
6
Emerging Principles of Regulatory Evolution
BENJAMIN PRUD’HOMME,*‡ NICOLAS GOMPEL,†‡ and SEAN B. CARROLL*
Understanding the genetic and molecular mechanisms governing the evolution of morphology is a major challenge in biology. Because most animals share a conserved repertoire of bodybuilding and -patterning genes, morphological diversity appears to evolve primarily through changes in the deployment of these genes during development. The complex expression patterns of developmentally regulated genes are typically controlled by numerous independent cis-regulatory elements (CREs). It has been proposed that morphological evolution relies predominantly on changes in the architecture of gene regulatory networks and in particular on functional changes within CREs. Here, we discuss recent experimental studies that support this hypothesis and reveal some unanticipated features of how regulatory evolution occurs. From this growing body of evidence, we identify three key operating principles underlying regulatory evolution, that is, how regulatory evolution: (i) uses available genetic components in the form of preexisting and active transcription factors and CREs to generate novelty; (ii) minimizes the penalty to overall fitness by introducing discrete changes in gene expression; and (iii) allows
interactions to arise among any transcription factor and downstream CRE. These principles endow regulatory evolution with a vast creative potential that accounts for both relatively modest morphological differences among closely related species and more profound anatomical divergences among groups at higher taxonomical levels.
It has long been understood that morphological evolution occurs through alterations of embryonic development (Gould, 1977; Raff and Kaufman, 1983). The key catalyst to the molecular study of morphological evolution has been the identification and functional characterization of developmental genes in animal model systems beginning in the 1980s. The development of specific body parts and organs was revealed to be orchestrated by networks of patterning genes that encode mostly transcription factors and cell-signaling molecules. It was then gradually realized that the formation of similar body parts and functionally equivalent organs in widely divergent animals is controlled by remarkably similar sets of orthologous pattern-regulating genes that have been conserved over hundreds of million years of evolution (Duboule and Dolle, 1989; Graham et al., 1989; Quiring et al., 1994; De Robertis and Sasai, 1996; Panganiban et al., 1997; Bodmer and Venkatesh, 1998; Carroll et al., 2004). However, the unexpected widespread genetic similarities presented a new paradox: if all animals are built by using similar genetic tools, how did their seemingly endless morphological diversity arise?
A vast body of comparative studies has revealed that morphological differences among taxa are correlated with differences in developmental gene expression patterns, which has supported the proposal that evolutionary modifications of gene expression (i.e., “regulatory evolution”) are the basis of morphological diversification (King and Wilson, 1975; Carroll, 1995). The question of morphological evolution then turned to how such spatial differences in gene expression arise. In principle, gene expression may evolve through changes in either the activity or the deployment of the proteins (primarily transcription factors) that govern gene expression, or in the regulatory sequences that modulate the expression of individual genes (at the DNA or RNA level).
Two clues to the general resolution of these alternatives were emerging from molecular developmental biology by the early 1990s. The first was the structural conservation and functional equivalence of key transcription factors, such as Hox proteins, which indicated that their biochemical activities were not diverging much, if at all (McGinnis et al., 1990; Halder et al., 1995). The second was the discovery of the unexpectedly complex and modular organization of the cis-regulatory regions of pattern-regulating genes (Stanojevic et al., 1991; Davidson, 2001). Most loci encoding pattern-
regulating proteins were found to include multiple individual cis-regulatory elements (CREs), with each CRE typically comprising binding sites for multiple distinct transcription factors and controlling gene expression within a discrete spatial domain in a developing animal. The realization that the total expression pattern of a gene was the sum of many parts, each directed by distinct CREs, marked a profound change in concepts of gene regulation. The modular arrangement of CREs also had clear implications for evolutionary genetics, because it suggested a mechanism for how selective changes in gene expression and morphology could evolve in one part of the body, independent of other parts (Carroll, 1995). The conservation of the biochemical activity of regulatory proteins, the divergence of their expression patterns across taxa, and the modular organization of CREs provided the basis for the general proposal that gene expression evolution, and therefore morphological evolution, would occur primarily through changes in cis-regulatory sequences controlling gene transcription (Carroll, 1995).
However, the evolutionary significance of the properties of CREs was not widely recognized at the time and, in our view, may still not be fully appreciated. We think there are several possible reasons for this (Carroll, 2005b). First, there is a much longer history of the analysis of coding sequences in evolutionary and population genetics. Second, the role of gene duplication has also long figured prominently in ideas about evolutionary novelty (Ohno, 1970). In contrast, the recognition of the complexity and evolutionary potential of CREs is more recent and has emerged primarily from molecular developmental genetics, outside of the primary literature of evolutionary genetics. And finally, there have been few detailed functional studies of CRE evolution. Most studies have focused on the functional conservation of CREs (Ludwig et al., 2000, 2005; Wratten et al., 2006). Until very recently, there have been very few direct empirical examples linking CRE evolution to morphological evolution (Belting et al., 1998; Wang and Chamberlin, 2002). As a result, beyond the growing acceptance of why regulatory evolution plays a role in morphological evolution, our understanding of how regulatory evolution occurs has been limited.
The elucidation of the mechanisms of CRE evolution in morphological diversification has required the identification of appropriate experimental systems. Because coding sequences are usually sufficiently conserved to identify orthologous sequences among different phyla, it was naïvely assumed initially that the same would hold true for CREs, and that functional comparison of divergent CREs from distantly related taxa would be possible. However, it was progressively realized that the turnover rate of noncoding DNA is much higher than for coding sequences, largely because of looser functional constraints, making orthologous sequence
identification and comparison much more difficult (Erives and Levine, 2004; Richards et al., 2005; Wittkopp, 2006) and often impossible, especially among higher arthropod taxa. To circumvent this difficulty, an alternative strategy has been to focus on rapidly evolving traits among closely related species or populations. The advantages of this approach are 2-fold: first, because the organisms under comparison diverged recently, it is expected that the number of genetic changes responsible for morphological divergence will be relatively modest and more readily distinguished from other changes not involved in morphological divergence. Second, in some cases, the relevant genetic changes can be investigated in their native ecological context and related to the potential adaptive role, if any, of morphological evolution.
Following this approach, recent studies have provided direct evidence of the role of CRE evolution in morphological evolution (Gompel et al., 2005; Jeong et al., 2006; Prud’homme et al., 2006). More importantly, these detailed functional analyses have revealed some surprising and previously unanticipated features of how gene regulation evolves at the molecular level that, we suggest, reflect general principles. The goal of this article is to articulate these emerging principles, namely how regulatory evolution: (i) proceeds using available preexisting genetic components, (ii) introduces discrete changes in gene expression thus minimizing deleterious effects and fitness penalties, and (iii) allows the association between any transcription factor and any downstream gene and thereby provides immense potential for evolutionary novelty. These principles explain both how and why regulatory sequence evolution is a pervasive, although not the exclusive, mechanism underlying morphological diversification.
PIGMENTATION PATTERNS AND GENE EXPRESSION AS MODELS OF REGULATORY EVOLUTION
Because morphological evolution is the product of the modification of the expression patterns of underlying genes, to understand how morphological changes arise, we must understand how changes in gene expression pattern arise. Pigmentation patterns in insects have been particularly amenable for these purposes for two reasons: first, many genes governing their formation have been characterized; and second, patterns are highly variable among closely related species, giving very different appearances to otherwise identical body parts (Wittkopp et al., 2003). For instance, the wings of some higher Diptera are largely identical with respect to their overall shape and venation patterns. Yet the various pigmentation patterns superimposed onto the common wing plan, ranging from a simple line, dot, or blotch to complex compound patterns make each species wing pattern largely different from the others (Fig. 6.1).

FIGURE 6.1 Wing pigmentation pattern diversity across higher Diptera. This plate illustrates the diversity of wing pigmentation patterns in the Acalyptratae, a large group of higher Diptera (Cyclorrhapha), which contrasts with the remarkable conservation of shape, dimension ratios, and venation patterns of these wings after >70 million years of evolution.
Pigmentation patterns result from the local conversion of precursor metabolites into pigment deposits by several enzymes (Wittkopp et al., 2003). The expression patterns of these enzymes, generally specified at an advanced developmental stage when the overall morphology closely resembles the adult layout, are the blueprints of the visible pigmentation patterns (Wittkopp et al., 2002a). Therefore, understanding how the expression patterns of the genes encoding these enzymes are established and change among species is key to understanding the formation and diversification of pigmentation patterns.
In principle, pigmentation patterns could evolve by changing the activity or spatial deployment of transcription factors that regulate pigmentation genes and/or by changes in the CREs of pigmentation genes themselves. Furthermore, such changes in CREs could entail either the modification of existing CREs or the de novo evolution of a CRE. Six cases of the gain or loss of pigmentation gene expression in fruit fly species have now been traced to the evolution of pigmentation gene CREs (Gompel et al., 2005; Jeong et al., 2006; Prud’homme et al., 2006). The frequency and details of CRE sequence evolution and the identity of the transcription factors involved in regulating these CREs and other case studies of morphological divergence among closely related species or populations (Belting et al., 1998; Sucena and Stern, 2000; Wang and Chamberlin, 2002; Sucena et al., 2003; Shapiro et al., 2004; Colosimo et al., 2005; Marcellini and Simpson, 2006) illustrate what we submit are general insights into the process of evolution by gene regulation.
USING AVAILABLE GENETIC COMPONENTS TO GENERATE NOVELTY
In some members of the Drosophila melanogaster species group, the males bear dark spots at the anterior tips of their wings, whereas in most other species, they do not (Kopp and True, 2002; Prud’homme et al., 2006). The evolution of the male wing spot thus presents a simple example of a novel pattern and poses a simple question: what changed between unspotted and spotted species? The difference in pigmentation patterns is reflected by differences in pigmentation gene expression. In particular, the product of the yellow (y) gene, which is critical for the production of black pigment (Walter et al., 1991), is expressed uniformly at low levels in the developing wing blade in unspotted species and in spotted species; it is also expressed at high levels where the spot will appear.
How did yellow expression evolve? The evolutionary divergence in Yellow expression results from functional changes in a CRE controlling y expression in the developing wing (the wing CRE). In unspotted species, this CRE, which is ≈1 kb long, drives a uniform expression pattern throughout the wing (Wittkopp et al., 2002b). In spotted species, the regulatory activity of this element has changed to also drive high levels of yellow in the spot area (Gompel et al., 2005). Therefore, in this instance, an ancestral CRE has been coopted and functionally modified to become a wing + spot CRE and to generate a novel pattern.
In theory, a spot CRE with a full complement of transcription factor-binding sites necessary to drive a wing spot pattern could have evolved anywhere in the yellow locus. However, a functional CRE usually requires a substantial number of inputs to generate a spatially restricted expression
pattern (Davidson, 2001). If a functional CRE were to evolve from naïve DNA, the evolutionary path to acquire all of the necessary transcription factor-binding sites, in a functional arrangement, would be relatively long, and it is difficult to see how selection might favor the intermediates. In contrast, a CRE that is functional in a given tissue already contains some of the sites necessary to direct gene expression in that tissue, and therefore it represents a more likely template to accommodate a new expression pattern in that tissue, because a relatively shorter evolutionary path would lead to functional novelty. Consequently, it seems more probable that a novel gene expression pattern in a tissue will arise from random mutations creating binding sites in the vicinity of an existing CRE driving expression in that tissue than from mutations in non-functional DNA.
Which trans-acting factor-binding sites have evolved in the wing CRE to create the spot pattern? In principle, this element could have evolved binding sites for a single transcriptional activator, which, in turn, had evolved to be expressed in a spot pattern. In fact, however, the formation of the yellow spot pattern entailed the evolution of binding sites for both activators and repressors involved in the building of the wing. In particular, the transcription factor Engrailed, present in cells in the posterior part of the wing, directly represses yellow expression, confining elevated yellow expression and, therefore, the formation of the pigmentation spot to the anterior region (Gompel et al., 2005).
The key point regarding the identity of Engrailed is that it is not a transcription factor specifically dedicated to pigmentation. Engrailed is a deeply conserved component of arthropod segmentation and appendage development, and its expression in the posterior compartment long preceded its involvement in the patterning of the pigmentation spot (Patel et al., 1989). Nevertheless, in this particular context, the evolutionary process took advantage of its presence and established a direct regulatory connection between Engrailed and a pigmentation gene, thus sculpting the contour of the pigmentation spot. In this instance, Engrailed has been recruited for a new function, without any change occurring in its activity, protein sequence, or expression.
The evolution of the wing spot illuminates a general mechanism by which a novel pigmentation pattern can be generated (Fig. 6.2). The development of the wing or any body part or organ is a sequential process controlled by an array of regulatory proteins (Carroll et al., 2004). As development proceeds, the expression of these proteins progressively delineates the wing layout, position of the veins, sensory organs, and so on. Collectively, the expression profiles of all wing-building transcription factors compose a complex mosaic of superimposed patterns or “trans-regulatory landscape” (Fig. 6.3). If and when combinations of binding sites for members of the trans-regulatory landscape evolve in the CRE of
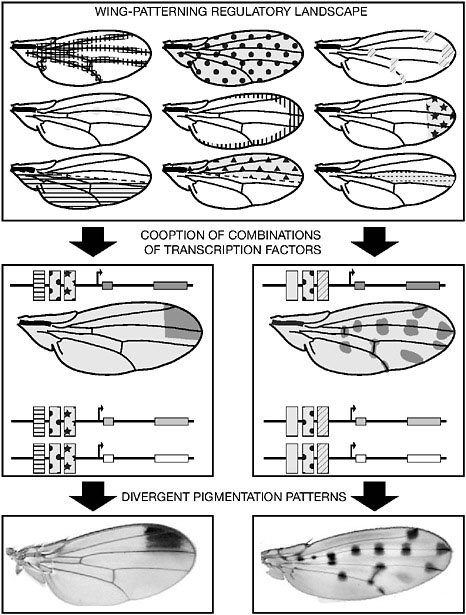
FIGURE 6.2 Regulatory evolution and wing pigmentation pattern diversity. A suite of transcription factors control the development of the fly wing, each one being expressed in a particular pattern. Altogether, these expression patterns constitute a wing trans-regulatory landscape, conserved among Drosophila species (Top). The recruitment of a subset of the trans-regulatory landscape components by pigmentation genes results in the corresponding redeployment of these genes (Middle) and ultimately in a novel wing pigmentation pattern (Bottom). The recruitment of different combinations of trans-acting factors in different fly species yields distinct pigmentation patterns. In Middle, rectangles with motif represent binding sites for different trans-regulatory landscape components.

FIGURE 6.3 A glimpse of the actual wing trans-regulatory landscape. The expression of GFP reports the expression patterns of various wing transcription factors at the time pigmentation genes are being expressed.
a pigmentation gene, then the expression profile of this gene may change. Because the formation of pigmentation patterns requires the coincident deployment of multiple pigmentation genes, it can be anticipated that these genes will fall under the control of a common suite of trans-acting factors.
In this view, diverse pigmentation patterns can arise from the evolution of regulatory connections among pigmentation gene CREs and different combinations of transcription factors (Fig. 6.2). In particular, more elaborate patterns can evolve through the progressive accumulation of regulatory links between components of the trans-landscape and pigmentation genes CREs. This gradual elaboration of complex patterns is reflected in the graded series of pigmentation patterns found in several fly lineages, where new elements are added in more derived species (e.g., the top three wings in the right column in Fig. 6.1). Hence, the complexity of the wing trans-regulatory landscape and the combinatorial nature of gene expression regulation are sufficient to account for the spectacular diversity of wing pigmentation patterns.
More generally, the evolution of wing patterns illustrates a fundamental principle of regulatory evolution: novel patterns arise more readily from the recruitment of available components, CREs, and transcription factors into new regulatory interactions rather than from the de novo creation of genes or CREs. Indeed, all of the diversity of wing pigmentation patterns illustrated in Fig. 6.1 may be accounted for by regulatory changes and would not require, in principle, any coding sequence changes among species.
CIS-REGULATORY EVOLUTION MINIMIZES FITNESS PENALTIES
Two processes shape the course of evolution: first, genetic mechanisms generate variations in different individuals of a population, regardless of their biological outcome and ecological consequences. Second, these variations are sorted out either by a selective process based on their relative consequences to reproductive success (fitness) or by random population sampling. Although we are mainly concerned with the genetic and molecular mechanisms of evolutionary innovations, the existence of selective pressures constantly sifting through the spectrum of emerging variations must be considered, because they constrain the scope of genetic changes permitted under natural selection.
The genetic changes contributing to morphological evolution can affect protein function through mutations in gene coding sequences or, instead, gene regulation, mainly through CRE evolution. What circumstances influence which of these changes is more likely to be tolerated under natural selection? The study of pigmentation pattern evolution has also proven insightful to address this question. During the course of Drosophila evolution, discrete pigmentation patterns have been gained by the ancestors of identified groups of flies, preserved in many descendant species, and occasionally lost in others.
In theory, the loss of a particular pigmentation pattern could occur by the loss of pigmentation gene expression or the disruption of pigmentation protein functions through mutations in their coding sequences. However, the latter kinds of genetic changes would have substantial collateral effects, affecting all pigmentation patterns and other processes in which these proteins are involved. Many fly pigmentation proteins are also involved in cuticle formation and the metabolism of dopamine, an essential neurotransmitter, and D. melanogaster yellow mutants are notorious for their poor mating success (Bastock, 1956; Chia et al., 1986; Walter et al., 1991; Drapeau et al., 2006). Hence, losses of pigmentation through changes in the coding sequences of pigmentation genes are unlikely to be tolerated by natural selection, because their fitness cost is too high.
Supporting this idea, three cases of loss of pigmentation patterns have involved the selective functional inactivation of a CRE of the yellow locus (Fig. 6.4). Both the male wing spots discussed above and male abdominal pigmentation on segments A5–A6 (see below) have been lost repeatedly in distinct lineages. In two independent cases examined, the loss of the wing spot involved the inactivation of the yellow spot element (Prud’homme et al., 2006). Similarly, the loss of yellow expression in A5–A6 results from the disruption of a specific CRE (Jeong et al., 2006). In each case, mutations altered the spatial distribution of the gene product in only one domain of the body, leaving the rest of the expression pattern and the protein activity intact. These examples illustrate that disruption of a dedicated CRE mini-
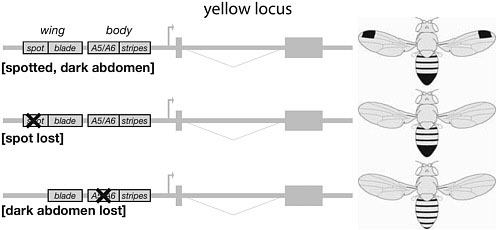
FIGURE 6.4 Cis-regulatory evolution circumvents pleiotropic effects. The yellow locus contains a series of CREs controlling the spatiotemporal expression of the gene. Four of them are represented on these schematics, driving expression in the wing blade, wing spots, segmental abdominal stripes, and male posterior abdominal segments. Some species have lost the wing spots or male abdominal pigmentation (Middle and Bottom, respectively) through the inactivation of the corresponding CRE. The selective disruption of a CRE does not affect other aspects of the expression pattern or the gene activity.
mizes the fitness penalties by affecting only one specific aspect of a gene’s function while leaving the other functions undisturbed.
Additional examples of the selective loss of gene expression are inferred to be associated with CRE evolution, including the loss of larval hairs in Drosophila (Sucena and Stern, 2000; Sucena et al., 2003) and pelvic reduction (Shapiro et al., 2004) and bony armor loss (Colosimo et al., 2005) in stickleback fishes. In all of these examples, there is one common denominator: the evolutionary changes involve mutations in a pleiotropic gene, i.e., a gene with multiple functions. A clear principle is emerging from the increasing number of case studies: pleiotropy imposes a genetic constraint on the type of changes that can be accommodated in morphological evolution. Highly pleiotropic genes (including most developmentally regulated genes) are more likely to contribute to morphological evolution through cis-regulatory changes than through coding sequence alterations. In contrast, known examples of pigmentation evolution resulting from the alteration of coding sequences affect genes involved in a single process, such as the overall body color in fish, mammals, or birds (Ritland et al., 2001; Theron et al., 2001; Eizirik et al., 2003; Nachman et al., 2003; Mundy et al., 2004; Hoekstra et al., 2006; Protas et al., 2006). Coding sequence changes
appear to be better tolerated in minimally pleiotropic genes. This principle of minimizing fitness penalties delimits the scope of what changes are permissible under natural selection and explains why CRE evolution is a pervasive mechanism underlying morphological diversification (Carroll, 2005b).
INTERACTION MAY EVOLVE BETWEEN ANY TRANSCRIPTION FACTOR AND DOWNSTREAM CRE
We have advocated that novelty in gene expression arises primarily through new regulatory interactions between existing CREs and transcription factors. But is it the case that any transcription factor may be coopted into regulating a CRE, or are there constraints on regulatory evolution imposed, for example, by the position components occupy in developmental gene networks? Development is often described as a hierarchical process governed by cascades of regulatory genes. The formation of body parts and organs typically begins with the expression of a particular combination of transcription factors in a small set of precursor cells. These proteins then direct the organization of the developing structure by regulating other pattern-regulating genes, the expression of which define smaller territories and progenitors of the multiple parts constituting the final structure. Ultimately, batteries of structural genes, including pigmentation genes, establish the terminal differentiation of the various cell types.
Intuitively, it may have been thought that proteins at a particular tier in the hierarchy primarily regulate genes in the next tier and so on, such that evolutionary modifications in regulatory connections occur mainly between two consecutive levels. However, studies of pigmentation pattern evolution in flies have revealed that regulatory evolution takes advantage of transcription factors throughout genetic hierarchies in an opportunistic way to generate new regulatory connections.
This notion is clearly illustrated by the evolution of abdominal pigmentation in the relatives of D. melanogaster. In many species, abdominal segments are pale with a stripe of black pigment. However, darkening of the entire terminal segments A5 and A6 has evolved in males of an ancestor of the melanogaster species group (Jeong et al., 2006). This pattern has been preserved in most species of the group but secondarily lost in others. As in the wing spot case, Yellow distribution prefigures the actual pigmentation pattern and is specifically expressed at high levels in the A5–A6 segments in species that are fully pigmented (Fig. 6.5).
How was the strong expression of yellow gained, and subsequently lost, selectively in A5–A6 segments? Dimorphic abdominal pigmentation in D. melanogaster is controlled by a genetic regulatory circuit that includes
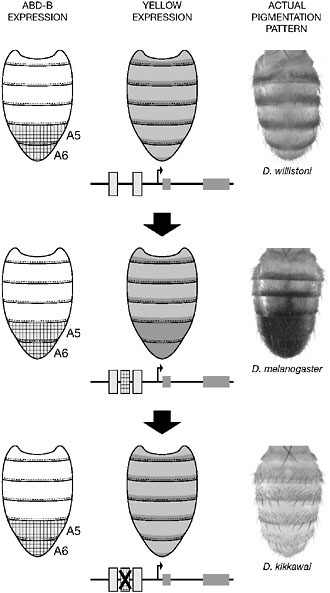
FIGURE 6.5 Regulatory changes underlying male abdominal pigmentation pattern evolution. In the D. melanogaster species group, the male abdominal pigmentation pattern is variable (Right). In the ancestral situation, here illustrated with Drosophila willistoni, both sexes carry an identical segmental stripe pattern. D. melanogaster males have evolved fully pigmented posterior segments (A5 and A6). This pattern has been secondarily lost in Drosophila kikkawai. These transitions result in part from changes in the regulation of the pigmentation gene yellow (Center). The gain and the loss of binding sites for the Hox protein Abd-B, a transcription factor expressed in posterior segments in Drosophila (Left), was involved in the gain and loss of expression of Yellow in the posterior abdomen, respectively.
the Hox protein Abdominal-B (Abd-B), which is expressed in terminal segments (Kopp et al., 2000). Initially, it was expected that Abd-B controls the formation of pigmentation pattern indirectly, through the regulation of another transcription factor (Kopp et al., 2000). Surprisingly, however, analysis of a yellow CRE revealed that expression in A5–A6 is directly controlled by Abd-B (Fig. 6.5) (Jeong et al., 2006). Evolution of Abd-B-binding sites in this CRE correlates with the evolution of pigmentation in A5–A6 and mutational inactivation of some of these Abd-B-binding sites is associated with the loss of yellow expression from A5–A6 and the loss of posterior pigmentation in one lineage (Fig. 6.5).
In the same way the evolution of the wing spot entailed the recruitment of the wing-building protein Engrailed, posterior abdominal pigmentation arose through the evolution of a direct regulatory link between a major body plan architect (Abd-B), lying at the top tier of the genetic hierarchy, and a far downstream structural gene (yellow). In these examples, deeply conserved body plan- and body-part-building transcription factors contributed to morphological evolution of closely related species through changes in the regulation of their sets of target genes.
The evolution of pigmentation patterns through coopting body-plan regulatory proteins illustrates a third principle of regulatory evolution: association between any transcription factor and a downstream CRE may evolve, irrespective of positions of these components in genetic hierarchies. This opportunistic nature of regulatory interactions contributes to the vast evolutionary potential of CREs. The three principles of regulatory evolution we have described explain the mechanisms through and circumstances in which regulatory changes are more likely to contribute to morphological evolution. These rules also have implications for understanding overall patterns of morphological change. Here we will expand our discussion to issues concerning the direction (i.e., gain versus loss) and magnitude of trait evolution over larger evolutionary time scales.
LOSSES ARE EASY, GAINS ARE HARDER
The evolution of form occurs in part through the gain and loss of morphological traits. However, it should be emphasized that the frequency of occurrence of gains and losses is very different. The pattern of trait turnover shows that the frequency of losses is generally much larger than that of gains. This is because a trait arising once in the common ancestor of a group of species is offered as many opportunities to be lost as there are descendant species in the group. The male wing spot and abdominal pigmentation pattern discussed above, for instance, have been lost independently at least five and three times, respectively (Jeong et al., 2006; Prud’homme et al., 2006). Furthermore, the loss of a trait could potentially
occur by the functional alteration of any of the loci involved in its formation. In the case of abdominal pigmentation losses, three different mutational paths, affecting distinct genes, have been followed during these evolutionary transitions (Jeong et al., 2006). For these reasons, losses of morphological traits are expected to be frequent and relatively “easy,” i.e., they have a simple genetic basis and may even occur in a single step.
In contrast, the gain of genetically complex traits appears harder, in that it requires the deployment of multiple gene products in a coordinated spatial and temporal manner. Obviously, this is unlikely to happen in a single step, because it requires potentially numerous changes at multiple loci.
The contrast between the paths of trait gain and loss is also manifested at the level of CRE evolution. The functional inactivation of a CRE can result from a few mutations or perhaps even a single point mutation, as exemplified by the disruption of the yellow spot element in a species that has recently lost its wing spot (Prud’homme et al., 2006). In comparison, the evolution of a new regulatory function, even through the cooption of an existing CRE, appears to require a relatively longer mutational path involving the acquisition of multiple transcription factor-binding sites. Furthermore, in documented cases where a new regulatory activity has evolved from cooption of an existing CRE (Jeong et al., 2006; Prud’homme et al., 2006), we observe that the two cis-regulatory activities reside in physically separable regions of DNA. These observations suggest that subfunctionalization of the ancestral CRE has occurred through additional changes that fine-tune gene expression in the domains governed by the now-separate elements (as discussed in Carroll et al., 2004, p. 222).
CONNECTING THE DOTS FROM PIGMENTATION PATTERNS TO BODY PLAN DIVERSIFICATION: THE COMPOUNDING OF REGULATORY CHANGES OVER EONS
A long-standing question in evolutionary biology has been whether the genetic and molecular mechanisms underlying morphological changes within populations (so-called “microevolution”) are sufficient to account for the differences in body patterns between species and at higher taxonomic levels (so-called “macroevolution”) (Huxley, 1942; Arthur, 2000; Leroi, 2000; Stern, 2000). We submit that an expanding body of evidence, including the examples described in the previous sections, is affirming that macroevolution is a matter of the very same genetic and molecular changes ongoing in populations, compounded over longer periods of time and large numbers of cladogenetic events.
The morphological differences among closely related species we have discussed above evolved by functional changes in CREs. The genetic
modifications underlying these changes are ordinary mutations of the same kind as those arising in natural populations in every generation (Rockman and Wray, 2002; Balhoff and Wray, 2005) and not rare genomic rearrangements or duplication events. Therefore, it appears the genetic changes generating morphological differences among species are of the same nature as the ones that arise within populations. One may wonder whether it is also the case for morphological divergences at higher taxonomic levels, or whether these rather large-scale morphological differences require distinct genetic mechanisms.
A large body of work has documented associations between major morphological differences, such as body-plan differences, with those in the expression pattern of Hox proteins or their downstream target genes (Averof and Akam, 1995; Burke et al., 1995; Averof and Patel, 1997; Belting et al., 1998; Cohn and Tickle, 1999; Carroll et al., 2004). One illuminating example where the mechanisms have been addressed in depth is the evolution of the two-winged dipteran body plan from four-winged ancestors by reduction of the hindwings. Many winged insects bear two pairs of wings attached to their second (T2) and third thoracic segments (T3). However, in Diptera, the hindwings have been modified into small balancing organs, the halteres. In Drosophila, the Hox protein Ubx, which is expressed in the T3 segment and appendages, controls the differentiation between wing and haltere (Weatherbee et al., 1998; Crickmore and Mann, 2006). Because Ubx is also expressed in the developing hindwings of four-winged insects (Warren et al., 1994), the evolutionary reduction of the hindwing in Diptera occurred under the control of the Ubx protein and did not result from a shift of the expression of Ubx.
In the Drosophila haltere, Ubx directly represses the expression of a set of wing-patterning genes (Galant et al., 2002; Hersh and Carroll, 2005). In contrast, in four-winged butterflies, Ubx does not repress those genes (Weatherbee et al., 1999). Hence, during dipteran evolution, this set of wing-patterning genes became Ubx-regulated. These genes became Ubx-responsive in the haltere by evolving Ubx-binding sites in CREs that direct gene expression in fore- and hindwings (Galant et al., 2002; Hersh and Carroll, 2005). However, Ubx is not a “wing repressor” protein per se, because in other non-dipteran insects, Ubx presumably regulates different sets of target genes in the hindwings (Warren et al., 1994; Tomoyasu et al., 2005). Therefore, the reduction of hindwings in Diptera results from changes in the regulatory connections between Ubx and downstream target genes that evolved changes in their wing CREs (Fig. 6.6).
Importantly, the evolutionary mechanisms of hindwing reduction comply with the regulatory principles we have described. The repression of wing-patterning genes in halteres exploits available CREs and transcription factors (Ubx). Because Ubx is involved in many develop-
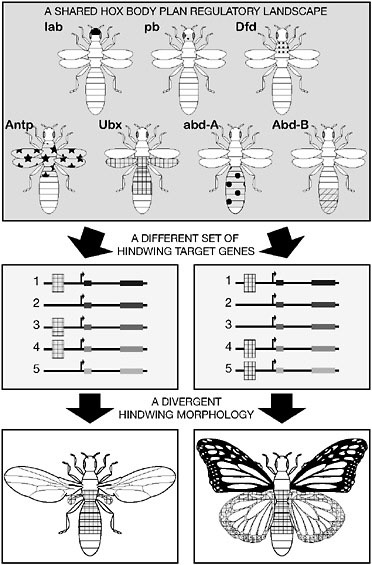
FIGURE 6.6 Body plan evolution by compounding regulatory changes. Hindwing reduction in Diptera results from changes in the regulatory connections between the Hox protein Ubx and downstream target genes (1 to 5). In Diptera, a suite of wing-patterning genes have evolved Ubx-binding sites in their CREs and, as a result, are repressed during hindwing development. In contrast, in four-winged butterflies, Ubx regulates a distinct set of target genes in the hindwing.
mental processes other than wing development, evolution of downstream wing-specific CREs enabled the selective changes in gene expression in the haltere while preserving other functions. Finally, multiple genes, at different levels of the wing genetic regulatory hierarchy, evolved Ubx regulation (Weatherbee et al., 1998; Hersh et al., 2007), suggesting that any gene of the wing developmental program can fall under the regulation of Ubx.
Thus, in the same way that abdominal pigmentation pattern evolved by changes in the regulatory connections between Abd-B and downstream pigmentation genes, the two-winged dipteran body plan evolved by changes in the regulatory interactions between Ubx and downstream wing-patterning genes. In this light, we see that the differences between the evolution of modest morphological traits, such as pigmentation patterns, and changes of larger magnitude, such as body-plan modifications, are not in the nature of the genetic changes but rather in their degree. Indeed, because several genes have evolved Ubx regulation, it is inescapable that hindwing reduction evolved progressively in a multistep process. More generally, it is reasonable to infer that large-scale morphological differences must typically arise by regulatory sequence mutations, presumably of small individual phenotypic effect, accumulating over time. Therefore, we submit that the same kind of genetic and molecular mechanisms are sufficient to account for both simple morphological changes and more profound body plan differences and that the principles of regulatory evolution we have delineated are general principles underlying morphological evolution.
Comparisons over large taxonomic distances have documented multiple examples of developmental gene duplications and coding sequence changes, and there is no doubt that these types of changes play a role in morphological evolution. However, what must be appreciated is the relative contribution of the different types of mechanisms to morphological diversity. It is well established that changes in gene number and regulatory protein motifs have been relatively few and far between during the >500-million-year span of animal evolution. In contrast, regulatory evolution, through regulatory sequence changes, is pervasive and constitutes the primary fuel of the continuous morphological diversification of lineages and traits in the “far between.”
CONCLUSION
A growing number of case studies exploring the mechanisms of morphological changes have provided direct evidence that CRE evolution plays a major role. From these examples, we have identified general rules regarding regulatory evolution, namely how regulatory evolution exploits available genetic components, irrespective of their hierarchical position
in gene networks to generate novelty, and minimizes fitness penalties. These rules offer a rationale explaining why regulatory changes are more commonly favored over other kinds of genetic changes in the process of morphological evolution, from the simplest traits diverging within or among species to body-plan differences at higher taxonomic levels.
Although progress has been made in understanding CRE evolution in a single gene, there are important outstanding issues that need to be addressed for a fuller picture of the origins of morphological diversity. In particular, two areas that have been largely unexplored seem now to be within reach. First, we need a dynamic picture of CRE evolution within populations. This entails, on the one hand, elucidating the contribution of mutation and recombination to the origin of variation in gene expression, and on the other hand, a sense of how genetic drift and selection shape the fixation of these variations over time (Rockman et al., 2005). Ultimately, such a dynamic picture of CRE evolution will help to reconstruct the mutational paths that lead to the origin of novel gene expression patterns.
Second, for complex genetic traits, we need to focus our attention not just on individual genes but on the complete set of genes involved in the formation, variation, and evolutionary divergence of the traits. Such a perspective is critical to understanding how sets of genes assemble into functional networks through the evolution of regulatory interactions (Tsong et al., 2003; Ihmels et al., 2005; Tsong et al., 2006) and thus shape morphological diversity and novelty.
ACKNOWLEDGMENTS
We thank Jeff P. Gruber and D. Young for help with Automontage imaging in Fig. 6.5 and Patricia Wittkopp for comments on the manuscript. N.G. is a Human Frontiers long-term fellow. This work was supported by the Howard Hughes Medical Institute (S.B.C.).




















