
2
Overview of Key Issues1
As indicators of biological function or state, biomarkers have many potential applications in research and medicine: they can provide information useful for the diagnosis, treatment, and prognosis of disease; they can indicate whether a drug is having an effect in an individual and whether side effects can be anticipated; and they can be used to screen populations for particular biological characteristics or environmental exposures. Biomarkers also have many potential applications in the development of drugs. As Janet Woodcock of the FDA pointed out, they can improve the predictability of drug development, and increase the value of preventative and therapeutic interventions by targeting individuals with a high probability of benefit and screening out those at high risk of side effects. Biomarkers can be used to screen compounds for toxicity before they enter clinical trials, to inform decisions about whether to develop a drug, to monitor the development of toxicity, to forecast adverse events given wider exposure, or to understand the mechanism by which a drug works.
Tests to assess the variability of a patient’s drug-metabolizing enzymes are already being used to adjust doses in individuals. Other biomarker-based tests are being used to determine whether an individual is at increased risk of having an adverse reaction to certain compounds, and to avoid treatment if the balance of benefit and risk is unacceptable. These kinds of applications can be expected to multiply rapidly.
Biomarkers can take many different forms. In preclinical screening, for example, they may entail studies of gene expression or cell systems. Animal studies can make use of genomic and proteomic techniques, thereby increasing the probability that initial administration to humans will be safe, or help establish the relevance of animal findings to humans. Biomarker findings in clinical trials and postmarket data also can provide information about mechanisms of drug toxicity or benefit and suggest the need for additional nonclinical studies to fully elucidate the relevant mechanisms. In a clinical setting, such information can be used, for example, to monitor reactions to drugs in individuals or to deselect individuals from trials who may be at risk from a treatment.
In considering the use of biomarkers for drug development, additional issues arise, said Alastair Wood of Symphony Capital, LLC. To be useful, a biomarker for toxicity found to be elevated by an investigational drug in preclinical studies must provide some level of confidence that carrying such a drug forward into clinical trials will produce toxicity in a proportion of patients. This proportion must be significant enough to alter decision making about developing the drug, to point to a different course of action in patient selection for clinical trials, or to necessitate more detailed studies prior to marketing so that safety signals can be assessed. Conversely, the absence of elevation of a biomarker should imply confidence that a safety problem will not occur in more than a known (low) proportion of patients. In this way, the use of a biomarker can provide risk assessment and risk mitigation, both to patients who are likely to receive the drug clinically and to the development program carrying that drug forward.
Beyond these broad considerations lie more detailed questions. If a biomarker is elevated in a small number of people in early clinical studies, what is the overall risk to any given individual or to a population? If the absolute degree of elevation is small, does this mean that the likely toxicity will be mild when the drug is given to a large population of patients, and/or does it mean that only a small proportion of patients will develop severe toxicity? Unfortunately, the answers to these questions are seldom known with any degree of certainty. Does the absence of a biomarker signal necessarily predict long-term safety?
The use of biomarkers potentially could address several major problems associated with drug development. The costs of new drug development have risen rapidly even as the number of new molecular entities (NMEs) submitted to the FDA has fallen (Figure 2-1). In addition, a number of drugs have been withdrawn from the market because of safety concerns. By enhancing the ability to assess whether drug candidates are promising early in development, biomarkers could reduce the costs of developing drugs and bringing them to the market, enhance the safety of new drugs, and improve
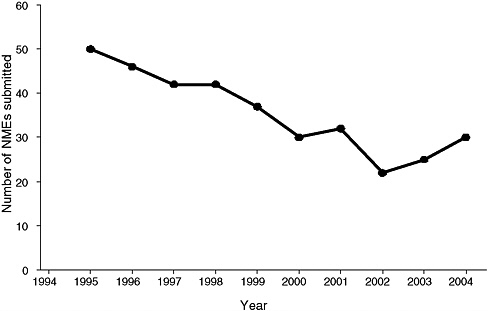
FIGURE 2-1 The number of new molecular entities (NMEs) submitted to the FDA has fallen since the mid-1990s.
SOURCE: Frantz, 2004.
the cost-effectiveness of drugs by targeting treatment to those patients with the best balance of risk and benefit.
A particularly valuable use of biomarkers would be to help bridge the gap between the preclinical and clinical development of new drugs. For example, a preclinical biomarker that produces similar results in tissue cultures or model organisms and in clinical use in humans might reliably predict human responses to a compound. Or a bridging biomarker might predict toxicity very early in humans—before harm occurs—and at very low doses. As the FDA white paper Innovation or Stagnation: Challenges and Opportunity on the Critical Path to New Medical Projects states, “A new product development toolkit—containing powerful new scientific and technical methods such as animal or computer-based predictive models, biomarkers for safety and effectiveness, and new clinical evaluation techniques—is urgently needed to improve predictability and efficiency along the critical path from laboratory concept to commercial product” (FDA, 2005, p. ii).
The remainder of this chapter reviews several important issues involved in the use of biomarkers in drug development: predictions based on biomarkers, validation vs. qualification, mechanisms vs. patterns, regulatory
approval of biomarkers, regulation of single biomarkers vs. panels of biomarkers, and measures of success. It concludes with a specific example: the use of biomarkers to improve the treatment of mental illness.
PREDICTIONS BASED ON BIOMARKERS
One critical issue involved in assessing the utility of biomarkers is how well they predict relevant outcomes. Measures of the performance of biomarkers include sensitivity, specificity, calibration, discrimination, and reclassification:
-
Sensitivity represents the proportion of truly affected cases (persons) in a screened population who are identified as being diseased by the test, and is a measure of the probability of correctly diagnosing a condition.
-
Specificity is the proportion of truly nondiseased persons who are identified as such by the screening test. For example, if a biomarker has high sensitivity but low specificity, most of the truly at-risk cases will be correctly identified, but many of the not-at-risk cases will also be identified as at-risk.
-
Calibration refers to the agreement between the predicted probability of an outcome and the actual probability when measured in a population.
-
Discrimination refers to the ability of a biomarker to distinguish those with a disease or event from those without. A biomarker could have excellent calibration with poor discrimination and vice versa.
-
Reclassification has become a critical issue in assessing biomarkers. It refers to the ability of a biomarker measurement to move the probability of an outcome beyond a threshold that leads to a different diagnosis, prediction of outcome, or clinical decision than would have been made based on prior information.
The synthesis of these measures is complex since biomarkers can be excellent for some purposes and mediocre for others, thereby complicating their use for decision making. One of the greatest challenges to the application of biomarkers in drug development is that numerous and conflicting arguments can be made for placing greater emphasis on specificity than sensitivity or vice versa. For example, one could argue that a biomarker that yields a high number of false negatives may fail in preclinical studies to detect problems with drugs that go on to produce toxicity in clinical studies. This lack of sensitivity not only puts patients at risk but also may result in the waste of future development costs. On the other hand, false positives
can be equally damaging by causing large numbers of potentially successful and safe drugs to be lost during development. Thus if sensitivity is too high at the expense of specificity, false positives will result in denying patients access to useful therapies. This complexity can be greatly exacerbated by the simultaneous use of multiple biomarkers in screening. For example, if every drug must be screened using 50 safety biomarkers, and if each biomarker has a false positive rate of 1 percent, up to half of all useful drugs will be wrongly eliminated during an early stage of development.
The acceptable sensitivity and specificity will vary from drug to drug and from indication to indication. For example, the safety requirements differ between a therapy for nasal allergy and a cancer drug. Wood stressed that a nuanced approach is needed to answer specific questions.
A major potential use of biomarkers is to predict and monitor the toxicity of a drug in a clinical trial. In these cases, an important issue is the extent to which a negative or a positive test has predictive value. In other words, if a person shows elevation of a biomarker and is deselected from a trial, how likely was that person to have actually experienced a clinically significant adverse event? Often the answer remains unknown, even when a drug is on the market, because the only way to fully articulate the performance of a biomarker is to measure the outcomes of the relevant population with an adequate sample size to generate reliable probability estimates.
Assays that can make such determinations may already be on the market with another indication or may need to be codeveloped with a drug. An example is the drug abacavir, whose use is limited by a significant incidence of adverse events. A randomized controlled trial demonstrated risk reduction with the use of a human leucocyte antigen (HLA) region marker for risk (HLA-B*5701), and this marker was recommended for use in a black box on the drug’s label. This diagnostic test had been well established because HLA markers are used for tissue typing.
With safety markers for new drugs, ethical considerations dictate ascertainment of the value of a test as early as possible in drug development. Explicit study designs are needed to answer safety questions, such as when to stop enrolling patients who test positive or to discontinue treatment in those with an elevated biomarker. It is critical to obtain definitive answers about safety while keeping participants in a trial as safe as possible.
VALIDATION VS. QUALIFICATION
Currently, there is a lack of clarity regarding several terms commonly used in the discussion of biomarkers. In particular, Woodcock urged that standard definitions be adopted for the terms “validation” and “qualification.” Validation, she said, should be used for analytic validation, which is a measure of how well a test detects or quantifies an analyte under various
conditions. Validation thus would require demonstration of the performance characteristics of an assay. In contrast, qualification is a measure of the use of a biomarker in a specific context. That context may be selecting or deselecting people for a clinical trial, monitoring drug-induced toxicity, or some other purpose. The amount of evidence needed to qualify a biomarker for a given purpose is related to the consequences of using the result to make decisions, such as whether to pursue the development of a drug or whether to withhold a drug from individuals in a clinical trial.
Analytic validation is necessary but generally not sufficient for a biomarker. It requires a stable platform and the establishment of standards that facilitate the linking of results across laboratories. Validation also requires study of variability among users and among laboratories. In addition, validation requires an understanding of the potential for drugs or other conditions to interfere with results. These are not the kinds of activities that generally earn tenure for faculty members, Woodcock observed, but they are critically important to understanding the performance of an assay. In contrast, qualification requires context-specific measurement of the performance of the biomarker in relation to an outcome or outcomes of interest.
MECHANISMS VS. PATTERNS
Another important issue for the development of biomarkers is the distinction between mechanistic understanding and pattern recognition. For some biomarkers, there may be a detailed understanding of the mechanism that links the use of a drug to the elevation of a biomarker and thence to the development of clinical toxicity. In other cases, a drug may produce an effect pattern—such as a pattern of gene activity on a microarray—but the mechanism linking the use of the drug to the change in the array and thence to an adverse clinical effect is either unknown or poorly understood. In these cases, decisions may have to be made on the basis of pattern recognition without a clear understanding of the mechanistic link.
When a mechanism is unknown, considerable work is required to define the level of specificity needed to influence decisions. Drug developers may not know what preclinical signals of toxicity to look for until clinical toxicity has been observed late in drug development or even in clinical use. For example, many kinase inhibitors now used clinically in oncology produce cardiac toxicity, perhaps because they inhibit a specific kinase in the heart. Without knowing whether that is indeed the mechanism or which specific cardiac kinase is responsible, however, mechanism-based biomarkers cannot be used to screen for this toxicity in preclinical studies. If the relevant kinase were discovered, a biomarker assay for that mechanism would enable rapid screening of drugs for toxicity. Therefore, understand-
ing of the mechanisms of toxicity offers the best chance of both developing safer drugs lacking that toxicity and defining useful biomarkers to detect toxicity early in drug development, while purely empirical assessment of biomarkers requires much larger samples with greater uncertainty.
An understanding of mechanism also can be critical in gauging the relevance of animal findings to humans. Many drugs are lost from development because of toxicity findings in animals that are seen infrequently or not at all in humans. Because the mechanism often is not understood, however, it is difficult to predict whether the same toxicity will occur in humans since there is no way to determine, other than by empirical observation in large numbers, whether the same systems are at play in human biology.
REGULATORY APPROVAL OF BIOMARKERS
Biomarkers being developed for commercial uses have several paths toward regulatory approval, each of which requires a different level of evidentiary data. For novel diagnostics, a premarket approval (PMA) application must be submitted, although the FDA can assign a “de novo classification” to a diagnostic test that streamlines the approval process. Other biomarkers used as in vitro diagnostics reach the market through a 510(k) application, which demonstrates that a product is “substantially equivalent” to some previous device. An important distinction between these mechanisms is that a PMA application must include data showing that the device is safe and effective, whereas a 510(k) application need only include data supporting the performance standards and validity of the device’s intended use. A third category of biomarkers reach the market as laboratory-developed tests that are not submitted to the FDA for approval but are marketed by laboratories overseen by the Clinical Laboratory Improvement Amendments (CLIA) program. Most commercially available genetic tests fall into this category.
If a biomarker or panel of markers is to be used to justify regulatory decision making, the assay used to measure that marker(s) must demonstrate validity and clinical utility. According to the FDA’s pharmacogenomic guidance document (FDA, 2005, p. 4), a valid biomarker is “a biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is an established scientific framework or body of evidence that elucidates the physiologic, toxicologic, pharmacologic, or clinical significance of test results.”
For in vitro diagnostics requiring a PMA, clinical utility must be demonstrated along with validity. Clinical utility could be demonstrated, for example, by adequate detection of an analyte if a clinical link is well-established in the literature. It also could be established through other means, such as the analysis of stored specimens. Again, the burden of proof
is proportional to the risk; thus, for example, prognostic claims for a test in the absence of a specific critical decision directly linked to the test result have less of a burden than other claims.
REGULATION OF SINGLE BIOMARKERS VS. PANELS OF BIOMARKERS
Marketing standards are the same whether a diagnostic is a single assay, a set of assays, or a panel of biomarkers. For example, in vitro diagnostic multivariate index assays (IVDMIAs) use the results from multiple analytes to create an “index,” “score,” or other measure. The method used to derive a score is often algorithmic and not clinically transparent. This is typical of several new technologies, such as the use of genomic or proteomic screens to produce a result.
The FDA has proposed a regulatory framework for IVDMIAs that involves submission to and review by the agency. Technical issues are often significant for an IVDMIA because of decisions about which analytes to include, how to weight those analytes, what cutoff values to use, how to handle changes to a test once it has been developed, and what quality control methods to apply. The FDA proposal has been controversial because of the conflict between the need for FDA review and the rapid evolution of the industry.
Multiplexed assays raise issues of effectiveness in addition to safety. For example, the National Cancer Institute is planning a prospective randomized trial for treatment or nontreatment of early-stage cancer based on a gene expression panel. In such cases, efficacy must be definitively tested in the intended population, and several trial designs for this purpose have been proposed in the literature.
MEASURES OF SUCCESS
A general issue in the use of safety biomarkers is how success should be defined. In the broadest possible terms, success is measured by improvement in the clinical safety of drugs being developed. As there is no way of preventing every drug that proves to have a toxic effect from proceeding into clinical trials, however, definitions and measures of safety must be established.
An unintended consequence of biomarker development may be a decrease in the number of available drugs. Once a biomarker has been developed and marketed, it may inhibit the development of drugs if it generates a positive signal that indicates potential future problems. Many companies would hesitate to proceed with the development of such a biomarker, even if there were a poor correlation between the biomarker and toxicity. One way to help establish definitions of success would be to look
back at drugs that have shown toxicity and identify which biomarkers were elevated in preclinical models. Such an approach would require that companies share compounds for study after clinical development or marketing has ended. This retrospective approach would be valuable as there is substantial knowledge of actual clinical experience with such drugs. In contrast, when elevation of a biomarker results in a company’s preemptive termination of development, there is limited evidence to evaluate.
Much of the publicity regarding drug safety has focused on the detection of events that are rare, such as acute hepatic failure, which recently was a cause for concern with the drug troglitazone. But a bigger problem, according to Wood, is the drug that produces an increased incidence of a frequent event, such as the Cox-2 inhibitors, which caused an increase in myocardial infarctions. A substantial increase in the rate of myocardial infarction with a drug could produce hundreds of thousands of cases, yet it could be difficult to detect the problem in preclinical work, especially if a mechanistic hypothesis were not available. In addition, the postmarket reporting system is ill qualified to detect an increased frequency of such events that are common in the background population.
The challenge, Wood concluded, is to develop safety markers that are reliable and validated across drugs and across companies, both prospectively and retrospectively. Regardless of whether the mechanism of action is known or unknown, it is necessary to develop systematic methods for exploring the biological and clinical implications. Thus, improved understanding of biomarkers must be coupled with improved epidemiological surveillance methods and randomized trials, when needed to elucidate modest differential effects of a drug on common outcomes. Meeting these needs will allow for the development of increasing numbers of drugs that are safer and less expensive to bring to market.
AN EXAMPLE: BIOMARKERS FOR TOXICITY OF PSYCHIATRIC DRUGS
Thomas Insel of the National Institute of Mental Health discussed the use of biomarkers in addressing a major problem in the United States, as well as globally—mental illness (see Box 2-1). Responses to both drugs and other types of therapy used to treat mental illness vary greatly. Today, there is no way to determine, a priori, which patients will respond well to which treatments or will experience adverse side effects with medication. The hope is that biomarkers will provide guidance for interventions at all stages of a mental illness. Biomarkers may even make it possible to predict future problems arising from mental illnesses such as schizophrenia and to use medications preemptively.
A major emphasis in recent years has been pharmacogenomics—the
|
BOX 2-1 The Toll of Mental Illness Mental illness is the leading cause of medical disability for people between the ages of 15 and 44. Mental illness is often chronic, can start early in life, is highly prevalent, and may be severely disabling. More than 30,000 suicides occur each year in the United States. By comparison, only three forms of cancer kill more than 30,000 people per year, and homicides and AIDS kill 18,000 and 20,000 people, respectively. Life expectancy for people with major mental illnesses is only 56 years, more than a quarter century less than the average. Most of this excess mortality is not due to suicide, but to general medical disorders that are secondary to the mental illness, such as pulmonary and liver disease. According to one estimate, for example, 44 percent of all cigarettes are smoked by people with mental illness. Although medications are widely used to treat mental illness—more than 200 million prescriptions per year are written for antidepressants, more than for any other class of drugs—currently available drug therapies are much less effective than desired. The total direct and indirect costs of mental illness in the United States are estimated at more than $300 billion, or more than $1,000 per American, yet only about $5 per American is spent on efforts to understand the causes, treatment, and potential preventive measures for these conditions. If these heterogeneous problems could be better understood and classified using biomarkers, substantial impact on mortality and morbidity in the U.S. population might be realized. SOURCE: Insel, 2008. Data: WHO, 2002. |
use of high-throughput resequencing to associate particular genetic variants with responses to medications. For example, variants in a protein that transports compounds across the blood–brain barrier can influence whether a medicine will be effective. Similarly, variants in neurotransmitter receptors can predict some of the variation in response. Thus far, however, the observed effects of genetic variants have been relatively small. In addition, the predictive power of genomics is limited by the heterogeneity of the disorders being treated and by individual variations in choice of treatment, response, toxicity, and adherence to a therapeutic regime.
A key problem has been predicting adverse effects in patients treated with psychiatric drugs. In a study involving 1,742 patients, 120 developed suicidal ideation while receiving antidepressants. Variants in two receptor genes were associated with increased thoughts of suicide, but these findings need to be replicated and extended.
While an individual marker may be informative, a combination of
several markers related to different parts of a pathway could be far more useful. Some of these markers may not be genetic—they may be “downstream markers” such as protein or metabolite levels in cells or the blood, or imaging of active brain regions. For example, imaging of a region of the brain known as “area 25” has revealed that it is overly active before treatment for depression and less active after treatment. This is the case whether the treatment consists of medication, cognitive-behavioral therapy, or even placebo. Conversely, in those who do not respond to an intervention, activity in this area does not decrease. This decrease in activity in area 25 thus appears to be necessary, and possibly sufficient, for the antidepressant response. Perhaps by combining a better understanding of brain circuitry from imaging with genetic and proteomic data, a panel of diverse biomarkers could be developed that would predict responses.
NIH supports research to discover potential biomarkers using a variety of approaches. The development and use of biomarkers can contribute to what Insel called the 3D pathway, which stands for discovery, development, and dissemination. Once potential indicators of clinical response or toxicity have been identified, these predictors need to be studied through prospective development studies. Finally, predictors need to be cost-effective so that they will be adopted and change the standard of care. Too often, powerful evidence-based interventions are neglected in medical practice because they either are not reimbursed or are not well understood.
Insel noted that, while biomarkers could have an enormous impact on the prevention, diagnosis, and treatment of mental illness, their benefits and costs need to be carefully weighed. The emphasis today is on making health care more efficient and less expensive, not more high-tech and more expensive.
REFERENCES
FDA (Food and Drug Administration). 2004. Innovation or stagnation: Challenges and opportunity on the critical path to new medical products. http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html (accessed October 17, 2008).
FDA. 2005. Guidance for industry: Pharmacogenomic data submissions. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126957.pdf (accessed October 17, 2008).
Frantz, S. 2004. FDA publishes analysis of the pipeline problem. Nature Reviews Drug Discovery 3:379.
Insel, T. 2008. Biomarkers for psychiatric drug toxicity. Speaker presentation at the Institute of Medicine Workshop on Assessing and Accelerating Development of Biomarkers for Drug Safety, October 24, Washington, DC.
WHO (World Health Organization). 2002. The world health report 2002: Reducing risks, promoting healthy life. Geneva, Switzerland: WHO.











