
1.6 (0.5-5.1) |
81.5 |
|
|
No exposure |
9 |
1.6 (0.5-5.4) |
62.8 |
|
|
<5 unit-yr |
9 |
1.4 (0.4-4.7) |
67.8 |
|
|
5-25 unit-yr |
5 |
1.3 (0.3-4.7) |
49.4 |
|
|
>25 unit-yr |
2 |
0.4 (0.1-2.3) |
58.4 |
|
|
Low intermittent exposure |
12 |
2.1 (0.6-7.5) |
63.3 |
|
|
Low continuous exposure |
9 |
2.2 (0.6-8.1) |
51.4 |
|
Henschler et al. 1995 |
Male German cardboard manufacturers, employed >1 yr |
5 |
7.97 (2.59-8.59) |
16.3 |
|
Anttila et al. 1995 |
Finnish workers occupationally exposed to TCE (U-TCA monitoring) |
|
|
|
|
|
Entire period since first measurement |
6 |
0.87 (0.32-1.89) |
70.4 |
|
|
0-9 yr |
1 |
0.53 (0.01-2.95) |
30.8 |
|
|
10-19 yr |
5 |
1.39 (0.45-3.24) |
47.2 |
|
|
20+ yr |
0 |
— (0.00-2.48) |
|
|
Axelson et al. 1994 |
Swedish men occupationally exposed to TCE (U-TCA monitoring) |
6 |
1.16 (0.42-2.52) |
59.6 |
|
Reference |
Study Population |
Exposed Cases |
Estimated RR (95% CI) |
Approximate Statistical Power to Detect a RR = 2 |
|
Raaschou-Nielsen et al. 2003 |
Danish workers exposed to TCE Men |
93 |
|
|
|
|
Women |
10 |
|
|
|
|
Duration of exposure (men) |
|
|
|
|
|
<1 y |
14 |
0.8 (0.50-1.4) |
96.6 |
|
|
1-4.9 yr |
25 |
1.2 (0.8-1.7) |
98.4 |
|
|
≥5 yr |
29 |
1.6 (1.1-2.3) |
97.0 |
|
|
Duration of exposure (women |
|
|
|
|
|
<1 yr |
2 |
1.1 (0.1-3.8) |
30.1 |
|
|
1-4.9 yr |
3 |
1.2 (0.2-3.4) |
37.1 |
|
|
≥5 yr |
3 |
1.5 (0.3-4.3) |
31.9 |
|
Zhao et al. 2005 |
Kidney in men Cumulative exposure score, lag 0 |
|
|
|
|
|
Low (0-3) |
6 |
1 |
|
|
|
Medium (>3-15) |
6 |
1.87 (0.56-6.20) |
43.8 |
|
|
High (>15) |
4 |
4.90 (1.23-19.60) |
18.7 |
|
Mortality |
|
|
|
|
|
Shindell and Ulrich 1985 |
White and nonwhite men and women in a manufacturing plant that used TCE as a degreasing agent |
Not reported |
|
|
|
Garabrant et al. 1988 |
Aircraft manufacturing workers, San Diego (about 37% of jobs had exposure to TCE) |
12 |
0.93 (0.48-1.64) |
90.9 |
|
Costa et al. 1989 |
Aircraft and aerospace components workers, Turin, Italy |
Not reported |
|
|
|
Reference |
Study Population |
Exposed Cases |
Estimated RR (95% CI) |
Approximate Statistical Power to Detect a RR = 2 |
|
Boice et al. 1999 |
Aircraft manufacturing workers in California |
|
|
|
|
|
Factory workers |
75 |
0.86 |
100 |
|
|
Routine exposure |
7 |
0.99 (0.40-2.04) |
71.3 |
|
|
Duration of exposure to TCE |
|
|
|
|
|
|
|
022 |
1 |
|
|
<1 yr |
6 |
0.97 (0.37-2.50) |
66.3 |
|
|
1-4 yr |
1 |
0.19 (0.02-1.42) |
60.3 |
|
|
≥5 yr |
4 |
0.69 (0.32-2.12) |
63.8 |
|
Blair et al. 1998 |
Male aircraft-maintenance workers in Utah |
|
|
|
|
|
No TCE exposure |
10 |
2.5 (0.7-8.9) |
50.7 |
|
|
<5 unit-yr |
8 |
2.0 (0.5-7.6) |
50.7 |
|
|
5-25 unit-yr |
1 |
0.4 (0.1-4.0) |
37.1 |
|
|
>25 unit-yr |
4 |
1.2 (0.3-5.7) |
44.9 |
|
|
Frequent peaks |
5 |
1.4 (0.3-5.7) |
47.0 |
|
Morgan et al. 1998 |
Aerospace workers in Arizona, TCE exposed subcohort |
|
|
|
|
|
Any exposure |
8 |
1.32 (0.57-2.60) |
65.5 |
|
|
Low exposure |
1 |
0.47 (0.01-2.62) |
33.3 |
|
|
High exposure |
7 |
1.78 (0.72-3.66) |
50.1 |
|
|
Peak exposures (reference=none/low) |
|
|
|
|
|
Medium/high |
8 |
1.89 (0.05-4.3) |
52.5 |
|
|
Cumulative (low) |
1 |
0.31 (0.04-2.36) |
43.9 |
|
|
Cumulative (high) |
7 |
1.59 (0.68-3.71) |
53.9 |
|
Henschler et al. 1995 |
Male German cardboard manufacturers, employed >1 yr |
2 |
3.28 (0.40-11.84) |
16.0 |
|
Greenland et al. 1994 |
White male U.S. transformer manufacturers, ever exposed to TCE |
|
NA 0.99 (0.30-3.32) |
|
|
Reference |
Study Population |
Exposed Cases |
Estimated RR (95% CI) |
Approximate Statistical Power to Detect a RR = 2 |
|
Ritz 1999 |
Mortality experience of male uranium-processing plant workers |
|
|
|
|
|
Total cohort |
8 |
1.17 (0.50-2.31 |
70.1 |
|
|
Bladder and kidney combined |
|
|
|
|
|
No. of yr exposed TCE level 1 |
|
|
|
|
|
<2 yr |
6 |
1 |
|
|
|
2-10 yr |
5 |
1.94 (0.59-6.44) |
37.9 |
|
|
≥10 yr |
2 |
0.76 (0.14-400.0) |
38.3 |
|
|
TCE level 2 |
|
|
|
|
|
<2 yr |
13 |
1 |
|
|
|
2-10 yr |
0 |
0 |
|
|
|
≥10 yr |
0 |
0 |
|
|
Sinks et al. 1992 |
Cardboard manufacturers, U.S. SMR |
1 |
1.4 (0.0-7.7) |
17.3 |
|
Chang et al. 2003 |
Kidney and other unspecified urinary organs |
|
|
|
|
|
Men |
0 |
0 (0.0-2.82)a |
|
|
|
Women |
3 |
1.18 (0.24-3.44) |
37.5 |
|
|
Men and women Duration of employment |
|
|
|
|
|
<1 yr |
1 |
0.62 (0.02-3.46)a |
27.8 |
|
|
1-5 yr |
2 |
3.08 (0.37-11.11)a |
16.6 |
|
Zhao et al. 2005 |
Kidney in men Cumulative exposure score, lag 0 |
|
|
|
|
|
Low (0-3) |
7 |
1 |
|
|
|
Medium (>3-15) |
7 |
1.43 (0.49-4.16) |
57.6 |
|
|
High (>15) |
3 |
2.03 (0.50-8.32) |
26.4 |
|
a95% CI calculated by the committee using standard methods from the observed and expected numbers presented in the original study. ABBREVIATIONS: CI, confidence interval; NA, not available; RR, relative risk; SMR, standardized mortality ratio. SOURCE: Adapted from IOM 2003. |
||||
as well as the confidence intervals are available and might be useful if error rates are known (Brenner and Gefeller 1993).
Statistical Power
Kidney cancer is a rare disease. In the United States, the age-adjusted rate of cancer of the kidney and renal pelvis is 12.6 per 100,000 people, and this rate varies by gender and race (SEER 2005; Table 3-6). The American Cancer Society (2006) estimates that nearly 39,000 patients will be diagnosed with kidney cancer in 2006, more than twice the number of patients expected to be diagnosed with liver cancer (see Chapter 4). Thus, only very large cohort studies would have a sufficient number of cases to provide adequate statistical power to estimate excess risks and exposure-response relationships. In particular, negative findings from cohort studies that have low statistical power are not likely to provide useful evidence, unless somehow the results are combined, for or against the hypothesis that trichloroethylene is a human carcinogen.
The statistical power of a study is reflected in the estimated variances and the associated confidence intervals. As well, in meta-analyses the power of a study is effectively accounted for in the weight (inverse of the estimated variance) used to calculate the summary relative risk and to estimate whether there is heterogeneity in the rate ratios between studies. In qualitatively assessing studies, it is nevertheless useful to assess formally the statistical power of each study. Statistical power is a function of the true relative risks of the study population; if exposures in the population are low then relative risks will be smaller. Thus, plots of statistical power versus different values of relative risk can be produced. The usefulness of such plots is illustrated by Beaumont and Breslow (1981) in an analysis of cohort studies of workers exposed to vinyl chloride monomer. They described a useful method for presenting the data by plotting the power of detecting a relative risk for
TABLE 3-6 Average Annual Incidence (per 100,000) of Kidney and Renal Pelvis Cancer in the United Statesa
various values of relative risk on the ordinate and the expected number of deaths on the abscissa. An alternative formulation is to plot the observed relative risk and confidence intervals versus calculated statistical power.
For cohort studies that compare an exposed population with an unexposed population,an approximate formula for power is given by:
where β is the Type II error
Z1-β is the Z value for power 1 - β,
Za is the upper 100th percentile of the standard normal distribution,
E is the expected number of deaths, and
RR is the estimated rate ratio or relative risk.
For each of the cohort studies considered by the committee, Table 3-5 provides the approximate statistical power to detect a RR of 2 or more, on the basis of a statistical signi .cance of 5% and the number of expected cases. (Power calculations could also be carried out for case-control studies.) Statistical power must be interpreted with knowledge of the exposure situation. For example, the study by Blair et al. (1998) had 82% power to detect RR values of 2, but the actual exposure in the cohort was so low that one would expect much smaller relative risks. However, the study did not have a large enough sample size to detect a smaller relative risk (the power to detect a RR of 1.5 was 39.6%). For this reason, plotting the power for various expected relative risks is useful. Even disregarding the degree of exposure, the power to detect excess risks was extremely limited in most studies. The essentially negative studies by Greenland et al. (1994), Axelson et al. (1994 [incidence]), Antilla et al. (1995 [incidence]), Blair et al. (1998 [incidence and mortality]), Morgan et al. (1998), Boice et al. (1999), and Hansen et al. (2001) were all underpowered.
Effects of Confounding
The impact of confounding by other risk factors also needs to be considered carefully in evaluating the evidence for kidney cancer. In the cohort studies, potentially confounding factors other than accounting for age, sex, and calendar year were not evaluated. The major risk factors for kidney cancer appear to be cigarette smoking, obesity, and ionizing radiation; some evidence suggests that phenacetin-containing drugs,diuretics, and exposure to asbestos might be associated with incidence (McLaughlin et al. 1996). The RR values for current smokers are in the range 1.5-2.0 and, unless smoking was strongly associated with exposure to trichloroethylene, it is unlikely that smoking, or any other risk factors, could lead to substantial bias.
Take a hypothetical situation in which a cohort of subjects is exposed
to trichloroethylene and the end point is incidence of kidney cancer. Assume that the observed standardized incidence ratio (SIR) is 2, but the prevalence of smoking in the cohort was not taken into account in its calculation, so that confounding could explain the observed effect. The following equation describes the relationship between incidence and exposure and the confounding effect of smoking:
where I is the incidence of kidney cancer, I0 represents the baseline rate, RRs,i represents the rate ratio for smoking (s) at level i (e.g., moderate, heavy), and ps,i is the proportion of individuals smoking at level i.
Given some realistic values of the proportion of workers smoking at different intensities and the relative risk of developing kidney cancer,the amount of bias can be calculated. Assume, for example, that there is no real association between kidney cancer and exposure to trichloroethylene, and only smoking increases the baseline incidence rate so that the RR for smoking is 2. Thus, the above equation reduces to I = I0 + pc. So, for example, if 50% of the population in the cohort smokes and 30% of the general population smokes, then the expected RR due only to smoking is 1.15, and this would not explain the observed RR of 2.
In most of these studies (Blair et al. 1998, Morgan et al. 1998, Boice et al. 1999, Ritz 1999), general population rates of kidney cancer were used as the comparison group, thereby indirectly standardized mortality ratios (SMR) or SIR were calculated. Such use of the general population as the reference population in most cases will lead to attenuated rate ratios because the workforce is generally healthier than the general population. This type of selection bias is one component of the “healthy worker effect” ((Fox and Collier 1976; Choi 1992; Li and Sung 1999; Baillargeon 2001). Rate ratios estimated from internal comparisons (e.g., comparing an exposed group with an unexposed group within the study population) will not have such a bias, although there can be effects from selection out of the study. There is no simple method to estimate the extent of the latter type of bias (for advanced methods, see Robins 1986, 1987a,b,c).
Exposure Assessments
Table 3-7 presents essential characteristics of the exposure assessments for selected cohort studies. Important factors in these studies that affect causal inference is whether subjects were actually exposed to trichloroethylene, whether exposures were to complex mixtures, whether the measurements were accurate (validity, reliability), and whether exposure-
TABLE 3-7 Characteristics of the Assessment of Exposure to Trichloroethylene in Selected Cohort Studies
|
Study |
Qualitative Assessment (industry, TCE use, coexposures, exposure prevalence X%, confounders) |
Information on Settings (location of exposures, area descriptors, jobs, tasks, exposure controls) |
Duration (source, data quality, period, duration of exposure, sufficient latency) |
Exposure Quantification (methods, relative errors, range over time & categories, prevalence of high exposures) |
Exposure Extrapolation (methods, assumptions, data sources [direct, indirect]) |
Dose Metrics (quantitative, semiquantitative, qualitative) |
|
Biomonitored populations |
||||||
|
Antilla et al. 1995 (2,050 males and 1,924 subjects ever monitored) |
Finnish mixed industries using TCE in 1950s and 1960s (also Perc and TCA). |
Limited general description; most people tested were degreasing or metal cleaners. |
About 1930-1982 In cohort 1967, mean follow-up was 18 yr; time since first test was assumed to be exposure duration. |
U-TCA |
None (Using the Ikeda [1972] relationship for TCE exposure to U-TCA, median exposures were <4-9 ppm.) |
Biomarker values |
|
Axelson et al. 1994 (1,421 males and subjects ever monitored) |
Swedish mixed industries using TCE. |
Limited general description. |
About 1930-1986 In cohort 1955, time since first test was assumed to be exposure duration. |
U-TCA |
None (Using the Ikeda [1972] relationship for TCE exposure to U-TCA, median exposures were <20 ppm.) |
Biomarker values |
|
Study |
Qualitative Assessment |
Information on the Settings |
Duration |
Exposure Quantification |
Exposure Extrapolation |
Dose Metrics |
|
Hansen et al. 2001 (803 males and female subjects ever monitored) |
Danish mixed industries using TCE, predominantly iron and metal product producers. |
Limited general description. |
Retirement records and job records from 1947 to 1989. |
U-TCA |
None (Using the Ikeda [1972] relationship for TCE exposure to U-TCA, median exposures were <2-7 ppm.) |
Biomarker values |
|
Aircraft workers |
||||||
|
Stewart et al. 1991 (exposure assessment); Spritas et al. 1991 (epidemiologic analysis; cohort 14,457) Blair et al. 1998 (epidemiologic analysis for reevaluation of cohort) |
Aircraft maintenance; solvents used for degreasing and cleaning; carbon tetrachloride until mid-1950s; TCE used up to 1970s: historical changes: about 1955-1978 TCE used in vapor degreasing; about 1955-1968 TCE used in hand degreasing; before 1955 Stoddard and carbon tetrachloride were used; about 50% exposed to TCE; coexposures to Stoddard solvent/ kerosene, 1,1,1-trichloroethane, gasoline, jet fuel, isopropyl alcohol; about 65% of men and 33% of women were exposed to TCE. |
Large maintenance facility at Hill Air Force Base, UT. Detailed records on setting and job activities, worker interviews; work done in large open shops; shops not recorded in personnel records; link of jobs with exposures was weak. |
From payroll records, 1940s-1990. In cohort if >1 yr in 1952-1956. Total duration likely to be accurate, but duration of specific exposures uncertain because of weak linkage of jobs and exposures; about 35 yr latency possible; exposures noted after 1951. |
Limited exposure monitoring data 1960-1990 for TCE. Plant JEM, rank order assignments by history; determined exposure duration during vapor degreasing tasks about 200 ppm/hr and hand degreasing about 20 ppm/hr; only 16% of total deaths occurred in subjects with >25 unit-yr (no data given on number of subjects with long high exposures). |
Similar jobs had similar exposures; limited data for early periods; exposure data for shops, but jobs in histories not linked to shops. Large misclassification of exposures in mixed solvent group. Median exposures were about 10 ppm for rag and bucket; 100-200 ppm for vapor degreasing. |
Cumulative exposure; calculated but results not reported; average exposure and duration of exposure. |
|
Boice et al. 1999 (cohort 77,965) |
Aircraft manufacturing; four different sites in Burbank, CA; TCE used 1960-1970s; 12% with TCE exposure (5,443); coexposures to other chlorinated solvents, ketones, alcohols, petroleum distillates, chromate, paints, cutting fluids, and fibers. |
Very limitedon work places; jobs in work histories collapsed into groups with “similar” exposures. |
Company records; good quality; provided data on start and end dates; median durations, exposure about 10 yr, latency about 30 yr. |
Workers assigned tofamilies: assembly, fabrication, processing, maintenance; smaller groups: processing operators and helpers (1,440) worked on vapor degreasers. |
None |
Years TCE exposure as routine or intermittent; job type and duration of exposure. |
|
Morgan et al. 1998 (cohort 20,508) |
Aerospace manufacturing but no details given on jobs or operations, or other exposures; knowledgeable employees assisted by company industrial hygienist, identified when and where TCE was used; degreasers about 1952-1977; about 23% exposed to TCE. |
Very limited;no data on buildings or operations; unclear if information on setting was used. |
Company records on each subject provided hire and end dates; two-thirds had >20 yr latency; limited data on duration of exposures, none on high exposures. |
No detail on protocol or possible errors; exposure range is uncertain; three categories assigned: high, work on degreasers (>50 ppm); medium, work near degreasers; low, occasionally near degreasers; no data on prevalence of high exposures. |
Long-term employees rated exposures by vague criteria; company industrial hygienist used the data to build an exposure matrix; no consideration of changes with time. |
Cumulative exposure by TCE weight times months; exposed to peaks was any time in high category; also a dichotomy of ever exposed, yes or no. |
|
Study |
Qualitative Assessment |
Information on the Settings |
Duration |
Exposure Quantification |
Exposure Extrapolation |
Dose Metrics |
|
Costa et al. 1989 (cohort 8,626) |
Aircraft manufacturing; many coexposures listed. TCE used up to 1970s; prevalence of TCE exposure not reported; many chemicals used over the years but not linked to subjects. |
One aircraft manufacturing site in Italy; no data on the site were given. |
Personnel records >1954; quality good; yr started and ended for each subject; median durations: employment about 10 yr, follow-up about 17 yr, only 25% had >25 yr |
None; used only four broad job groups: blue collar 75% of total, technical staff, administrative clerks, and white collar. |
None |
Job groups |
|
Garabrant et al. 1988 (cohort 14,067) |
Aircraft manufacturing; TCE usage not described; plant with detailed records on small sample, and interviews, assigned about 37% TCE exposed TCE used up to 1970s. |
Major process categories listed with percentage of subjects in each from a sample of work histories. |
Work records, quality good; >4 yr work Latency <30 yr; mean follow up was 15.8 yr |
None for the cohort |
None, no link with jobs or exposures. |
Duration of employment |
|
Mixed settings |
||||||
|
Raaschou-Nielsen et al. 2003 (exposures in Raaschou-Nielsen et al. 2002) |
Mixed companies |
Limited general description from national sources. |
In cohort 1952 |
Samples from national program; iron and metal: quantitation not used in mortality study. |
Regression relationship was developed by industry. GM: 60 ppm 1947-1959; 49 ppm 1960- 1969; 20 ppm 1970-1979 |
|
|
Ritz 1999 |
Uranium-processing workersusing TCE in 1950s and 1960s, also cutting fluids, kerosene, company records. |
|
>3 yr, 1951-1996 In cohort 1951 |
Exposure monitoring data, plant JEM, semiquantitative assignments by industrial hygienists and employees. |
None |
|
|
Chang et al. 2003 |
Large electronics plant; used TCE and Perc (found in local well water). |
Taiwan; no direct information was provided on presumed TCE and Perc exposures. |
Short duration about 10 yr |
None |
None |
Years of employment |
|
Sinks et al. 1992 |
Paperboard manufacturing, TCE present in materials, usage not described. |
TCE usage not described. |
Work histories, quality good, duration in depts.; sufficient latency. |
None |
None |
Duration indepartment |
|
Study |
Qualitative Assessment |
Information on the Settings |
Duration |
Exposure Quantification |
Exposure Extrapolation |
Dose Metrics |
|
Henschler et al. 1995 |
Cardboard manufacturer in Arnsberg, Germany. TCE used as degreasing solvent, small amounts of TCA and Perc also used. |
Highly detailed on TCE use, area descriptors (hot machines and areas, large amount of TCE used, jobs, tasks) extensive cleaning; no controls. |
Company data, questionnaire, interviews, median 34 yr observation, sufficient latency. |
None (neurological symptoms reported, >200 ppm likely). |
None in paper (Cherrie et al. [2001] extrapolated based on details in paper: peaks >2,000 ppm long-term 100-200 ppm). |
Exposed vs. unexposed |
|
ABBREVIATIONS: JEM, job-exposure matrix; Perc, tetrachloroethylene; TCA, trichloroacetic acid; TCE, trichloroethylene; U-TCA, urinary metabolite of trichloroethylene (and Perc, trichloroethane, and 1,1,1-trichlorethane). |
||||||
response relationships were estimated. For some studies, whether subjects were actually exposed to trichloroethylene is doubtful (Shindell and Ulrich 1985; Garabrant et al. 1988; Sinks et al. 1992; Greenland et al. 1994). In some studies, biological monitoring for urinary trichloroacetic acid was conducted. The half-life of urinary trichloroacetic acid is on the order of 100 hours (Muller et al. 1974) and it represents total acute exposures (occupational and other sources) to trichloroethylene, tetrachloroethylene, and trichloroacetic acid in the environment (e.g., drinking water). In any risk assessment, close attention must be paid to the validity and reliability of the exposure assessments in all the included studies.
As indicated above, there is good reason to exclude all the dry-cleaning studies because there was apparently little use of trichloroethylene (Stewart and Dosemeci 2005). In addition, the cohort study of Swedish Airforce personnel exposed to aircraft fuel (Seldén and Ahlborg 1991) should be excluded, as it is likely that they were exposed to no more than trace amounts of trichloroethylene.
Specific Issues with Certain Cohort Studies
Studies from Arnsberg Area of Germany
Considerable controversy has surrounded the studies by Henschler et al. (1995) and Vamvakas et al. (1998) conducted in the Arnsberg area of Germany. Recent papers by Brüning et al. (2003) and Pesch et al. (2000a) cover the same region in Germany but at a later time. The region was a center of metal machining and processing operations. This concentration of operations resulted in a high prevalence of workers with intense exposures to trichloroethylene.
The finding of a cluster of cases prompted the study by Henschler et al. (1995). Because the study population included the area with the cluster, the observed relative risks for kidney cancer were much higher than in any other study (SMR, 7.97; 95% CI, 2.59-18.59; five cases), compared with incidence rates from the Danish Cancer Registry. Because of its very high SMR, this study has been the subject of much scrutiny. The study population comprised a cohort of 169 workers reportedly exposed to trichloroethylene at a cardboard manufacturing plant. The workers were employed for at least 1 year between 1956 and 1975. The comparison populations comprised a matched cohort of 190 workers from the plant who were not exposed to trichloroethylene as well as the general population of Denmark and Germany, as reflected by rates of cancer incidence from the Danish Cancer Registry and mortality from a German registry. Follow-up of both populations was from 1956 until the end of 1992. Trichloroethylene was used in degreasing operations from 1956 to 1975 and no specific exposure assessments were
carried out. However, extensive detail about the nature of the operations permitted extrapolations of possible exposures (Cherrie et al. (2001).
The Henschler et al. (1995) study has been criticized for a number of reasons, including the following:
-
A cluster of cases of kidney cancer were used to design the study, and they were included in the analysis (see Swaen 1995; McLaughlin and Blot 1997). As a consequence, statistical inference is not valid unless the cluster is removed, but removing the cluster would reduce substantially the study’s statistical power. An additional consequence of having an incorrect estimate of statistical precision is that the study could not be included in a formal meta-analysis and care must be taken in interpreting risk estimates in informal analyses. Although estimates of rates and relative rates are valid, the main issue is how to calculate an appropriate variance so that statistical tests can be conducted and confidence intervals estimated correctly. The issue is that the nominal P values do not apply as this working population was selected because of the cluster. The option of excluding the cluster of cases could be implemented if the cluster occurred at the beginning of the risk period and did not include all cases; then theses cases and person-time could be excluded, yielding a valid estimate of risk and statistical precision. McLaughlin and Blot (1997) suggested that the first three cases diagnosed in 1990 and 1991 might represent the original cluster; in that case, the SMR would be approximately 3.2.
-
Fourteen subjects who were lost to follow-up were excluded, although they should have been included in the person-year calculations (Swaen 1995). The likely effect of this exclusion is that the number of person-years is underestimated; the expected number of deaths is less than it should be, so the observed risk ratio is overestimated. The committee concludes that, given the small number of subjects, the extent of underestimation is likely not great.
-
Different methods were used to assign underlying causes of death in the cohort and reference population (Swaen 1995). This could have introduced a positive bias (away from the null) if the hospital or physician records were more likely to detect cases of the cancers of interest than the medical institutions serving the general population. The committee concluded that this effect could not have explained the entire excess risk.
-
The follow-up of some subjects might have been outside the range of the stated follow-up period (Bloemen and Tomenson 1995). The committee did consider this a pertinent criticism, as it would have resulted in an overestimation of person-time and expected deaths and thereby would have led to an underestimate of the relative risks.
-
The intensity of exposure to trichloroethylene in this occupational
-
environment was unclear (Swaen 1995; Cherrie et al. 2001). However, given the information the paper provided on the job activities and work locations, the committee judges that the magnitude of the exposures can approximated. Cherrie et al. (2001) extrapolated the exposures on the basis of the data presented and a simple engineering model of the workplace. The analysis indicated that the subjects in this study likely had substantial peak exposures to trichloroethylene above 2,000 ppm and probably had sustained long-term exposures above 100 ppm.
-
Use of the Danish Cancer Registry to calculate expected values might not be appropriate if rates of renal cell cancer in the region surrounding the plant are very different from those in Denmark. The age-standardized rate in the late 1990s among men in Denmark was 10.6 and in Germany it was 12.3 (Ferlay et al. 2004). If these differences in rates apply when the study was carried out, this would imply that the expected number of deaths would have been inflated by about 14% (and the rate ratio underestimated by that amount).
-
Could there be unmeasured confounding? The low SMR for lung cancer (mortality SMR = 1.4 in the exposed cohort) suggests that it is unlikely that smoking would play an important role.
Although clear methodological difficulties are associated with this study, which leaves doubt about the veracity of the results, if exposure to trichloroethylene were as high as suggested (Henschler et al. 1995) then part of the excess could reflect a causal process at high exposures. The committee concludes that it is neither prudent nor useful to ignore the findings of this study, but they must be evaluated within the context of the available literature, particularly in the context of other studies from the same base population.
New Studies Published in 2005
The committee reviewed one report that was published during its deliberations (Zhao et al. 2005). This investigation was a cohort study of 6,107 male workers at the Santa Susana Field Laboratory (Boeing) in California, employed between 1950 and 1992. The follow-up period was until the end of 2001. This study is one of the few to conduct a detailed assessment of exposure that allowed for the development of a job-exposure matrix that provided rank-ordered levels of exposure to trichloroethylene, benzene, polycyclic aromatic hydrocarbons, mineral oil, and hydrazine. A monotonic increase in mortality rates due to kidney cancer by increasing levels of cumulative exposure was found. This relationship was accentuated with adjustment for other occupational exposures, although the confidence intervals were increased. This phenomenon is not unusual in occupational
studies as exposures are usually highly correlated and adjustments often inflate standard errors without removing any bias. Thus, these adjustments must be interpreted cautiously.
Case-Control Studies
The main methodological points associated with case-control studies are the accuracy of the outcome, estimates of exposure, statistical power, confounding, and selection bias. Tables 3-8 and 3-9 provide the essential design characteristics and selected results of case-control studies on kidney cancer and occupational exposure to trichloroethylene. Each of these studies would need to be evaluated more fully as to the likely level of exposure and the proportion of subjects exposed to trichloroethylene. In particular, this is necessary for the pooled analysis by Mandel et al. (1995), in which only occupations were reported (based on studies by McCredie and Stewart [1993], Schlehofer et al. [1995], and others). Including estimates of statistical power for the case-control studies, as was done in Table 3-5, might be useful.
In contradistinction to the cohort studies, in most of the case-control studies the diagnosis of cancer was confirmed histologically (except, perhaps, in the studies by Ashengrau et al. [1993] and McCredie and Stewart [1993]). Thus, few subjects who did not have a tumor would be included. In addition, many of these studies had a large number of cases (e.g., more than 200) and, if the prevalence of exposure to trichloroethylene were sufficiently high, then there would be adequate power to detect positive associations should trichloroethylene be a risk factor for kidney cancer. However, a number of small case-control studies lacked the statistical power to detect modest associations with exposure to trichloroethylene (Jensen et al. 1988; Harrington et al. 1989; Sharpe et al. 1989; Aupérin et al. 1994; Vamvakas et al. 1998; Parent et al. 2000; and Brüning et al. 2003). Graphs similar to that suggested earlier for cohort studies of effect versus power could be developed for these case-control studies (see Beaumont and Breslow 1981). Calculations of power for case-control studies have been published (Self and Mauritsen 1988; Self et al. 1992) and are available in some software packages (e.g., Egret SIZ).
Confounding
As discussed in the section on cohort studies, risk factors for kidney cancer include cigarette smoking, obesity, and ionizing radiation; phenacetin-containing drugs, diuretics, and exposure to asbestos may also be associated with incidence of kidney cancer (McLaughlin et al. 1996). Studies by Asal et al. (1988), Partanen et al. (1991), McCredie and Stewart (1993), Aupérin et al. (1994), Chow et al. (1994), Mellemgaard et al. (1994), Mandel et al.
TABLE 3-8 Description of Case-Control Studies That Present Associations Between Kidney Cancer and Possible Exposure to Trichloroethylene
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Asal et al. 1988 |
Cases from 29 hospitals in Oklahoma diagnosed and confirmed in 1981-1984; hospital controls selected from the same hospitals and matched on age, sex, race, hospital, and date of admission; population-based controls selected through RDD. No response rates provided. |
315 renal cell carcinoma |
313 hospital 336 population |
Dry-cleaning work; painter or paint-manufacturing work |
In-person interview assessing occupations (job titles) and industrial exposures. |
Logistic regression |
Weight,age, alcohol consumption, occupation, smoking, snuff use, coffee consumption, kidney stones, hypertension, other medical factors |
|
Jensen et al. 1988 |
Cases, under age 80 yr, reported to the Danish Cancer Registry from Copenhagen and the surrounding island of Sjaelland in 1979-1982, with 90% histologic verification; controls selected from hospital where cases arose, excluding those with urinary tract and smoking- related diseases; controls matched for hospital, sex, and age. Response rates: 99.0% of cases, 100.0% of controls. |
96 renal, pelvis, and ureter |
288 |
Painter or paint-manufacturing work |
In-person interviews with questionnaire assessing personal habits and occupational history (job or industry titles and self-reported exposures). |
Logistic regression Although controls matched to cases (ratio 3:1) on hospital, age, and sex, a matached analysis was not conducted. |
Sex, lifetime tobacco smoking |
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Harrington et al. 1989 |
Cases diagnosed and histologically confirmed in 1984-1985 and reported to the West Midlands Regional Cancer Registry (UK); controls randomly selected from practitioner records and matched for age, sex, ethnicity, location, and socioeconomic group. No response rates provided. |
54 renal (adeno-carcinoma) |
54 |
Solvents |
In-person interviews with questionnaire assessing lifetime occupational history (job titles); exposure indexes determined by occupational hygienist or chemist, but not defined explicitly Values used: “unexposed,” index <1; “intermediate,” 1-99; “exposed,” >100. |
Matched analyses |
Matching variables |
|
Sharpe et al. 1989 |
Cases diagnosed at one of four Montreal-area hospitals in 1982-1986 and one of five other hospitals in 1982-1987; cases were histologically confirmed and alive at time of chart review; controls selected from suspected renal cell carcinoma cases, but final diagnoses were not cancer; matched 1:1 for sex, age (+5 yr), and urologist. Response rate: 97% overall. |
164 renal prevalent cases |
161 |
Organic solvents |
History of exposure to hydrocarbons obtained through mailed questionnaire and supplemented by telephone interview (self-reports). |
Unadjusted analysis, but matched-unadjusted |
None |
|
Partanen et al. 1991 |
Cases, age over 20 yr, identified through the Finnish Cancer Registry in 1977-1978; controls randomly selected from the Population Register Centre matched for year of birth, sex, and survival status. Excluded subjects who did not work. Response rates: 69% of cases, 68% of controls. |
408 renal cell |
819 |
Nonchlorinated solvents |
Mailed questionnaire or phone interview (direct or proxy) assessing lifetime occupational history (job or industry titles); industrial hygienist coded and assigned summary indicators of specific exposures. |
Conditional logistic regression |
Matching variables, smoking, coffee consumption, obesity |
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Aschengrau et al. 1993 |
Cases reported to the Massachusetts Cancer Registry, diagnosed in 1983-1986 among residents of five upper Cape Cod towns; living controls were selected from HCFA records and RDD; deceased controls identified by the state Department of Vital Statistics and Research files. Response rates: 80.6% of cases, 75.9% of HCFA controls, 73.9% of RDD controls, 78.8% of next of kin of deceased controls. |
35 kidney |
777 |
Tetrachloro-ethylene |
Exposure dose estimated in areas of contaminated drinking water, accounting for location and years of residence, water flow, pipe characteristics. |
Logistic regression |
Sex, age at diagnosis, vital status, educational level, usual number of cigarettes smoked, occupational exposure to solvents, specific cancer risk factors controlled for in respective analyses |
|
McCredie and Stewart 1993 |
Cases, age 20-79 years, among residents of New South Wales in 1989-1990 identified from the New South Wales Central Cancer Registry and from physicians; controls selected from electoral rolls and matched on age distribution. Response rates: 68% renal cell cases; 74% renal pelvis cases; 74% controls. Included in the combined analysis of Mandel et al. 1995 |
489 renal cell 147 renal pelvic |
523 |
Dry-cleaning industry work, solvents |
Questionnaire (in-person interview or mailed with telephone follow-up) to assess employment in specific occupations and industries (job or industry titles), self-reported. |
Logistic regression |
Age, sex, interview method, cigarette smoking, body mass index, education, analgesic use |
|
Aupérin et al. 1994 |
Cases of renal cell carcinoma diagnosed in France, 1987-1991, confirmed histologically. Controls matched by age, sex, hospital, and interviewer and comprised nonmalignant and malignant disease. Response: about 100% |
196 cases (138 men, 58 women) |
347 161 cancer 186 other diseases |
Occupation or industry |
In-person interview in hospital, occupational history. |
Conditional logistic |
Matching variables, education, smoking, body mass index |
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Mellemgaard et al. 1994 |
Cases, age 20-79 years, identified from the Danish Cancer Registry and pathology departments in 1989-1992 with histologic confirmation; controls selected from the Central Population Register and matched for age and sex. Response rates: 80% of cases, 79.2% of controls. |
368 renal cell |
396 |
Dry-cleaning work, solvents |
In-person interviews with questionnaire assessing most recent and longest-held occupation (job titles) and exposure to specific agents (self-reports). |
Logistic regression |
Age, body mass index, smoking |
|
Mandel et al. 1995 |
Cases, age 20-79 years, from six international sites, diagnosed and confirmed in 1989-1991 using cancer registries or surveillance of clinical and pathology departments; controls selected from population registers, electoral rolls, residential lists, HCFA records, or RDD, depending on the site; controls matched on age and sex. No response rates provided (included studies of McCredie and Stewart 1993 and Schlehofer et al. 1995). |
1,732 renal |
2,309 |
Occupationaltitles, including dry cleaning solvents, dry-cleaning work |
In-person interviews to assess lifetime occupational history (job titles) and exposure to specific agents (self-reports). |
Logistic regression |
Age, center, body mass index, cigarette smoking |
|
Schlehofer et al. 1995 |
Cases, age 20-75 years, identified through 10 urology departments in the Rhein-Neckar-Odenwald area of Germany in 1989-1991 with histologic confirmation; controls randomly selected from population register and matched on age and sex. Response rates: 84.5% of cases, 75% of controls. Included in combined analysis of Mandel et al. 1995. |
277 renal cell |
286 |
Chlorinated solvents |
In-person interview with questionnaire assessing exposure (in excess of 5 years) from list of specific substances (self-reports). |
Logistic regression |
|
|
Parent et al. 2000; Siemiatycki 1991 |
Male cases, age 35-75 years, diagnosed in 1 of 19 large Montreal-area hospitals in 1979-1985 and histologically confirmed; controls identified concurrently at 18 other cancer sites; age-matched, population-based controls were also chosen from electoral lists and RDD. Response rates: 82% of all cases, 71% of population controls. |
142 renal cell |
2,433, consisting of 533 population controls and 1,900 subjects from other cases of cancer |
294 agents, including TCE; results for TCE not published but obtained from authors. |
In-person interviews (direct or proxy) with segments on work histories (job titles and self-reported exposures); analyzed and coded by a team of chemists and industrial hygienists (about 300 exposures on semiquantitative scales). |
Logistic regression |
Age, body mass index, cigarette smoking, respondent status |
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Vamvakas et al. 1998 |
Cases who underwent nephrectomy in 1987-1992 in a German hospital; controls selected in 1993 from accident wards of three nearby hospitals. Response rates: 79.5% of cases, 75% of controls. |
58 renal |
84 |
Trichloroethylene |
In-person, unblinded interviews by physicians (direct or proxy) with structured questionnaire assessing occupational history (job titles) and specific agent exposures (self-reports). Ranking of exposures based on assessment of duration, frequency, concentration, and prenarcotic symptoms. Not clear how exposure was assessed. |
Logistic regression |
Age, sex, smoking, body mass index, blood pressure, intake of diuretics |
|
Chow et al. 1994; Dosemeci et al. 1999 |
White cases, age 20-85 years, with histologically confirmed diagnosis identified through the Minnesota Cancer Surveillance System in 1988-1990; controls identified through RDD (age 21-64 years) and HCFA records (age 65-85 years), stratified for age and sex. Response rates: 87% of cases, 86% of controls. Occupational analysis: 55% of cases, 83.6% of controls. |
438 renal cell carcinoma |
687 |
Trichloroethylene, tetrachloro-ethylene, solvents in general |
In-person interviews (direct or proxy) with questionnaire assessing occupational history; job titles were coded and merged with a job-exposure matrix from the National Cancer Institute. |
Logistic regression |
Age, smoking, hypertension, body mass index |
|
Pesch et al. 2000a |
Cases in large hospitals in five regions in Germany in 1991-1995 with histologic confirmation; controls randomly selected from local residency registries matched on region, sex, and age. Response rates: 88% of cases, 71% of controls. |
935 renal cell |
4,298 |
Trichloro-ethylene, tetrachloro-ethylene, organic solvents |
In-person interviews of lifetime occupational history using questionnaire to assess job titles and self-reported exposures, exposures ascertained by job-exposure matrices. |
Conditional logistic regression |
Matching variables, smoking |
|
Reference |
Description of Study Population |
Number of Cases |
Number of Controls |
Relevant Exposure(s) |
Determination of Exposure |
Analysis |
Adjustment for Confounding |
|
Brüning et al. 2003 |
Cases of renal cell carcinoma diagnosed in Germany (same region as in Vamvakas and Henschlerstudies), 1992-2000, and study conducted in 1999-2000. Controls, matched by sex and age (+5 yr), selected from surgery wards and departments of geriatrics in same area. Response rates: 82.7% of cases, not stated for controls. |
134 prevalent cases |
401 |
Occupation orindustry, specific exposure from JEM, self-reported exposure |
Occupational history and specific to TCE, self-reports of exposure to TCE and perchloroethylene, reports of prenarcotic symptoms for peak exposures. Used job-exposure matrix of Pannett and the CAREX system to infer exposures. |
Conditional logistic (based on frequency-matched) |
Matching variables, smoking |
|
ABBREVIATIONS: HCFA, health care financing administration; JEM, job-exposure matrix; RDD, random digit dialing; TCE, trichloroethylene. SOURCE: Adapted from IOM 2003. |
|||||||
TABLE 3-9 Selected Results from Case-Control Studies of Kidney Cancer and Occupational Exposure to Trichloroethylene
|
Reference |
Study Population |
Exposed Cases |
Estimated RR (95% CI) |
|
Siemiatycki, 1991; Parent et al. 2000 |
Kidney cancer, men, Montreal Exposure assessed by experts from job descriptions |
|
|
|
Any exposure |
4 |
0.8 (0.4-2.0)a |
|
|
Substantial exposure |
2 |
0.8 (0.2-2.6)a |
|
|
Dosemeci et al. 1999 |
Ever exposed to TCE, according to National Cancer Institute job-exposure matrix |
|
|
|
|
Residents of Minnesota |
55 |
1.30 (0.9-1.9) |
|
|
Men |
33 |
1.04 (0.6-1.7) |
|
|
Women |
22 |
1.96 (1.0-4.0) |
|
Vamvakas et al. 1998 |
Residents of Germany with long-term exposure |
19 |
10.8 (3.36-34.75) |
|
Pesch et al. 2000a |
Participants in multiple centers in Germany German job-exposure matrix |
|
|
|
|
Trichloroethylene (men) |
|
|
|
|
Medium |
135 |
1.1 (0.9-1.4) |
|
|
High |
138 |
1.1 (0.9-1.4) |
|
|
Substantial |
55 |
1.3 (0.9-1.8) |
|
|
Trichloroethylene (women) |
|
|
|
|
Medium |
28 |
1.2 (0.8-1.8) |
|
|
High |
29 |
1.3 (0.8-2.0) |
|
|
Substantial |
6 |
0.8 (0.3-1.9) |
|
|
Job task-exposure matrix approach Trichloroethylene (men) |
|
|
|
|
Medium |
68 |
1.3 (1.0-1.8) |
|
|
High |
59 |
1.1 (0.8-1.5) |
|
|
Substantial |
22 |
1.3 (0.8-2.1) |
|
|
Trichloroethylene (women) |
|
|
|
|
Medium |
11 |
1.3 (0.7-2.6) |
|
|
High |
7 |
0.8 (0.4-1.9) |
|
|
Substantial |
5 |
1.8 (0.6-5.0) |
(1995), Vamvakas et al. (1998), and Parent et al. (2000) controlled for at least smoking and body mass index. Because it is unlikely that exposure to trichloroethylene is associated with these factors, the committee judges that they do not significantly affect the estimates of risk. However, it might be useful in the risk assessment to distinguish and compare estimates of risk between studies in which confounding was and was not accounted for.
One possible source of bias that affects all case-control studies to some degree is the nonrepresentativeness of the study population to the target population. This can occur through sampling variation (essentially a ran-
dom error, which is reflected in the estimates of variance and confidence intervals) and also through systematic effects in which subjects are recruited in a nonrandom way that might depend on exposure status (selection bias). In most case-control studies, the sampling fraction for cases is close to 100%, so that any selection biases would be manifest only if selection probabilities in the control series varied with exposure. However, low response rates can affect the validity of the findings, especially if they are also associated with exposure. The committee did not inspect the case-control studies for possible selection biases, but this is a necessary step in the risk assessment. In addition, the committee did not evaluate every study with regard to response rates, except noting low response rates in some studies (e.g., Partanen et al. 1991; McCredie and Stewart 1993) and those studies in which response rates were not stated (Asal et al. 1988; Harrington et al. 1989; Mandel et al. 1995).
Exposure Assessment
Table 3-10 presents the essential characteristics of exposure for selected case-control studies. The committee has general concerns about studies that use self-reports of exposures to occupational agents, especially in large urban centers where the prevalence of any one exposure would be low (Sharpe et al. 1989; Mandel et al. 1995; Schlehofer et al. 1995). In contrast to personal characteristics, such as age and race, for which subjects’ reports are the gold standard, most individuals do not know what agents they used, especially if the agent occurred as part of a mixture or if a wide range of materials were used. On the other hand, most workers probably would know if they used a degreasing solvent.
The committee recognizes that there are special situations in which subjects are aware of their exposure circumstances. Questions about observable, objective facts tend to be answered accurately, such as “was there visible smoke in the air?” In workplaces where one or two materials are widely used, workers often know their common names, such as TCE, “Perc” (tetrachloroethylene), or Stoddard solvent, because there are limited choices and it makes a difference in the workplace which one is used. It is likely that workers enrolled in the study by Vamvakas et al. (1998) knew what solvent they were using; trichloroethylene was the solvent of choice for degreasing because it evaporates rapidly.
As with the cohort studies, an important factor that affects causal inference is how exposure is measured and assigned (validity, reliability) and whether exposure-response relationships are measurable. In studies in which analyses were conducted only by job title (Asal et al. 1988; Jensen et al. 1988; McCredie and Stewart 1993; Aupérin et al. 1994; Mellemgaard et al. 1994; Mandel et al. 1995), there would have been substantial mis-
TABLE 3-10 Characteristics of the Assessment of Exposure to Trichloroethylene in Selected Case-Control Studies
|
Study |
Qualitative Assessment |
Information on Settings |
|
Vamvakas et al. 1998 (follow-up to Henschler study) (pending legal cases for workers’ compensation—possible bias?) |
Small companies making metal parts in Arnsberg area (no cases from Henschler case group); TCE used for cleaning; high prevalence of companies in area; some use of Perc; qualitative assessment by occupational hygienists and physicians, interviews. |
Detailed info on TCE use, area descriptors—hot dip baths, small work areas, jobs, tasks—extensive cleaning; no controls. |
|
Brüning et al. 2003 (follow-up to Vamvakas study) |
Small companies making metal parts in Arnsberg area (no cases from Vamvakas case group); TCE used for cleaning; high prevalence of companies in area; some use of Perc; qualitative assessment by occupational hygienists. |
Detailed info on TCE use, area descriptors—hot dip baths, and small work areas, jobs, tasks—extensive cleaning; no controls. |
|
Brauch et al 1999; 2004 (molecular study of von Hippel-Lindau gene mutations in subjects highly exposed to TCE) |
TCE used up to 1970s. Questionnaire by personal interview; secondary questions if TCE reported; employer records, hygienists from insurance companies. |
Same plants as in Vamvakas study. Detailed info on TCE use, area descriptors—hot dip baths, small work areas, jobs, tasks—extensive cleaning; no controls. |
|
Pesch et al. 2000a,b (incidence study) |
Broad community study of five regions in Germany (included Arnsberg area). |
Limited data on exposure setting; self-assessed exposure. |
|
ABBREVIATIONS: JEM, job-exposure matrix; Perc, tetrachloroethylene; TCE, trichloroethylene. |
||
classification that would have attenuated rate ratios. In addition, in some studies exposure was attributed by expert assessments or through the use of job-exposure matrices, but the assessments were restricted to exposures to any solvents without identifying which were used (Harrington et al. 1989; Partanen et al. 1991; Dosemeci et al. 1999). In other studies, subjects reported whether they were exposed to specific agents (Sharpe et al. 1989; Mandel et al. 1995; Schlehofer et al. 1995; Vamvakas et al. 1998; Pesch et al. 2000a), and in some of those studies (e.g., Sharpe et al. 1989) rate ratios could have been overestimated because cases overreported exposure. In other studies, trichloroethylene was assessed specifically (Dosemeci et al.
|
Duration |
Exposure Quantification |
Exposure Extrapolation |
Dose Metrics |
|
Questionnaire, interviews, median 33 yr observation, sufficient latency. |
None (frequency and severity of neurological symptom reports used to estimate high exposures; >200 ppm likely). |
None in paper (Cherrie et al. [2001] extrapolated based on details in paper: peaks 400-600 ppm, long-term about 100 ppm). |
Combination of symptom severity and frequency and duration of exposures ranked +, low; ++, medium; +++, high. |
|
Questionnaire, interview job histories and next of kin; median 33 yr observation, sufficient latency. |
None (self-reported exposures, and two JEMs, CAREX, and British; >200 ppm likely for symptom reports). |
None in paper (Cherrie et al. [2001] extrapolated based on details in paper: peaks 400-600 ppm, long-term about 100 ppm). |
Job and industry groups with exposures from JEM; self-reported exposure and symptom frequency. |
|
Questionnaire, interview job histories and next of kin. |
Scheme developed for cases by Vamvakas et al. (1998). |
None in paper (Cherrie et al. [2001] extrapolated based on details in paper: peaks 400-600 ppm, long-term about 100 ppm). |
Combination of symptom severity and frequency of exposures; ranked +, low; ++, medium; +++, high. |
|
Questionnaire, interview job histories, agent use, and tasks. |
Broad German and British JEMs, and local JEM based on job titles and tasks using self-reported information. |
Expert judgment for who was exposed. No quantitative estimates. |
Ever exposed; agent index based on duration, intensity, and probability of exposure. |
1999; Parent et al. 2000; Pesch et al. 2000a; Brüning et al. 2003) and those studies may be more informative. In any risk assessment, the validity and reliability of the assessments of exposure have to be investigated closely.
Two small case-control studies conducted in the Arnsberg area of Germany found excess risks for exposure to trichloroethylene (Vamvakas et al. 1998; Brüning et al. 2003). The Vamvakas et al. (1998) study compared 58 renal cell cancer cases diagnosed between 1987 and 1992 with controls to provide an independent assessment of the Henschler et al. (1995) study. None of the cases in this study was included in the Henschler study. The odds ratios (OR) increased dramatically (OR = 1, 6.61, 11.92, and 11.42)
with increasing exposures (categories of “no exposure,” “+,” “++,” and “+++”, respectively, based on exposure types, duration, and extent of prenarcotic symptoms). As summarized in Table 3-11, there were large differences in the severity of symptoms and duration of exposure by the amount of exposure. There were also large differences in the source of exposure, with the use of hot dip tanks (major sources of trichloroethylene vapors) predominating in the highest category and largely rag-and-bucket cleaning (limited local sources of trichloroethylene vapors) in the lowest category, which are consistent with large differences in the intensity of exposure. Thus, there is a high degree of consistency between symptom severity and reports of exposures.
Another case-control study by Brüning et al. (2003) extended the period of observation from 1992 to 2000 and used the same rating scheme for exposure to trichloroethylene but used an independent set of cases and controls drawn from a wider geographic area. The validity of the data gathered by questions about neurological symptoms, which were asked of subjects in the Vamvakas et al. study, is a concern because legal proceedings were in progress to compensate workers for damage to their health. Two important considerations suggest that the workers’ reports are valid. First, the Vamvakas et al. scale did not rely only on neurological symptoms but
TABLE 3-11 Prenarcotic Symptoms and Exposure Duration and Intensity Associated with Rated Exposure Levels in the Vamvakas et al. (1998) Study
|
Descriptor |
Rated Exposure Level |
||
|
+ |
++ |
+++ |
|
|
Prenarcotic symptoms (number, # categorya) |
1, #1 |
4, #3 |
8, #3 |
|
3, #0 |
9, #2 |
2, #2 |
|
|
Frequency |
1, daily |
4, daily |
5, daily |
|
(number, daily or times per week) |
3, none |
4, 2 times per wk 5, 1 time per wk |
1, 3 times per wk 4, 2 times per wk |
|
Exposure types |
— |
2 hot clean, |
7 hot dip tanks, |
|
|
— |
9 cold dip tanks, |
1 welding on tanks with residues, |
|
|
3 rag and bucket |
1 rag and bucket, |
|
|
|
1 polishing |
1 dry clean |
2 cold dip tanks |
|
Total duration (range) |
Mean, 1,850 hr |
Mean 4,141 hr |
Mean, 28,800 hr |
|
(1,100-2,500 hr) |
(650-9,800 hr) |
(2,300-78,000 hr) |
|
|
aSymptom grades were as follows: #0, none; #1, light symptoms (light dizziness, modest headaches); #2, moderate symptoms (light daze, clear dizziness, headaches); #3, severe symptoms (daze vertigo, severe headaches, and nausea, which did not permit the subject to remain exposed). |
|||
also included an assessment of duration and exposure intensity associated with the particular activities. Second, there are a variety of sensory cues that taken together can distinguish low and high vapor concentrations in addition to neurological symptoms. For example, at low exposures the vapors are colorless, nonirritating, and not pungent, but with high concentrations (greater than several hundred ppm) there are a variety of sensory cues: the vapors are irritating, have a strong odor, and were found to produce reduced performance and central nervous system symptoms in human volunteers during experiments in the 1960s (Stopps and McLaughlin 1967; also see Chapter 6). Thus, the workers’ neurological symptoms were associated with other less subtle sensory responses, and they were only one dimension of the exposure evaluation. More importantly, overreporting would introduce misclassification, which would reduce the association between the symptoms and exposure.
The study by Vamvakas et al. (1998) has also been criticized in the literature; the essential observations are as follows:
-
Omission of cases from the Henschler et al. (1995) study (noted Mandel and Kelsh 2001). The effect of including these cases would lead to even higher estimates of risk.
-
Including only cases who worked in small industries and not applying the same criteria to controls (noted by Mandel and Kelsh 2001), which might lead to an underestimate of exposure among the control subjects and, thus, the odds ratios may have been overestimated. The committee shares the concern about this type of selection bias.
-
Cases were selected from one hospital but controls were selected from other hospitals in the area (noted by Green and Lash 1999; Mandel and Kelsh 2001). The committee is sanguine with regard to the selection of controls from other hospitals as it accepts the argument that these hospitals specialized in the type of care that they provided.
-
Prevalent cases (1987-1992) and controls (residual noncases) were selected in 1993 and interviews were conducted in 1993 (noted by Green and Lash 1999). McLaughlin and Blot (1997) suggested that survival in this period of time was 50% to 60%. Thus, some cases might have died in the interim before they could be interviewed and would have been excluded. This could have led to an inaccurate estimate of the exposure distribution. On the other hand, the control subjects who were enrolled when the interviews were conducted might not represent the true exposure distribution of the target population through time. In particular, exposures among the controls could have been underestimated if exposure diminished with time and if the selection of controls did not fully represent the actual distribution in the past (e.g., through changes in the population—immigration or emigration). Although this is a conjecture and the effects are difficult to predict,
-
a sound design would have attempted to minimize such distortions. Thus, the committee is concerned about the possibility of a selection bias in this study and about the quality of the data obtained from subjects diagnosed in the past, especially if self-reported exposures were partially the basis of the assessments of exposure to trichloroethylene.
-
Interviewers (physicians) were aware of subjects’ case status and surrogate respondents were used for deceased cases (but not for controls) (noted by Mandel and Kelsh 2001). One might expect that exposure of case subjects could have been overestimated because physicians more aggressively sought symptoms and exposure reports, although this was not possible for the deceased cases. It is unclear what overall effect this would have on the findings, although an analysis excluding the deceased cases would be useful.
-
Although the concentrations of exposure are unknown, the committee’s analysis of the data in the Vamvakas et al. (1998) study, presented in Table 3-12 (see Appendix D for more detailed analysis of the this study), makes it clear that the severity of symptoms and the severity and duration of exposures were all substantial and consistent for the cases, and the controls as a group had fewer symptoms and lower exposures. The committee disagrees with the conclusions of some critics (Green and Lash 1999; Cherrie et al. 2001; Mandel and Kelsh 2001) that it was unclear how exposure to trichloroethylene was assessed. Table 3-12, prepared from the data of Vamvakas et al. (1998), clearly shows that graded differences on several scales are consistent with the ratings. Thus, a clear ordinal scale is present. However, the precise magnitude of exposures associated with these ratings is difficult to assess. Cherrie et al. (2001) separately estimated the exposure intensities with a suitable engineering model, which estimated peak exposures in the range of 500 ppm and averages about 100 ppm. The committee agrees with this assessment (see Appendix D). These exposures were consistent with the symptom reports in laboratory studies (Stopps and McLaughlin 1967).
-
The control subjects were younger than the cases, implying a different potential for exposure to trichloroethylene (noted by Green and Lash 1999); therefore, risks could have been overestimated. If amounts of exposure have decreased and workers entered the workforce at about the same age across calendar periods, this could lead to an underestimate of exposure among controls, thereby leading to overstated risk ratios. However, responding to that criticism, the authors noted that there were no changes in exposure before 1986, when the allowable exposure was regulated. Given that the small enterprises have the highest exposures (Raaschou-Nielsen et al. 2002) and are usually the last to respond to regulations because of their limited resources, large employers are the initial focus of regulator activity. Further, the comment that younger workers would have lower exposures is
TABLE 3-12 Trichloroethylene Exposure Summary for the Arnsberg Area Studies
|
Study |
Peak Exposures |
Long-term Exposures |
Notes |
|
Henschler et al. 1995 |
>2,000 ppm, machine cleaning with neurological symptoms; about 100 ppm continuous cold cleaning. |
100 ppm 100 ppm |
Cherrie et al. (2001) estimates |
|
Vamvakas et al. 1998 |
400-600 ppm hot cleaning with neurological symptoms. |
100 ppm |
Cherrie et al. (2001) estimates |
|
Brüning et al. 2003 |
400-600 ppm hot cleaning, with neurological symptoms. |
100 ppm |
|
-
not generally true because apprentices usually do the least skilled, dirtiest jobs; in the United States and Europe, younger workers have the highest exposures.
-
The authors did not find the two main accepted risk factors for renal cancer (smoking and obesity) (Mandel and Kelsh 2001). The main risk factors appear to be weakly associated with renal cancer, so not identifying these associations could be due to chance, lack of statistical power, or possibly to homogeneity of the population.
The study by Brüning et al. (2003) was carried out in a broader region of southern Germany, which included the Arnsberg region, by the same team of investigators but covered the calendar period 1992-2000 and a different set of cases and controls. Again, prevalent cases of renal cell carcinoma were identified in 1992-2000 and interviews were conducted in 1999-2000. Controls were identified and interviewed in 1999-2000 and were recruited from noncancer patients having surgery and from a local department of geriatrics. The geriatric department was used to enroll controls for patients who were older. Exposure was assessed on the basis of occupational history and self-reports of exposure to trichloroethylene and tetrachloroethylene, reports of prenarcotic symptoms for peak exposures using the same scheme as that in the Vamvakas et al. study, and the job-exposure matrix of Pannett and the CAREX system to infer exposures. The committee judges that exposure range was likely similar to that in the Vamvakas et al. study, although they were drawing cases from a wider base population, which had a lower prevalence of exposures. For “ever exposed” to trichloroethylene, the investigators observed an OR of 2.47 and almost a 6-fold increase in risk among subjects who had daily occurrences of narcotic symptoms.
Some criticisms of this study are similar to those of the Vamvakas et al. study: (1) use of prevalent cases and residual noncases; (2) questions about
the specific secondary study base that was used (surgery, geriatric clinics) and how representative it was of the target population; (3) whether interviewers were blinded and whether any of the authors were interviewers; and (4) whether surrogate respondents were interviewed as controls for deceased cases. In addition, although subjects were matched, there were noticeable differences in age (median age of cases, 68 years; median age of controls, 66 years).
The study by Vamvakas et al. exhibited a very large estimated OR of 10.8. The committee has concerns that the true exposure distribution of the target population was underestimated in the enrolled control series (criticisms 2, 4, 5, and 7 described above). Given the very large estimates of risk, a sensitivity analysis is warranted if these data are to be used in a risk assessment. The follow-up case-control study of Brüning et al. showed an OR of 2.47 for the same type of self-reported exposure to trichloroethylene but a broader more heterogeneous base population; the OR for jobs involved in metal degreasing that had potential exposure to trichloroethylene and tetrachloroethylene was 5.57. As the committee evaluated that the assessment of exposure in this study was similar to that of Vamvakas et al., this lower odds ratio might indicate bias in the Vamvakas et al. study or statistical variation between studies because the Brüning et al. study included a broader base population than that of the Vamvakas et al. and the Henschler et al. (1995) studies, which could have entailed a greater extent of misclassification of exposures. Despite these issues, the committee was impressed that three studies of the Arnsberg region of Germany, with very high apparent exposures and different base populations, showed a significant elevation of risk.
If there is doubt about the validity of a study, then the risk assessment can be conducted by including and excluding that study and determining the sensitivity of the findings. With regard to the study by Henschler et al., which shows much higher risks than the others, sensitivity analyses are warranted and their need can be argued by analogy to the standard practice in epidemiology of assessing the effects of outliers; in risk assessment, this would be equivalent to the assessment of heterogeneity, except that the issue of a biased variance in the Henschler et al. study needs to be addressed.
Genetic Mutations and Kidney Cancer
Brüning et al. (1997a) described a possible somatic mutation in the von Hippel-Lindau (VHL) tumor suppressor gene in the etiology of renal cell carcinoma arising from exposure to trichloroethylene. Brauch and coworkers have reported additional studies (Brauch et al. 1999; 2004). Brauch et al. (1999) compared somatic mutations among 44 cases with documented exposure to trichloroethylene in metal-processing plants in the Arnsberg
TABLE 3-13 Number of Exposed and Unexposed Patients with VHL Gene Mutations
|
Trichloroethylene Exposurea |
VHL Mutational Status |
|||
|
None |
1 |
≥2 |
Total |
|
|
High, +++ |
2 |
4 |
11 |
17 |
|
Medium, ++ |
6 |
15 |
3 |
24 |
|
Low, + |
3 |
0 |
0 |
3 |
|
No documented occupational exposureb |
31 |
42 |
0 |
73 |
|
aAt the beginning of the discussion of the Brauch et al. (1999) paper it is noted that “The present study relies on the standardization of TRI [trichloroethylene] exposure levels of the RCC patients (10) … ,” where reference 10 is the Vamvakas et al. (1998) study; many of the coauthors are the same for both papers. Also in the paper by Brauch et al. (2004), p. 303, Table 1, a footnote notes that exposure data came from Vamvakas et al. (1998) subjects, and the case numbers of both Brauch et al. (1999, 2004) studies overlap. This indicates that the cases and their exposure assignments were obtained from the Vamvakas et al. (1998) study. bThe controls were drawn from other parts of Germany (populations assumed to be without the high prevalence of exposure to trichloroethylene) and evaluated by the same interview and questionnaire protocols. SOURCE: Adapted from Brauch et al. 1999. |
||||
region of Germany with 107 cases who had no such occupational exposure. The results of the study are summarized in Table 3-13. In addition, mutations at nucleotide 454 were found in 7 of the 17 high-exposure subjects and six of the 24 medium-exposure subjects, but no mutations were found in the three low-exposure patients or in 107 unexposed subjects.
Few details were presented about how subjects were selected, although they may have been recruited from the living subjects of the Vamvakas et al. (1998) case group. Whether there was blinding of exposure status in the assessments of somatic mutations was not stated.
In a second study, Brauch et al. (2004) reanalyzed cases from the Vamvakas et al. (1998) study for mutations in the VHL somatic gene. Thirty-eight of the original 58 patients with renal cell carcinoma were analyzed, and the authors used the original Vamvakas et al. exposure classification. Of the 17 exposed cases 15 had mutations and among the 21 unexposed cases 2 were found to have mutations (OR = 71.3). Because it was unclear to the committee why only this subset of cases was analyzed, a simple sensitivity analysis was conducted in which it was assumed that all 20 cases who were excluded were exposed but did not have any mutations. This analysis, which assumes extreme selection bias, still led to an OR of 6.5. An advantage of
this “case-only” analysis is that it does not require use of the control series (analogous to a case-only study in gene-environment interactions); it was unclear whether there was blinding of exposure status when the molecular analyses were conducted.
Role of Metabolism in Trichloroethylene-Induced Renal Tumors
Extensive studies of trichloroethylene metabolism, coupled to its potential mechanism of action in nephrocarcinogenicity, have been reported (reviewed by Brüning and Bolt 2000). Trichloroethylene induces renal toxicity and renal tumors in rats (Maltoni et al. 1988; NTP 1988, 1990; EPA 2001). The nephrocarcinogenic effects of trichloroethylene are more pronounced in male rats, compared with female rats and were absent in male and female mice.2 Studies of trichloroethylene metabolism in rodents and humans support a role for bioactivation in the development of nephrotoxicity and nephrocarcinogenicity after exposure to trichloroethylene (Lash et al. 1995, 2001a,b, 2002, 2003; Lash 2004). Trichloroethylene is metabolized by two competing pathways: oxidation by CYP450 and conjugation with glutathione (discussed earlier in this chapter; see Figure 3-1). Glutathione conjugation of trichloroethylene results in formation of S-(dichlorovinyl)glutathione, which is metabolized by enzymes of the mercapturic acid pathway (γ-glutamyl transpeptidase, aminopeptidase) to S-(1,2-dichlorovinyl)-L-cysteine, which is then metabolized by cysteine conjugate β-lyases, leading to the formation of electrophilic chlorothioketenes and sulfoxides. Concentrations of trichloroethylene in renal cortical homogenates have been reported to be generally two- to three-fold higher than in liver homogenates, and both oxidative and glutathione conjugation products were found in the liver and kidneys (Lash et al. 2006). These results are consistent with in vitro studies showing metabolism by kidney tissue. Males had substantially higher urinary excretion of S-(1,2-dichlorovinyl)-L-cysteine, suggesting greater metabolism by the glutathione pathway. It should be noted that results were reported for only three animals per time point and interpretation of the data is complicated by anomalous dose-concentration time profiles for trichloroethylene and its metabolites. The nephrotoxicity and nephrocarcinogenicity of trichloroethylene have been linked to the formation of S-(1,2-dichlorovinyl)-L-cysteine derivatives.
S-(1,2-Dichlorovinyl)-L-cysteine and its mercapturic acid metabolite N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine have been identified in the urine of humans exposed to trichloroethylene, providing evidence for the glutathione-dependent bioactivation of trichloroethylene in humans. Metabolism of trichloroethylene via the mercapturic acid metabolic pathway is consistent with the fact that the male rat is a sensitive species, because reduced glutathione (GSH) conjugation, γ-glutamyl transpeptidase, and cysteine conjugate β-lyase activity are all significantly higher in male than in female rats (Lash et al. 2002). Moreover, pharmacokinetic analysis of human volunteers after exposure to trichloroethylene (50 or 100 ppm) revealed that blood S-(dichlorovinyl)glutathione concentrations were 3.4-fold higher in males than in females, whereas clearance half-time values for systemic clearance of S-(dichlorovinyl)glutathione were similar in both genders (Lash et al. 1999). In the liver, metabolism of trichloroethylene via the mercapturic acid metabolic pathway is quantitatively less than via the CYP450-dependent metabolic pathway. However, the glutathione-dependent pathway becomes more pronounced when the oxidative metabolism of trichloroethylene is saturated in the case of high-dose exposure. Cummings and Lash (2000) demonstrated that human kidney tissue forms GSH conjugates with a Km (0.58 mM) in the range of Km values for oxidative metabolism by rodent microsomes (0.38 mM for mice, 0.07 and 0.48 mM for rats; Table C-2). They reported minimal or nondetectable P450-mediated trichloroethylene metabolism in human kidney tissue.
Genotoxicity
Trichloroethylene causes a significant increase in the incidence of renal tumors in rats when administered orally and a marginal incidence of renal tumors when administered via inhalation; on the basis of limited evidence for carcinogenicity in humans and sufficient evidence for carcinogenicity in experimental animals, the International Agency for Research on Cancer (IARC 1995b) classified trichloroethylene as a probable carcinogen in humans (group 2A). Moore and Harrington-Brock (2000) reviewed the genotoxicity of trichloroethylene and its glutathione-derived metabolites, and Brüning and Bolt (2000) reviewed the results of genotoxicity tests and concluded that trichloroethylene is, at most, a weak genotoxicant but noted that S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine have genotoxic effects including mutagenicity in the Ames test, unscheduled DNA synthesis, and formation of adducts in vitro with adenine, cytosine, and guanine. In the preliminary screening phase, the standard battery of genotoxicity tests might be unable to identify tissue-specific carcinogens, if the test system lacks the enzymes needed to form the toxic metabolite, and
certainly does not provide any information on the possible species specificity of the test compound (Brambilla and Martelli 2004; Moore and Harrington-Brock 2000). Recently, Robbiano et al. (2004) applied both in vitro and in vivo assays to measure genotoxicity to kidneys of rodents and repeated the assays in primary cultures of human kidney cells. Six chemicals known to induce kidney tumors in rats, including trichloroethylene, were examined for their ability to induce DNA fragmentation and the formation of micronuclei in primary cultures of rat and human kidney cells and in kidneys of intact rats. Each chemical was tested at three to six concentrations (four 2-fold dilutions for trichloroethylene); the highest concentration tested produced a less than 30% reduction in survival. Significant dose-dependent increases in the frequency of DNA single-strand breaks and alkali-labile sites (as measured by the Comet assay) and in micronuclei frequency were obtained in primary kidney cells from male rats and from humans of both genders, with subtoxic concentrations of trichloroethylene. Among the six test compounds (benzofuran, bromodichloromethane, captafol, nitrobenzene, ochratoxin A, and trichloroethylene), trichloroethylene and bromodichloromethane exhibited the lowest DNA-damaging and micronuclei-inducing potencies (with ochratoxin A exhibiting the highest) in rats and humans. In agreement with these findings, statistically significant increases in the average frequency of both DNA breaks and micronucleated cells were observed in the kidneys of rats given a single oral dose (half the lethal dose to 50% of rats) of the six test compounds. For all these effects, the magnitude of the response was among the greatest for trichloroethylene. The results of this study also showed that the six rat kidney carcinogens produced genotoxic effects in primary cultures of human kidney cells that were quantitatively and qualitatively similar to those observed in primary cultures of rat kidney cells. Taken together, these findings provide evidence that trichloroethylene is genotoxic in short-term genotoxicity assays in kidney cells isolated from rats and human donors.
However, the authors noted limitations in the experimental design that limit interpretation and the significance of the above studies (Robbiano et al. 2004). These limitations include (1) examination of trichloroethylene on cells from only three donors, (2) considerable variation in the frequency of DNA lesions induced in the cells, and (3) the possibility that kidney cells derived from kidney cancer patients could be more sensitive to DNA-damaging activity due to a more marked expression of enzymes involved in the metabolic activation of kidney procarcinogens and suppression of DNA repair processes. Therefore, the results of the genotoxicity studies must be considered solely as indicating that trichloroethylene might be genotoxic to the human kidney; the authors suggest that the designation of “inadequate evidence for carcinogenicity to humans” might not be tenable in the absence of sufficiently powered and carefully controlled epidemiologic studies.
Mode of Action
Role of von Hippel-Lindau Tumor Suppressor Gene
Most of the studies thus far reviewed on the subject of the renal carcinogenicity of trichloroethylene rely on either epidemiologic approaches or on studies of trichloroethylene metabolism and toxicity. The development of DNA technology and the discovery of tumor suppressor genes opened up a new route of investigation on the potential carcinogenic effects of trichloroethylene. Mutation or inactivation of the p53 tumor suppressor gene is a common genetic alteration in human cancers. However, the p53 gene is not a target in human and rat renal cell carcinoma (Reiter et al. 1993; Nishiyama et al. 1995). Inactivation of the VHL tumor suppressor gene in humans is responsible for the hereditary VHL cancer syndrome, predisposing affected individuals to a variety of tumors in specific target organs. More than 80% of sporadic renal cell carcinoma, but not papillary renal cell carcinoma, is associated with inactivation of the VHL gene (Gnarra et al. 1994). The VHL gene is only infrequently involved in extrarenal neoplasms, despite the broad range of VHL mRNA expression (including brain, adrenal, prostate, and lung), suggesting that its function as a tumor suppressor gene is specific for kidney epithelial cells (Walker 1998). The protein product of the VHL gene appears to regulate cell cycle arrest (transition from G1 to G0) by stabilizing the cyclin-dependent kinase inhibitor p27 (Soucek et al. 1998). Although the VHL gene, which is commonly mutated in human renal cell carcinoma, does not appear to be involved in rat renal cell carcinoma (Walker et al. 1996), it shares a common downstream effector (p27 that controls cell cycle progression) with the TSC2 gene, a genetic target of renal cell carcinoma development in the rat. Because VHL is not a target gene in rodent models of chemical-induced or spontaneous renal carcinogenesis, future animal studies should use models in which target genes share common downstream signaling pathways with VHL.
One paper has linked the VHL gene to chemical-induced carcinogenesis. Shiao et al. (1998) demonstrated VHL gene somatic mutations in N-nitrosodimethylamine-induced rat kidney cancers that were of the clear cell type. The clear cell phenotype is rare in rat kidney cancers, but it was the only the clear cell cancers that showed VHL somatic mutation. This provided an additional link between VHL inactivation and clear cell kidney cancer.
Brauch et al. (1999, 2004) analyzed renal cancer cell tissues for mutations of the VHL gene and reported increased occurrence of mutations in patients exposed to high concentrations of trichloroethylene. In the first study (Brauch et al. 1999), subjects were identified from an occupational trichloroethylene exposure registry. They found multiple mutations in 42%
of the exposed patients who experienced any mutation and 57% showed loss of heterozygosity. A hot spot mutation of cytosine to thymine at nucleotide 454 (C454T) was found in 39% of samples that had a VHL mutation and was not found in renal cell cancers from nonexposed patients or in lymphocyte DNA from either exposed or nonexposed cases or controls. As discussed earlier, little information was given on how subjects were selected and whether there was blinding of exposure status during the DNA analysis.
In the second study, Brauch et al. (2004) investigated 38 renal cell carcinoma patients from a previous German case-control study performed by Vamvakas et al. (1998). Brauch et al. compared different renal cell carcinoma patient groups (trichloroethylene-exposed versus nontrichloroethylene-exposed patients). The Vamvakas et al. study had described differences in renal cell carcinoma risks between trichloroethylene-exposed (n = 17) and nonexposed patients (n = 21). Brauch et al. (2004) extended the analysis by comparing age at diagnosis and histopathologic parameters of tumors as well as somatic mutation characteristics in the VHL tumor suppressor gene. Renal cell carcinoma did not differ with respect to histopathologic characteristics in both patient groups. Comparing results from trichloroethylene-exposed and nonexposed patients revealed clear differences with respect to (1) frequency of somatic VHL mutations, (2) incidence of C454T transition, and (3) incidence of multiple mutations. The latter is an indication that the effect of trichloroethylene is not limited to clonal expansion of cells mutated by some other agent. The C454T hot spot mutation was exclusively detected in tumors from trichloroethylene-exposed patients, as were multiple mutations. Also the incidence of VHL mutations in the trichloroethylene-exposed group was at least 2-fold higher than in the nonexposed group.
Brauch et al. were not able to analyze all the samples from the Vamvakas study, in part because samples were no longer available. Using the data described by Brauch et al. (2004) (VHL mutation found in 15 exposed and 2 nonexposed individuals, and VHL mutation not found in 2 exposed and 19 unexposed individuals), an OR of 71.3 is calculated. The most extreme example would be to assume that all 20 cases who were excluded were exposed but did not have mutations in VHL (VHL mutations were found in 15 exposed and 2 unexposed individuals and VHL was not found in 22 exposed and 18 unexposed individuals), which leads to an OR of 6.5, which remains significant.
Collectively, the data support the concept of a genotoxic effect of trichloroethylene leading to VHL gene damage and subsequent occurrence of renal cell carcinoma in highly exposed subjects. All the evidence, taken together, provides a consistent and plausible mechanism for a causal relationship and is strongly supportive of trichloroethylene being a human carcinogen after long-term exposure to high doses, such as occupational
exposures described in both studies conducted in Germany (Henschler et al. 1995; Vamvakas et al. 1998).
The VHL gene is commonly altered in kidney tumors, especially those with the clear cell phenotypes. The alterations include loss of the entire or a large part of the gene (>90%) and small base changes (30% to 60%), including insertions, deletions, and point mutations (Shiao 2004). These changes can lead to reduction of protein expression, protein truncation, and incorrect amino acids incorporated into the protein (also called missense mutation). Consequently, wild-type constitutive functions of VHL are inactivated, with the subsequent potential to initiate and to promote tumor development. However, different mutations might have distinct tumorigenic potentials. Frequent and diverse VHL mutations in sporadic renal cell carcinoma provide a sizable mutation spectrum that has been used to correlate with environmental exposures. The rationale of using genetic signature as a marker of environmental exposure has been strengthened by in vitro and in vivo studies. Correlating of specific mutations within the VHL gene with certain environmental exposures could lend support to the potential mutagenicity of an agent. Identification of DNA damage unique to exposure is necessary to provide strong evidence for the mutagenic potential of an environmental agent. Many types of DNA damage have been shown to induce unique signatures of gene mutations (see Table 3-14).
A worldwide mutation database compiling VHL mutations in sporadic renal cell carcinoma showed that missense mutations compose about 29% of all mutations; a large majority of base changes (71%) are nonmissense, including insertions, deletions, and frameshift alterations (see Table 3-15). When bases were determined, G:C to A:T, A:T to G:C, and A:T to C:G composed 48% of the changes. Similar mutation spectra have been obtained
TABLE 3-14 Mutation Spectra Indicative of Environmental Exposures and DNA Damage
|
Base Change |
Possible Causes |
|
Transition |
|
|
G:C to A:T |
Deamination of 5-methyl-C or C; alkylation of G at O6 position |
|
A:T to G:C |
Deamination of A; alkylation of T at O2 or O4 position |
|
Transversion |
|
|
G:C to T:A |
Mispairing of A with 8-OH-G or with apurinic G |
|
A:T to T:A |
Mispairing of A with apurinic A site |
|
A:T to C:G |
Misincorporation of 8-OH-G; error-prone repair of O2- or O4-alkyl T |
|
G:C to C:G |
Mispairing of G with oxidatively damaged G |
|
SOURCE: Shiao 2004. Reprinted with permission; copyright 2004, National Cancer Institute at Frederick. |
|
TABLE 3-15 VHL Mutations in Sporadic Renal Cell Carcinomas
|
|
Trichloroethylene Exposure |
|||||
|
Brüning et al. 1997a |
Brauch et al. 1999 |
Brauch et al. 2004 |
UMDa |
|||
|
Yes |
Yes |
No |
Yes |
No |
Unknown |
|
|
Number of patients |
23 |
44 |
73 |
17 |
21 |
|
|
Patients with mutations |
23 (100%) |
33 (75%) |
42 (58%) |
14 (82%) |
2 (10%) |
|
|
Number of mutations |
23b |
50 |
42 |
24 |
2 |
222 |
|
Missense |
1 |
27 (54%) |
NA |
17 (71%) |
2 (100%) |
64 (29%) |
|
Nonmissense |
3 |
23 (46%) |
NA |
7 (29%) |
0 (0%) |
158 (71%) |
|
G:C to A:T |
1 |
21 (78%) |
NA |
12 (71%) |
1 (50%) |
21 (25%) |
|
C to T at 454 |
|
(13) |
(0/107) |
(9) |
(0) |
(0) |
|
G:C to T:A |
|
0 |
NA |
0 |
0 |
19 (22%) |
|
G:C to C:G |
|
5 (19%) |
NA |
4 (24%) |
0 |
16 (19%) |
|
A:T to T:A |
|
1 (4%) |
NA |
1 (6%) |
0 |
9 (11%) |
|
A:T to G:C |
|
0 |
NA |
0 |
1(50%) |
14 (16%) |
|
A:T to C:G |
|
0 |
NA |
0 |
0 |
6 (7%) |
|
aUniversal Mutation Database (Beroud et al. 2000). bBy single-strand conformation polymorphism (4 sequences confirmed). ABBREVIATION: NA, not applicable. |
||||||
from cells and animals treated with alkylating agents, such as nitrosamines found in tobacco smoke and potent human and animal renal carcinogens. The involvement of alkylating agents in the causation of renal cell carcinoma is further supported by the isolation of O6-methylguanine and other alkylated DNA-damaged bases. However, mutation spectra after exposure to trichloroethylene or analog compounds, in cells and animals, have not been consistent. Nonetheless, increases in GC to AT and GC to TA mutations have been observed in bacteria. Muller et al. (1998) identified cytosine adducts from haloketene and halothioketene products of trichloroethylene; these are structurally similar to hydroxylamine cytosine adducts that result in C to T mutations (Budowsky 1976). Increases in VHL missense mutations, predominant in G:C to A:T base changes, and a hot spot of mutation at nucleotide 454, correlated with trichloroethylene exposure (Brauch et al. 1999). The three reports of trichloroethylene exposure from the same group suggest that trichloroethylene increases VHL mutations and generates a unique genetic signature of trichloroethylene exposure, which leads to the development of renal cell carcinoma. Although the findings linking trichloroethylene to renal cancer are of great consequence and relevance, further
confirmation of mutagenicity and carcinogenicity at the molecular level is required to confirm the initial observations. As discussed earlier, consensus for the mutagenicity of trichloroethylene in mammalian cells remains to be established. If the mutation spectra in bacteria are considered, one would expect to see increases of both G:C to A:T and G:C to T:A mutations in trichloroethylene-exposed humans. However, a disproportionate number of G:C to A:T VHL mutations were reported (Brauch et al. 1999). Because alkylating agents, present in patients exposed to tobacco smoke, diuretic treatment for hypertension, and long-term dialysis for end-stage renal failure, also induce the same G:C to A:T base changes, analysis of prior trichloroethylene studies need to adjust for these risk factors. The temporal relationship between various mutations in the VHL gene and renal tumor progression needs to be examined more critically to unequivocally evaluate the cause-and-effect relationships. The mutagenicity of trichloroethylene should also be validated in additional cohorts. Further, the tumorigenic potentials of various VHL mutations need to be integrated, because mutated bases need not always be carcinogenic.
It remains debatable whether alterations in VHL alone are sufficient to trigger tumorigenic processes in the kidney, especially since experiments failed to detect any tumors in VHL knockout mice (Gnarra et al. 1997; Haase et al. 2001). Studies attempting to link the VHL gene to kidney tumor development are continuing in a variety of experimental models (Shiao et al. 1997, 1998; Walker 1998). However, there does not appear to be an experimental animal model with which to investigate the effects of trichloroethylene-induced mutations in the VHL gene and kidney tumor development.
Role of Nephrotoxicity in Trichloroethylene Renal Cancer
In animal studies, renal cancer occurs at high doses and is preceded by nephrotoxicity affecting the proximal tubule (NTP 1988, 1990). This has led to the proposal that nephrotoxicity is a prerequisite for the development of renal tumors and that exposures below nephrotoxic concentrations pose no risk of cancer. That is, there is a threshold exposure below which nephrotoxicity, and therefore renal cancer, will not occur (Brüning and Bolt 2000; Harth et al. 2005). In this scenario, nephrotoxicity, and subsequent cell division repairing that damage, functions as a promoter, allowing the expression of mutations (either spontaneous or induced by exposure to other agents, such as smoking and diuretics) within the renal cortex. Alternatively, trichloroethylene is a complete carcinogen, with nephrotoxicity as the promoter for cells initiated by a trichloroethylene metabolite. There is evidence that trichloroethylene is genotoxic to human cells (Robbiano et al. 2004).
Nephrotoxicity is almost certainly secondary to formation of a toxic metabolite, and species differences in the extent of formation of that toxic metabolite could render humans less likely to develop nephrotoxicity and therefore cancer. The CYP2E1 and -3A5 isoforms that metabolize trichloroethylene have polymorphisms within national populations, resulting in considerable interindividual differences of enzyme expression. On a practical level, the population diversity in bioactivation and detoxification abilities could effectively obscure any threshold.
Investigations of nephrotoxicity in human populations have been pursued and the results show that highly exposed workers experience a tubular type of proteinuria, evidence of damage to the proximal tubule (Brüning et al. 1999a,b; Bolt et al. 2004). What is not clear is the magnitude of exposure needed to produce kidney damage. The fact that proteinuria was found in workers exposed to trichloroethylene concentrations that were not measured but were described as current occupational exposures (Green et al. 2004) is inconsistent with nephrotoxicity occurring only at high exposures that are not relevant to current occupational exposures.
FINDINGS
Although the committee was not charged with performing a risk assessment, it became clear from the epidemiologic evidence that there were sufficient data to make a recommendation about whether the findings of the mortality and incidence studies provided support for or against the hypothesis that exposure to trichloroethylene was associated with the induction of kidney cancer. There is strong evidence that exposure to high doses of trichloroethylene is associated with increased rates of kidney cancer. In particular, support for this conclusion derives from findings of increased risks in a cohort study (Henschler et al. 1995) and in case-control studies from the Arnsburg region of Germany (Vamvakas et al. 1998; Pesch et al. 2000a; Brüning et al. 2003). The committee notes that, as the designs of these case-control studies improved with time, increased risks were still observed. In addition, the finding of a mutation in the VHL somatic gene adds strength to these observations, although it would be useful if this finding were replicated in other settings. Of considerable interest was the finding of an increased risk among workers of a cardboard manufacturing plant in the United States (Sinks et al. 1992), who might have had exposures comparable to that in the study by Henschler et al. (1995). Other studies with appropriate power to detect risks from relatively low exposures also showed increased risks, notably the studies by Dosemeci et al. (1999), Raaschou-Nielsen et al. (2003), and Zhao et al. (2005).
Supporting this conclusion is the concordance between studies on humans and experimental animals for the site of tumors and occurrence of
toxicity. In bioassay studies, rats developed tubular toxicity before tumors developed. Nephrotoxicity preceding cancer also appears likely in humans, although nephrotoxicity assessments in human studies were not made until after the development of renal cancer and were based on only one parameter.
The committee reviewed studies on two modes of toxicity proposed to be linked to cancer—accumulation of α2µ-globulin and PPAR agonism. The committee concluded the evidence demonstrates these modes do not occur for trichloroethylene-induced renal cancer. The committee also concluded that trichloroethylene causes an increase in the urinary excretion of formate but notes the disparities between formate production and toxicity contradicts the conclusion that accumulation of formate is a mode of action for trichloroethylene nephrotoxicity.
Studies with experimental animals and human tissues support the conclusion that trichloroethylene, via one or more of its metabolites, is genotoxic. In animal studies, trichloroethylene appears to be a weak genotoxicant. The studies with human tissues used a small number of samples and, therefore, the committee notes this weakens the weight of evidence.
In the kidney, trichloroethylene can act as a complete carcinogen (at the stages of both tumor initiation and tumor promotion and progression) in a dose-dependent manner. Different types of kidney cancer can be triggered by different genes. After the discovery of the VHL tumor suppressor gene, it became recognized that homozygous inactivation of the VHL gene was linked to the occurrence of renal clear cell carcinoma, the renal carcinoma preferentially induced by trichloroethylene. In exposed subjects, the genotoxic effect of trichloroethylene likely results from bioactivation pathways leading to renal VHL gene damage and renal cell carcinomas. The findings of experimental, mechanistic, and epidemiologic studies lead to the conclusion that trichloroethylene can be considered a potential human carcinogen.
RESEARCH RECOMMENDATIONS
-
Because sulfoxide metabolites are more potent nephrotoxicants than their parent S-conjugates, more research is needed on the extent of formation of S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine sulfoxides by human tissues (liver and kidney), the extent to which these reactions occur in vivo, the enzymes involved, and their interindividual variability, including the role of genetic polymorphisms. The toxicologic significance of trichloroethylene or S-(1,2-dichlorovinyl)-L-cysteine S-conjugate sulfoxidation products also should be evaluated.
-
High frequencies of missense mutations in the VHL gene do not constitute a cause of renal cell carcinoma; the tumorigenic potential of missense
-
mutations in the VHL gene should be determined. The potential of specific missense mutations in the VHL gene contributing to tumor initiation and progression should be determined.
-
Although correlation of VHL mutations to trichloroethylene exposure and renal cell cancer are persuasive, the findings need to be validated in other populations and geographic areas. Because many risk factors for renal cell carcinoma generate mutation spectra similar to that of trichloroethylene, coexposure to trichloroethylene with other risk factors needs to be seriously considered and accounted for in future epidemiologic studies.
-
Mechanistic studies should include field studies of populations exposed to trichloroethylene to assess the range of metabolic pathways used and relative amounts of metabolites from each pathway as a function of exposure intensity and enzymatic genotypes. This information will greatly help in the interpretation and extrapolation of information from rodents to humans.
-
Additional studies of nephrotoxicity in workers exposed occupationally to trichloroethylene should be performed. It is important that actual exposures are measured and not estimated using biological markers that are subject to large interindividual differences.
-
No analytic community studies were included in the committee’s assessment of kidney cancer. Given the importance of contamination of water supplies by trichloroethylene, it is important that sufficiently robust studies (with sufficient statistical power and exposure assessments) be conducted in the general population where such exposures might be occurring.
-
Any follow-up epidemiologic study must have a wide range of exposures, preferably to the range of the Vamvakas and Henschler studies to provide an anchor in that range where effects were seen. There may be opportunities for studies of populations in developing countries in Asia and Eastern Europe, where high exposures to trichloroethylene may not have been controlled. Strong, quantitative exposure assessments will be critical for these studies to be useful for resolving the remaining dose-response issues.















































































