
2
The Safety of Vaccines and Vaccination Practices
The starting point for the contemporary vaccine safety system was the National Childhood Vaccine Injury Act (NCVIA) of 1986.1 Enacted in the face of mounting public concern over both the safety of certain licensed childhood vaccines and the economic viability of the vaccine industry (Mariner, 1992), the act had two principal objectives. The first objective was to ensure that even as the public’s health is protected through immunization, a system exists to compensate the small number of individuals who suffer injury thought to be caused by vaccines without the delays and costs associated with tort litigation. Simple fairness requires a mechanism to compensate those thought to be injured by vaccines that are properly manufactured and administered, that are recommended for universal use, and in some cases required by states for school entry to protect public health.2 The other principal objective of the law was to create a climate of safety through adoption or expansion of optimal public health and clinical practices (e.g., monitoring vaccine safety, provision of printed patient information3) and the application of the best science to vaccine safety.
The fact that the founding of the National Vaccine Program (and by extension its executive entity, the National Vaccine Program Office [NVPO]) was among the desired outcomes in an act focused on vaccine safety is not
|
1 |
Public Law 99-660, codified at 42 U.S.C. 201; see Appendix C. |
|
2 |
All states allow medical exemptions from school-entry vaccination requirements, 48 states allow religious exemptions from vaccination, and 19 states allow philosophical exemptions as well (NCSL, 2009). |
|
3 |
The law specifically requires the provision of Vaccine Information Statements. |
coincidental. The act lists the program’s nine responsibilities4 with regard to intra-governmental coordination and coordination with stakeholders; most refer to the safety of vaccines and to adverse events. NVPO’s coordinating role was and remains an essential part of the vaccine safety system established by the act. This chapter examines how the plan could enhance the vaccine safety system; a more extensive discussion of the coordination required to implement the National Vaccine Plan is provided in Chapter 6.
The first part of this chapter provides an overview of four major components of the 1986 legislation and their current status: (1) vaccine safety surveillance and research; (2) information and communication about vaccine safety (discussed in Chapter 3); (3) the program of compensation for injuries thought to be caused by vaccine; and (4) the National Vaccine Program and Plan. The second part of the chapter is organized around four recommendations about priority actions for vaccine safety in the National Vaccine Plan.
In the history of vaccine development and regulation, concern has focused on both vaccine efficacy (and correlates of clinical protection) and vaccine safety. Both vaccine efficacy and vaccine safety are relative: no vaccine is 100 percent effective or 100 percent safe. The use of vaccination has reduced the incidence of disease (and therefore the immediate threat to any individual) and, concomitantly, the burden of fear of disease-related morbidity, disability, and death. The lower risk of disease has understandably led to higher expectations of vaccine safety. The story of polio vaccine illustrates the evolving nature of a vaccine’s risk-benefit balance and of the understanding of that balance as additional information on possible adverse events accrues and as disease incidence changes. In the 1950s when poliomyelitis was endemic in the United States, the benefit of live, attenuated poliovirus vaccine for the individual and the community was judged to greatly outweigh the risk of vaccine-associated paralysis, which occurred at a rate of about 1 case per 2.4 million doses distributed (CDC, 2009b). By 2000, when polio had been eliminated from the Western Hemisphere, this risk of vaccine-associated paralytic polio was judged no longer acceptable in the United States. An enhanced inactivated polio vaccine (IPV), first licensed by the Food and Drug Administration (FDA) in 1987, was ultimately recommended by the Advisory Committee on Immunization Practices (ACIP) in 2000 for exclusive use in routine immunization (CDC, 2000; Moylett and Hanson, 2004).
Interventions given to healthy persons to prevent disease are required to have a low risk-to-benefit ratio when compared to therapeutic interventions. Many childhood vaccines that are recommended for universal use by ACIP are required by states for attendance in licensed day care facilities and public schools, and thus administered to large segments of the population (e.g., nearly the entire annual birth cohort of more than 4 million children). Some adult vaccines are also universally recommended, others are recommended for specific occupations (e.g., health care workers) and, in some cases, required by employers. A substantial proportion of the adult population receives influenza vaccine each year (for example, between two-thirds and three-quarters of adults 65 years of age or older received influenza vaccination in 2008; CDC, 2006).
The process of anticipating, detecting, and quantifying the risks of rare adverse events following immunization presents an enormous challenge. Vaccine studies submitted as part of Biological License Applications to FDA’s Center for Biologics Evaluation and Research (CBER) have historically included several thousand individuals. Rare but serious adverse events may follow vaccination, sometimes at rates in the range of one in a million vaccine recipients. Even vaccine trials including 100,000 or more participants may not have adequate statistical power to detect such rare adverse events. Delaying licensure after efficacy has been shown in order to amass additional evidence related to rare adverse events associated with a candidate vaccine would result in continuing cases and deaths due to the preventable disease.
After FDA licensure, as knowledge about a vaccine’s safety increases when large numbers of individuals are immunized, additional safety assessment becomes possible, complementing pre-licensure data. Vaccine safety researchers both at FDA and outside government have emphasized the equal importance of adequate pre-licensure study and post-licensure surveillance for “signals” of adverse events. It is challenging to detect a true “signal” of a vaccine safety problem amidst the considerable “noise” of coincidental, only temporally related events.
Licensure of second generation rotavirus vaccines offers a clear example in which pre-licensure studies put a special emphasis on vaccine safety because of knowledge about the risk of intussusception acquired after introduction of the first licensed rotavirus vaccine. The large studies undertaken for the second-generation rotavirus vaccines—70,000 subjects for RV5 (bovine-based, RotatTeq) and nearly 75,000 for RV1 (human-based, Rotarix)—reflect a specific safety concern related to the first rotavirus vaccine (Ellenberg et al., 2005; GSK, 2008; Heyse et al., 2008).
The FDA Amendments Act of 2007 has strengthened CBER’s authority to require post-licensing studies. FDA may require the manufacturer to conduct post-licensure studies of vaccine safety that meet certain specifica-
tions (e.g., design, size). FDA monitors a wide range of safety data from the systems described below. The Centers for Disease Control and Prevention (CDC), in addition to joint management of the Vaccine Adverse Events Reporting System (VAERS), implements rapid epidemiologic evaluation of possible safety signals, such as the evaluation of intussusception following the use of RRV-TV (rhesus-based, RotaShield).
A CASE STUDY OF VACCINE SAFETY SYSTEM FUNCTIONING
The federal vaccine safety system’s response to evaluate reported adverse events following immunization with RRV-TV entailed an extensive effort. The response spanned at least 19 states, involved 40 of CDC’s Epidemic Intelligence Service officers, and drew on the capabilities of federal, state, and local public health agencies and health care organizations to locate and verify vaccination histories and outcomes in infants given the rotavirus vaccine and to undertake scientific analysis.5 A timeline of events culminating in the withdrawal of the ACIP recommendation for rotavirus vaccine is provided in Table 2-1.
The experience with RRV-TV illustrates comprehensively the safety system’s components, their capabilities, and their functioning. Subsequent efforts to develop, license, and monitor the safety of RRV5 and RV1 were informed by lessons learned from the first vaccine and led to changes in study design and regulatory expectations for rotavirus vaccines (e.g., a requirement for an unprecedented expansion of the size of the Phase III clinical trials; targeted post-licensure surveillance).
At the time it licensed RV1 in 2008, FDA required a large post-licensure observational safety study in the United States to assess the potential serious risk of intussusception and other serious adverse effects (specifically Kawasaki disease, hospitalizations due to acute lower respiratory tract infections, and convulsions) in vaccine recipients. Requirements included a study sample size of 44,000 vaccinated subjects (to be adjusted based on the background rate of intussusception), and a study design “to detect an increased relative risk of intussusception due to vaccine with a relative risk of 2.5 or greater and with 80 percent power” (FDA, 2008a). The study began June 2009 and is expected to end in 2012.
TABLE 2-1 Rotashield Vaccine Timeline (1999-2001)
|
Date |
Event |
|
|
Before licensure |
In the absence of rotavirus vaccines, there are 3 million cases of rotavirus infection per year (in children under age 5); for 500,000 cases, medical attention is sought, and 60,000 to 70,000 are hospitalized. According to Rennels (2000), rotavirus gastroenteritis caused 25 pediatric deaths per year. There is no known association between wild rotavirus infection and intussusception. The rotavirus vaccine manufacturer sponsors 27 clinical trials in 9 countries involving more than 10,054 children who received the vaccine (Rennels, 2000). Study data are submitted to FDA as part of the Biological License Application process. ACIP forms a rotavirus working group, and the group’s review of serious adverse events in the pre-licensure trials finds 5 cases of intussusception in children who received the vaccine and 1 case among 4,633 children receiving placebo (Rennels, 2000; Rennels et al., 1998). |
|
|
August 31, 1998 |
Rotavirus vaccinea (RRV-TV) is licensed by FDA for use in infants (CDC, 1999). Vaccine package insert includes reference to intussusception as potential adverse event (Rennels, 2000). However, background rates of intussusception are not statistically different from those identified during pre-licensure study (Rennels et al., 1998; see below). |
|
|
|
Intussusception in the following groups |
Rate (%) |
|
|
Study placebo recipients |
0.022 |
|
|
Study vaccine recipients (all doses) |
0.05 |
|
|
Study vaccine recipients (dose proposed for licensure) |
0.024 |
|
|
Health plan population 1995-1996 (California Kaiser Permanente study) |
0.074 |
|
|
General population 1991-1995 (New York State) |
0.05 |
|
|
FDA requires Phase IV (post-licensure study) of adverse events (CDC, 2004). Intussusception search code is added to VAERS database (Rennels, 2000). |
|
PART I:
COMPONENTS OF THE 1986 LEGISLATION
Reporting and Investigating Adverse Events: Assessing Causality
Post-licensure vaccine safety surveillance is an important component of the vaccine safety system that begins operation for a given vaccine at the time it is licensed by FDA and health care providers begin to administer it. Surveillance for adverse events following immunization—based on reporting by the public, health care providers and manufacturers—is conducted by two entities: VAERS and the Vaccine Safety Datalink (VSD). The military also provides vaccination to its personnel, and the Department of Defense operates its own military immunization program and the Vaccine Healthcare Centers Network (2009) that provide “expert consultative services for vaccine adverse events case management and reporting; research in vaccine safety and quality assurance; and healthcare provider/patient education and training programs.”
Before the 1986 NCVIA was enacted, reports of adverse events following immunization were captured through two different systems. One was a system administered by FDA and intended to gather spontaneous vaccine adverse event reports from manufacturers, pharmacists, health care providers, and the military. The other system, the Monitoring System for Adverse Events Following Immunization established in 1978, was administered by CDC and intended to collect reports from parents whose children received publicly purchased vaccine. As a result of the law, the two reporting systems were integrated into VAERS, which began operating in November 1990 and currently receives approximately 30,000 reports annually from manufacturers, health care providers, and vaccine recipients or their parents or guardians (CDC, 1990; HHS, 2008b). Approximately 85 percent of reports received by VAERS describe mild events, while 15 percent describe serious adverse events (life-threatening, requiring hospitalization, or resulting in death) (CDC, 2009a). (See Figure 2-1 for an overview of the federal vaccine safety system.)
The VAERS system has strengths and weaknesses. A major strength is that anyone may submit a vaccine adverse event report to VAERS including consumers. Weaknesses of VAERS include incomplete reporting of adverse events, varying quality and completeness of individual reports, and several important biases (Iskander et al., 2006; Varricchio et al., 2004). Although the system is capable of capturing rare and unusual adverse events following immunization, and CDC staff use sophisticated data mining techniques to maximize the usefulness of VAERS data to detect safety signals (Iskander et al., 2006), reports to VAERS may simply indicate a perceived relationship to the vaccine, usually based on a coincidental temporal association between vaccine administration and the adverse event. To assess causality, one needs to compare the expected rate of the reported condition in a comparison
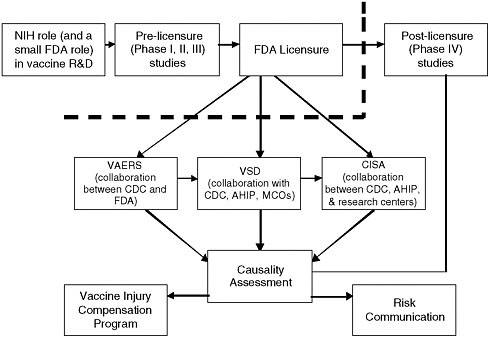
FIGURE 2-1 In December 2008, HHS and NVPO released a detailed overview of the federal vaccine safety system in some detail, with some reference to non-government stakeholders (HHS, 2008). In addition to the federal agencies charged with various components of researching, regulating, monitoring, and communicating about vaccine safety, many other stakeholders, including vaccine manufacturers, academic researchers, health care providers, public health agencies, and the public make important contributions.
NOTE: The thick dotted line represents the point at which vaccines enter the market, become recommended for use, and become increasingly used in the population. Risk communication is discussed in more detail in Chapter 3 of the present report. Adapted from HHS (2008a). For the sake of simplicity, does not reflect Department of Veterans Affairs (VA) and Department of Defense (DoD) contributions to the federal vaccine safety system (see discussion in text). Acronyms: CISA: Clinical Immunization Safety Assessment; MCOs: Managed Care Organizations; NIH: National Institutes of Health.
group. Because there are no comparison groups available for VAERS, data the system collects are almost always only one part of the information needed to assess whether or not there is an association between a vaccine and an adverse event. Due to incomplete reporting and lack of appropriate comparison groups, neither the incidence of an adverse event nor the relative risk of the event in vaccinated individuals can be calculated (Varricchio et al., 2004). Thus, VAERS data cannot ordinarily establish that an epidemio-
logic, much less, a causal, association exists between the suspected adverse event and immunization.
The VSD program represents a model for government-academia-health care delivery system collaboration involving CDC, FDA, AHIP, and university-based researchers. VSD utilizes databases from eight managed care organizations that provide medical care for 8.8 million children and adults. Because VSD has the capability of generating appropriate comparison groups it can also analyze data that establish an epidemiologic association that can provide stronger evidence for causality than that provided by case reports. VSD conducts active surveillance for adverse events of interest and for any adverse event resulting in a health care contact within a time period of interest following vaccination (i.e., VSD data can be searched systematically for specific events or time periods). After the introduction of new vaccines, VSD investigators develop hypotheses, often on the basis of reports to VAERS or data from pre-licensure trials, and the VSD database facilitates their investigation. In the past several years, VSD has developed a new capability—rapid cycle analysis of its database, that enables VSD researchers to conduct near real-time (weekly) active surveillance of vaccine safety.
One of VSD’s strengths is the link to electronic medical records and access to medical charts for clinical information and vaccination histories. Limitations of the VSD include its sample size, which, though large, may not be adequate to detect association for extremely rare adverse events, for example those that occur in one in a million individuals. VSD is currently conducting monitoring of adverse events for the following vaccines: meningococcal conjugate vaccine, Tdap, MMRV, seasonal influenza, quadrivalent HPV, combination DTaP-Hepatitis B-IPV, and RRV5. VSD is also preparing for active surveillance using Rapid Cycle Analysis for the vaccine against novel influenza A (H1N1), RRV1, DTaP-IPV, and DTaP/IPV/Hib vaccines.
Another component of the vaccine safety system is the Clinical Immunization Safety Assessment (CISA) network that is a collaborative effort between CDC, AHIP, five academic medical institutions, and one managed care organization. CISA investigators conduct intensive clinical study of cases of adverse events following immunization, in an effort to better understand the complex relationship to vaccines and inform the development of guidance for clinicians on the management of serious adverse events (Halsey et al., 2009). Some of CISA’s work leads to publication (Slade, 2009).
One of CISA’s collaborators is the Department of Defense’s network of Vaccine Healthcare Centers (VHC). CISA’s role and expertise are complementary to those of VSD. While VSD takes an epidemiologic approach to assessing causality, CISA’s approach is focused on understanding the pathogenesis of adverse events following immunization at the individual level, as the only component of the vaccine safety system that conducts clinical and
basic science research and that provides consultation (via phone and e-mail) to providers managing adverse events following immunization (Halsey et al., 2009; Slade, 2009).
FDA’s role in monitoring vaccine safety (together with its effectiveness, as they cannot be considered in isolation from one another [IOM, 2007]) spans a vaccine’s entire lifespan. The regulatory role begins when a vaccine developer approaches the agency to discuss preliminary plans for a Biologic License Application and request an Investigational New Drug protocol to allow clinical trials with humans. That role continues during regulatory review, through the point of licensure, which today includes requirements for post-licensure surveillance (i.e., Phase IV) studies, and for as long as the vaccine is manufactured or used. As noted earlier, FDA collaborates with CDC in managing the VAERS system and in overseeing post-licensure safety research. The processes by which the pre-licensure review of a vaccine fully anticipates and informs the post-licensure phase have undergone some strengthening in response to the FDA Amendments Act. For example, FDA has gained authority to require post-licensure studies and specific plans to minimize and manage risks posed by medical products including vaccines.
FDA is in the early stages of establishing the Sentinel Initiative, a system for large-scale surveillance of medical product safety, including vaccines. The initiative is intended “to link and analyze healthcare data from multiple sources” by accessing (and analyzing) data from 25 million patients by July 1, 2010, and from 100 million patients by July 1, 2012 (FDA, 2008b). FDA is supporting researchers to evaluate various methodologic approaches and other dimensions of the use of very large databases to evaluate medical product safety. A national discussion among both federal partners and non-government stakeholders about vaccine safety research priorities could also inform investigations based on the sentinel system. This will require greater coordination between CDC, FDA, and other federal agencies, and the committee hopes, strong coordination with national health information technology efforts. The committee noted that the plans of the Office of the National Coordinator for Health Information Technology (ONCHIT) include objectives on linkage with immunization registries and recognition of immunization status as an important component of electronic health records. Some ONC documents, such as the “Meaningful Use Matrix” (intended to guide the meaningful use of electronic health records to engage patients, provide real-time access to all medical information, and support quality and safety as well as improved access and the elimination of health care disparities [ONCHIT, 2009]) also include references to FDA’s Sentinel Initiative. Although there are considerable barriers to the successful development and implementation of health information technology, the committee hopes that what it has noted is an indication that health information technology planning at the highest levels of government is coordinated with the
national medical product safety surveillance effort. At the time of this writing, CDC, FDA, and VSD have developed and are implementing a system to monitor the safety of the H1N1 pandemic influenza vaccine. The network for Post-Licensure Rapid Immunization Safety Monitoring (PRISM) has been established to link immunization registries in a number of states with the databases of large health maintenance organizations. This system considerably expands VSD’s sample size and could perhaps provide some idea of how the Sentinel Initiative may function (HHS et al., 2009).
Finally, in addition to its role in VAERS, VSD, and CISA, CDC also is the nation’s lead public health agency, able to respond rapidly to the emergence of a vaccine safety question with the expertise needed to assess urgent public health issues such as disease outbreaks or serious vaccine safety concerns. CDC is able to deploy epidemiologists and other experts to conduct case-control interviews, conduct laboratory research, work with state public health personnel and health care providers, and carry out other activities needed to intensively investigate a potential serious adverse event following immunization. The experience with the first rotavirus vaccine, RRV-TV, described above, is an example of the federal and state public health capabilities in quickly responding to and elucidating the meaning of a vaccine safety “signal” captured through VAERS or by other means (e.g., active surveillance through VSD). After a vaccine’s licensure, CDC’s efforts to ascertain vaccine safety (and effectiveness) complement those carried out by FDA, in addition to activities conducted jointly, such as VAERS and VSD.
Information and Communication
The social context of vaccine safety has changed in the decades since the 1986 act was signed into law. As immunization has resulted in vastly lower rates of some diseases and entirely eliminated other diseases, the direct relationship between vaccine and disease prevention has become less and less visible to the public. Today diseases such as polio, diphtheria, and congenital rubella syndrome no longer top the list of fears parents have for their children’s health; polio has been eradicated from the Western Hemisphere, and other diseases may be mere memories or only rare occurrences. A major decrease in the rate of a vaccine-preventable disease may alter the risk-benefit analysis for a vaccine targeting that disease. Other changes in the past two decades include social and cultural transformations that have shaped public attitudes toward vaccination both positively and negatively. These include the emergence of active and engaged patients, and the rapid availability of vast amounts of information via the Internet, and the emergence of organized groups opposed to immunization. The committee believes that one major challenge in communicating about vaccines relates to their dual identity as a medical intervention to protect an individual against
disease and a public health intervention to prevent community-wide outbreaks and protect those in the community who cannot be vaccinated due to age or health status, or those who do not mount an adequate immune response to vaccines.
The committee asserts that this duality—the personal and public benefits of vaccines—has not been clearly conveyed in the process of communicating about immunization. This is a serious omission because it hinders necessary public discussion of the implied conflict between two widely held values: protecting the health of the community and individual freedom of choice. As the incidence of a transmissible vaccine-preventable disease declines questions arise about what is the optimal balance of individual choice and responsibility to the community. Communication on these topics needs to be expanded and strengthened as our nation’s public health becomes increasingly dependent on vaccine-induced immunity to diseases still prevalent elsewhere in the world and therefore are only a plane ride away from U.S. communities in addition to diseases that remain endemic in the United States. Also, the many efforts undertaken by the nation’s vaccine safety system are frequently invisible to the public and communication about vaccine risk would be strengthened by thorough and clear information about the science of vaccine safety and about the mechanisms that are in place to detect and respond to potential vaccine safety problems. Chapter 3 in this report provides more extensive discussion of communication about vaccines and vaccination.
The Compensation Program
As noted in the Introduction to this report, a central purpose of the 1986 law was compensation of individuals thought to have been injured by vaccines. The rationale underlying the Vaccine Injury Compensation Program (VICP) is that when individuals have been injured by an appropriately manufactured and administered vaccine as part of a public health program they should be compensated. Prior to the creation of the VICP by the passage of the NCVIA in 1986, individuals who believed they had been injured by a vaccine had to seek compensation through litigation in the tort system. Escalating litigation costs related primarily to claims of vaccine-related injuries inadvertently caused by properly manufactured and administered live oral polio virus vaccine and whole cell pertussis vaccines threatened the vaccine industry’s economic viability and thereby the supply of vaccine required to continue public health immunization programs. Congress established VICP “[b]ecause society mandates the use of vaccines, through state laws for school enrollment” and therefore “it is reasonable and appropriate that society take responsibility for unavoidable adverse outcomes associated with the use of vaccines” (Evans, 2006:S132).
As described in the Introduction, petitioners to the program who meet basic preconditions (e.g., demonstrating that they received the vaccine they claim caused the injury) may obtain compensation by two pathways, depending on whether the alleged injury is listed on the VICP injury table. If they claim an injury listed on VICP’s injury table, they need not demonstrate a causal link between the injury, and the vaccine listing in the table attests that there is accepted scientific evidence of a causal link between the injury and the vaccine. The “off-table” pathway allows individuals who believe they have been injured by a vaccine to be awarded compensation by establishing by only a preponderance of evidence that their injury is related to a vaccine even though the weight of available evidence may fall far short of the scientific standard for establishing a causal link between the vaccine administered and the alleged injury. On-table injuries can be resolved without litigation,6 since causality has been established. Off-table injuries are more complex to address, and in the absence of scientific consensus about causality U.S. Court of Federal Claims special masters must weigh the evidence. Currently most claims under the Vaccine Injury Act follow the off-table pathway. Because the special masters interpret that the intent of Congress was to compensate all individuals thought to have been injured by ACIP-recommended childhood vaccines, inevitably some individuals will be compensated whose injuries were not in fact caused by a vaccine.
Analysis of the claims relating to off-table injuries may have the potential to generate hypotheses deserving scientific study that could yield evidence of causal associations that would then warrant modification of the vaccine injury table. There have been delays in updating injuries listed in the table and undertaking the necessary research to update the table. This has generated concern that there is a lack of government commitment to understanding the full extent of vaccine risks even as it pursues universal immunization as a public good. Delays also have resulted in the large proportion of off-table awards based only on a preponderance of the often scant evidence available, perhaps heightening public worry about vaccine safety.
NVPO and the Plan
The Introduction to this report provides a history of NVPO and the National Vaccine Plan. NVPO’s potential contributions to coordination on vaccine safety are discussed in Part II below.
PART II:
RECOMMENDATIONS ABOUT PRIORITY ACTIONS IN THE PLAN
The committee’s process of deliberation identified four priority actions in Goal 2:
-
Development of a coordinated national vaccine safety research agenda;
-
Development of a coordinated, structured process to address vaccine safety signals promptly on detection and track them through until resolution (including a way to consider and address safety concerns raised through the Vaccine Injury Compensation Program);
-
Establishment of a vaccine safety advisory group capable of supporting coordination among federal agencies and with stakeholders, and soliciting and receiving public and stakeholder input on matters related to vaccine safety (the topic of transparency about policy making and other communication issues is discussed in Chapter 3); and
-
Allocating resources adequate to support basic, clinical, and epidemiologic research pertinent to vaccine safety.
A Coordinated and Transparent Process to Address Vaccine Safety Signals from Detection to Resolution
Although there are many effective components of the vaccine safety system (as evident in the detection and investigation of safety concerns related to rotavirus, MMRV,7 and influenza vaccines, among others), the system can be enhanced through coordination. Coordinating vaccine safety within government agencies (also ideally involving stakeholders) could facilitate application of advances in vaccine safety science, the timely and efficient response to safety concerns, and the best use of resources. As one example, there currently is no formal and coordinated process for referring safety problems to VSD or CISA. The process is largely ad hoc and it appears that CISA, for example, relies on multiple informal mechanisms such as personal contacts with providers in a CISA center’s immediate community to generate research questions. Although the committee was not asked to and did not evaluate the functioning or effectiveness of CISA or VSD, the committee believes that these programs play crucial roles in the vaccine safety system, and ensuring their optimal functioning is necessary for an effective vaccine safety system.
Furthermore, the committee understands that there is no organized mechanism to address scientific questions that arise in the course of adju-
dicating cases before the U.S. Court of Federal Claims. Analysis of claims may generate research questions that can both address gaps in vaccine safety science and reduce the complexity of the compensation process. Creating such a mechanism (or expanding the currently limited frequency with which issues are referred from the compensation program to the hypothesis-testing VSD investigators or to the CISA researchers) would “close the loop,” incorporating information that emerges from injury cases into the vaccine safety scientific research agenda as an additional source of adverse event detection or hypothesis generation.
In recent years, the public debate about vaccines has extended to vaccine safety research, and questions have arisen about how decisions are made about what research and surveillance activities are undertaken. This is, in part, a communication challenge because the decision-making processes undertaken by CDC together with academic and health care researchers are generally not visible to the public. The approach to assessing causality when a concerning adverse event is temporally associated with a vaccine includes several criteria that evaluate factors such as methodologic rigor, and consistency and frequency of findings.8 Data obtained from case reports, case series, or court cases are sufficient only for the purpose of hypothesis generation. The possible association with vaccine must then be evaluated in a scientifically rigorous manner to answer the question.
Recommendation 2-1: The National Vaccine Plan should establish a process to identify potential vaccine safety hypotheses for further basic, clinical, or epidemiologic research through annual review of data from VAERS, VSD, CISA and the VICP, and from information available from sources outside the United States.
The compensation program fills a crucial need, and it is important to strengthen its effectiveness. The program needs to preserve its ability to compensate those possibly injured by vaccines, despite a lack of scientific certainty about the etiology of adverse events. The committee believes that scientific hypotheses raised by the compensation program could inform the safety research agenda, help to spur additional research, and potentially
bring resolution to some of the uncertainties encountered in the compensation program. (Decisions in the VICP provide a crucial opportunity [and imperative] to communicate about vaccine safety. This is discussed in more detail in Chapter 3.)
The Research Agenda
The NVAC draft report on the Immunization Safety Office (ISO) agenda (May 28, 2009) stated that “there is a strong need for a federal vaccine safety research agenda that encompasses research undertaken by non-ISO CDC offices, FDA, and National Institutes of Health (NIH) and requires increased collaboration and coordination between all federal agencies with a stake in vaccine safety” (NVAC, 2009b:9).
This statement is similar to comments made at the committee’s April 2009 stakeholder meeting on the vaccine safety component of the draft National Vaccine Plan. Stakeholders noted that there are important gaps in basic, clinical, and epidemiologic science relating to vaccine safety at several different levels. The wide range of specific research questions that could be considered includes: safety of concurrently administered and combination vaccines; vaccine safety in special or vulnerable populations; individual susceptibility (urea-cycle defects, other inborn errors of metabolism); and safety of rechallenge (administering another dose in individuals who experienced a serious adverse event following vaccination with a given vaccine).
There is no coordinated national vaccine safety research agenda that includes the combined input of federal agencies and external stakeholders. CDC’s ISO has a research agenda, and it has sought the help of NVAC and NVPO to obtain broad public input on updating that agenda. The thorough process of developing guidance for ISO undertaken by NVAC with NVPO support has included a broad level of stakeholder engagement through meetings with the public and expert stakeholders, and solicitation of comments on the agenda through notices in the Federal Register. NVPO and NVAC have been reviewing the large volume of material generated by these efforts, and the process has culminated in a report to ISO summarizing the input received and making recommendations on the research agenda (NVAC, 2009b).
ISO is responsible for only a part of the vaccine safety research conducted in the United States, whether by ISO staff or by collaborators, such as VSD and CISA investigators and sites. Although the ISO agenda represents the future work of a considerable proportion of the federal vaccine safety system, a national vaccine safety research agenda is needed to help guide and coordinate the efforts of all federal agencies and non-government and industry stakeholders that are responsible for one or more aspects of vaccine safety research. Furthermore, the ISO agenda is largely focused on
epidemiologic and clinical assessments of whether there is a causal connection between adverse events following immunization and the vaccines that were administered, and that agenda includes only a small amount of basic science investigation (conducted by the CISA network).
The NIH website states “vaccine safety is an integral component of all National Institute of Allergy and Infectious Diseases (NIAID) vaccine research and development” but provides no specifics. A review of the list of study sections that conduct peer review of proposals to NIH institutes shows that, although there is one study section on vaccines and another on HIV vaccines, there is no study section on vaccine safety (this confirms an observation made at the committee’s April 2009 stakeholder meeting). There clearly is a role for NIH-supported research regarding vaccine safety, but remarkably, given its importance to the nation’s public health, vaccine safety research (other than basic, for example, pathophysiology or genomics) does not appear to be included among the many types of research NIH undertakes or supports.
Given the increased interest in and scientific endeavors to support patient-centered medicine, and questions that have arisen about how vaccination may or may not affect certain groups (e.g., children with inborn errors of metabolism), it is important that the U.S. vaccine safety system address these questions. Contributions to vaccine safety science could come from institutes other than NIAID (which has primary responsibilities for vaccine-related research) and could involve the basic science research efforts of the National Institute of Neurological Disorders and Stroke, the National Institute of Arthritis and Musculoskeletal and Skin, the National Institute of Child Health and Human Development, the National Institute of Diabetes and Digestive and Kidney Diseases, National Human Genome Research Institute, and the National Cancer Institute. There has been a high level of public interest in and speculation about potential links between vaccines and certain diseases of the immune and nervous systems, such as Guillain-Barré syndrome, autism, ADHD,9 and asthma, and the institutes listed above support a variety of studies that might shed some light on biological mechanisms, genetic factors, and other characteristics that may have relevance to vaccine safety.
Recommendation 2-2: The National Vaccine Plan should emphasize the development and publication of a framework for prioritizing a national vaccine safety research agenda that spans all federal agencies and includes all stakeholders, including the public.
The scientific criteria of such a framework for prioritization might include, but are not limited to:
-
Assessment of the nature and extent of existing evidence for a possible association of an adverse event with a vaccine.
-
Determination of the individual or public health burden of potential adverse events following immunization.
-
Consideration of the feasibility of scientifically rigorous study of a safety concern.
-
Assessment of biological plausibility of a causal association between an adverse event and a vaccine.
Coordination and Vaccine Safety
One of the five dimensions of the vaccine safety system outlined by the 1986 act refers to the overarching National Vaccine Program, and by extension, to its operating arm, NVPO. The HHS Comprehensive Review of Federal Vaccine Safety Programs and Public Health Activities (2008a) states that NVPO coordinates HHS vaccine safety activities and explains that NVAC provides a forum for the exploration of vaccine safety policy issues that arise among HHS agencies (see Box 2-1). Although the legislation was clearly intended to address the need for intra-departmental coordination of
|
BOX 2-1 Role of the National Vaccine Program Described in the 1986 Act “[C]oordinate and provide direction for research carried out in or through the National Institutes of Health, the Centers for Disease Control, the Office of Biologics Research and Review of the Food and Drug Administration, the Department of Defense, and the Agency for International Development on means to induce human immunity against naturally occurring infectious diseases and to prevent adverse reactions to vaccines.”a |
vaccine safety activities, meaningful, effective coordination to address gaps in the science vaccine safety, as one example, has not been achieved. This may, at least in part, be due to the factors (lack of funding, human resources, authority, and visibility) that have prevented NVPO from functioning as intended by the 1986 law. Additional discussion of NVPO and interagency coordination is provided in Chapter 6.
Multiple government agencies and private sector entities handle aspects of vaccine safety, therefore, developing and implementing a national-level safety research agenda requires coordination among federal agencies, such as NIH and FDA, and with stakeholders (such as health care providers who work with special populations, and vaccine manufacturers) to assume joint responsibility for and work collaboratively on some of the great challenges in vaccine safety research. The committee found evidence that the system can work well to address safety concerns. However, achieving coordination among government agencies, understanding and addressing the public perception of the safety system’s competence and transparency, and allocating resources for vaccine safety research that are commensurate with the expanding task (i.e., surveillance and study of the safety of a growing number of licensed vaccines currently in use) all remain major challenges. Factors that may have slowed the pace of progress in vaccine safety include the absence of broader NIH participation in vaccine safety research; NVPO’s lack of authority and resources needed to fully perform the coordinating role (with respect to vaccine safety issues, among others) called for by legislation; and the lengthy process over the past several years of finding a permanent home in CDC for ISO, and until recently, lack of stable, permanent ISO leadership.
The absence of interagency coordination on vaccine safety was recognized in a 1998 Task Force on Safer Childhood Vaccines report that recommended that the Interagency Vaccine Group (IAVG) formed in the 1980s take on as a primary responsibility the coordination of vaccine safety activities, and that NVPO function as the secretariat for the IAVG in that area (see Box 2-2). The current IOM committee was struck by the Task Force report’s continuing relevance more than a decade later. The committee recognized that the task force was seeking to fill a major gap in the coordination necessary to ensure an effective vaccine safety program. The task force discussed the Interagency Vaccine Group, an internal government entity that is still in operation, and described a potential role for it in strengthening coordination of vaccine safety activities, from communication, to monitoring and surveillance, to safety research. The Task Force report also stated that “[o]verall coordination of programs involving both broad vaccine issues and vaccine safety is the responsibility of the Vaccine Interagency Group [sic]
|
BOX 2-2 An Example of Coordination Within HHS: Role of the Interagency Vaccine Group The 1998 Task Force on Safer Childhood Vaccines defined IAVG’s role as follows:
|
of the National Vaccine Program Office” (NIAID, 1998).10 More recently, the NVAC State of the Program 2008 report, stated that “[f]ederal efforts have suffered from a lack of coordination and oversight of vaccine-related activities, highlighted most recently by the need for the Secretary of HHS to empanel a special interagency vaccine safety working group to better address this growing concern” (NVAC, 2009a).
NVAC’s observation raises two related but distinct points about federal vaccine safety activities. Despite some potential overlap, coordination and oversight are somewhat different functions, with different rationales, and likely different solutions. Coordination refers to working together effectively to define priorities, to achieve a shared vision and to resolve strategic issues that exceed one agency or stakeholder’s capabilities. Operationally, coordination may mean working to avoid wasteful duplication of effort and inefficient use of funds. Oversight is defined as “watchful care” and typically refers to the function of assuring accountability and propriety. In the realm of vaccine safety, there is a history of dialogue about independent oversight of vaccine safety monitoring and research as a response to concern about a perceived inherent conflict of interest in government in general and CDC specifically, given its responsibility both for preventing disease through the optimal use of vaccines and for monitoring post-licensure vaccine safety in the population (CDC, 2005; Cooper et al., 2008; Salmon et al., 2004).
The committee recognizes the desire to strengthen confidence in the safety system, and is aware of some of the arguments employed. The topic of the placement of an entity conducting vaccine risk management as opposed to risk assessment was discussed at the committee’s April 2009 stakeholder meeting. The committee deems only the matter of intragovernmental coordination (HHS, its agencies, and DoD) as directly germane to its task and to the preparation and implementation of the National Vaccine Plan (the primary instrument for effecting coordination in the National Vaccine Program). The matter of independent oversight falls outside this IOM committee’s scope of work.
The IAVG’s functioning in the area of vaccine safety does not necessarily match the description in the 1998 task force report, and there is no reason why it should. The committee believes that the job description developed by the task force remains relevant, that it calls for a different type of entity (in addition to IAVG in its ongoing role), and that such a role would ideally be performed in a setting that permits meetings that are open to the public.
This IOM committee believes that a federal advisory group has the potential to play a crucial role both as a facilitator of coordination (especially with stakeholders, and as a supporter of NVPO’s role in coordinating within government), and also as a somewhat independent source of guidance on vaccine safety issues. NVPO provides staff support and works very closely with NVAC, the advisory committee established to advise the Secretary of HHS on matters related to National Vaccine Program. The role played by the NVAC Vaccine Safety Working Group in reviewing policy matters related to vaccine safety appears to have contributed an independent and credible perspective on vaccine safety. With NVPO support, the group also has engaged the public in thoughtful dialogue about challenging matters of vaccine safety policy. While a working group structure provides useful flexibility, its activities may be less transparent than those of a subcommittee, as subcommittees are required to follow the Federal Advisory Committee Act.
Recommendation 2-3: The National Vaccine Plan should include the establishment and scope of work of a permanent NVAC vaccine safety subcommittee to:
-
provide guidance on the activities described in Recommendations 2-1 and 2-2 in a public and transparent manner;
-
provide guidance about the identification and evaluation of potential safety signals; and
-
publish on a biennial basis a review of potential safety hypotheses; current vaccine safety activities including those of pre-and post-licensure studies, VAERS, VSD, and CISA; and planned priorities for research.
The NVAC subcommittee could be informed of potential safety signals and the actions planned to investigate the signal and related public communication. Public representation on the subcommittee is crucial, and the committee notes that NVAC has set strong precedent in including public or consumer representatives (all of its recent committee and working group rosters attest to this; refer to Appendix E for a short history of HHS public engagement activities related to vaccines).
Funding for Vaccine Safety Research
A major theme in the stakeholder comments about Goal 2 of the draft National Vaccine Plan was the inadequacy of funding for vaccine safety research (IOM, 2009). This concern has been voiced elsewhere by other commentators in the field (for example, Cooper et al., 2008). The committee believes that there are two major areas where vaccine safety research
TABLE 2-2 Comparison of Immunization Safety Office and Vaccines for Children Program Funding
|
Year |
ISO Funding |
Vaccines for Children Program Funding |
|
2004 |
$21.8 million |
>$1 billion |
|
2005 |
$22.8 million |
$1.2 billion |
|
2006 |
$21.7 million |
$1.7 billion |
|
2007 |
$21.5 million |
$1.9 billion |
|
2008 |
$21.7 million |
~$3 billion |
|
SOURCES: Personal communication, C. Johnson, CDC, July 7, 2009; Shefer, 2008. |
||
warrants additional support. First, the CDC Immunization Safety Office needs more funding and staff to conduct its work. The second area pertains to NIH research and would necessitate a partial reorientation of some of the agency’s research priorities to ensure a greater balance between classic investigator-initiated research, which is a crucial engine of vaccine innovation, and research prompted by public health concerns specifically focused on vaccine safety, including some level of directed research, and not simply limited to very basic or early clinical research.
There are strong obstacles to such a reorientation in NIAID (the NIH institute with primary responsibilities for vaccines), especially in the absence of a strong coordinating entity within the National Vaccine Program that can help align program-wide needs (such as vaccine safety research) with solutions. The committee reviewed the lengthy list of NIH-funded vaccine-related research projects and found that a small proportion appear to have some relevance to safety, and an even smaller subset have safety as a primary objective. As a simple illustration, a search of the database of NIH-funded vaccine-related research yielded 24 studies (out of 3,003) that referred to safety in the title (6 from NIAID, 18 funded by other institutes), and the vast majority appear to be pre-licensure Phase I or II studies. This paucity of research on vaccine safety is congruent with stakeholder comments at the committee’s April 2009 meeting, where the low level of NIH funding for vaccine safety research was a major theme. The IOM committee contacted NIAID to inquire about the status of the Program Announcement for Research to Advance Vaccine Safety (first introduced in 2008), what and how many proposals had come in, and what proposals were funded. The institute’s response was to refer the committee to the RePORTER database to search for the two relevant funding codes.11 The committee did so in August 2009 but found no information about funded research pertaining to the two program announcements.
Funding for vaccine safety research conducted and supported by CDC is also limited. Although the childhood and adolescent immunization schedule has grown between 2004 and 2008 (with the addition of two rotavirus vaccines and two vaccines against human papilloma virus, and new combination vaccines, among others), the budget for CDC’s Immunization Safety Office has not. Table 2-2 is provided to illustrate that while funding of vaccine purchases for the Vaccines for Children entitlement program (that may be a reasonable proxy for government expenditures on vaccines) has increased three-fold between 2004 and 2008, the ISO budget has remained unchanged. Funding for vaccine safety monitoring and research has not grown commensurate with the widening task (e.g., a growing list of recommended vaccines) and parallel investment in the vaccine supply. Thus, despite the fact that the universe of potential vaccine safety questions and signals can be expected to expand with the addition of new vaccines, the funds available to support, for example, VSD’s Rapid Cycle Analysis and CISA’s in-depth clinical studies of vaccine adverse event pathogenesis, have not increased to match the growing responsibilities of ISO.
The committee believes that the current climate of support for science-based policy and for reforming health care offers opportunities not only to enhance coordination and increase the visibility of vaccine safety activities, findings, and policy decisions, but also to strengthen the funding allocated to the crucial tasks of monitoring and studying the safety of licensed vaccines. Stakeholders such as academia, industry, and the public could contribute to the vaccine safety system and are important to include in dialogue about the national vaccine safety research agenda discussed above and in devising innovative mechanisms to fund important research that currently does not have well-established funding mechanisms to address it. With regard to academia and its contributions to the safety research agenda, stakeholder comments identified a need to comprehensively integrate education about vaccines and immunization in professional education, and also to train the next generation of vaccine safety researchers in relevant disciplines.
Recommendation 2-4: The National Vaccine Plan should incorporate concrete steps to expand and strengthen vaccine safety research, including:
-
enhanced funding for CDC’s Immunization Safety Office activities, including support of extramural research;
-
enhanced funding for FDA’s safety monitoring activities; and
-
expansion of NIH vaccine safety activities to include research portfolios, funding through requests for proposals, program announcements, and creation of a study section dedicated to vaccine safety research.
Funding could be allocated to each federal agency to support activities that implement the identified priorities as appropriate to each agency’s research capabilities and strengths.
CONCLUDING OBSERVATIONS
A dearth of vaccine safety research initiatives to address public concern about vaccine safety will not strengthen public confidence in the immunization system. In the legal arena for example, absence of an adequate body of good scientific evidence and a mere preponderance of the scant, often flawed evidence available may result in compensation of off-table injuries that may not be causally related to vaccines, adding to public uncertainty about the safety of vaccines.
As mentioned earlier in this chapter, most discussions about the safety of vaccines raise questions about communication, and one of the important topics in vaccine communication is vaccine safety. The links between Goals 2 and 3 in the National Vaccine Plan were also very evident at the committee’s information-gathering meetings with national stakeholders. Communication, or “informed vaccine decision making,” as the topic is framed in the draft plan, is discussed in detail in Chapter 3. It is important to recognize that given the current social and cultural climate, many discussions about vaccine safety will have a strong undercurrent of references to public confidence in the system.
REFERENCES
CDC (Centers for Disease Control and Prevention). 1990. Current trends vaccine adverse event reporting system—United States. MMWR 39(41):730-733.
CDC. 1999. ACIP Rotavirus Vaccine Recommendation. Available: http://www.cdc.gov/mmwr/Preview/Mmwrhtml/00056669.htm [accessed September, 2009].
CDC. 2000. Poliomyelitis prevention in the United States: Updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 49(RR05):1-22.
CDC. 2004. Suspension of rotavirus vaccine after reports of intussusception—United States, 1999. MMWR 53(34):786-789.
CDC. 2005. Blue Ribbon Panel Meeting, Summary Report, June 3 and 4, 2004. Available: http://www.cdc.gov/od/ads/brpr/brprsumm.htm [accessed January 27, 2009].
CDC. 2006. Influenza and pneumococcal vaccination coverage among persons aged >65 years—United States, 2004–2005. MMWR 55(39):1065-1068.
CDC. 2009a. Frequently Asked Questions about VAERS. Available: http://vaers.hhs.gov/vaers.htm [accessed August 13, 2009].
CDC. 2009b. Poliomyelitis. Available: http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/polio.pdf [accessed August 2009].
Chang, E.J., K.M. Zangwill, H. Lee, and J.I. Ward. 2002. Lack of association between rotavirus infection and intussusception: Implications for use of attenuated rotavirus vaccines. Pediatr Infect Dis J 21(2):97-102.
Cooper, L.Z., H.J. Larson, and S.L. Katz. 2008. Protecting public trust in immunization. Pediatrics 122(1):149-153.
Ellenberg, S.S., M.A. Foulkes, K. Midthun, and K.L Goldenthal. 2005. Evaluating the safety of new vaccines: Summary of a workshop. AJPH 95(5):800-807.
Evans, G. 2006. Update on vaccine liability in the United States: Presentation at the National Vaccine Program Office workshop on strengthening the supply of routinely recommended vaccines in the United States, 12 February 2002. Clin Infect Dis 42(Suppl 3):S130-S137.
FDA (Food and Drug Administration). 2008a. April 3, 2008 Approval Letter. Available: http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm133541.htm [accessed December 9, 2009].
FDA. 2008b. The Sentinel Initiative: National Strategy for Monitoring Medical Product Safety. Available: http://www.fda.gov/downloads/Safety/FDAsSentinelInitiative/UCM124701.pdf [accessed September, 2009].
Folb, P.I., E. Bernatowska, R. Chen, J. Clemens, A.N.O. Dodoo, S.S Ellenberg, C.P. Farrington, T.J. John, P.H. Lambert, N.E. MacDonald, E. Miller, D. Salisbury, H.J. Schmitt, C.A. Siegrist, and O. Wimalaratne. 2004. A global perspective on vaccine safety and public health: The global advisory committee on vaccine safety. AJPH 94(11):1926-1931.
GSK (GlaxoSmithKline). 2008. GSK Receives Favourable Recommendation by FDA Advisory Committee for Rotarix [rotavirus, live, oral]. Available: http://www.gsk.com/media/pressreleases/2008/2008_pressrelease_0006.htm [accessed December 9, 2009].
Halsey, N., K. Edwards, C. Dekker, R. Baxter, N. Klein, P. LaRussa, C. Marchant, and R. Engler. 2009 (February). Clinical Immunization Safety Assessment Network (CISA): A Summary of CISA Activities by the Principal Investigators. Atlanta, GA: CDC.
Heyse, J.F., B.J. Kuter, M.J. Dallas, and P. Heaton. 2008. Evaluating the safety of a rotavirus vaccine: The REST of the story. Clin Trials 5(2):131-139.
HHS (Department of Health and Human Services). 2008a. A Comprehensive Review of Federal Vaccine Safety Programs and Public Health Activities. Available: http://www.hhs.gov/nvpo/nvac/documents/vaccine-safety-review.pdf [accessed January 2009].
HHS. 2008b. About the VAERS Program. Available: http://vaers.hhs.gov/about/index [accessed December 9, 2009].
HHS, AHRQ, CDC, FDA, HRSA, Indian Health Service, NIH, Department of Defense, and Department of Veterans Affairs. 2009. Federal Plans to Monitor Immunization Safety for the Pandemic 2009 H1N1 Influenza Vaccination Program. Available: http://www.flu.gov/professional/federal/monitor_immunization_safety.html [accessed November 2009].
IOM (Institute of Medicine). 2007. The Future of Drug Safety. Washington, DC: The National Academies Press.
IOM. 2009 (April 14). Transcript, Fourth National Stakeholder Meeting of the IOM Committee on Review of Priorities in the National Vaccine Plan. Washington, DC: IOM.
Iskander, J., V. Pool, W. Zhou, R. English-Bullard, and the VAERS Team. 2006. Data mining in the US using the Vaccine Adverse Event Reporting System. Drug Saf 29(5):375-384.
Kramarz, P., E.K. France, F. Destefano, S.B. Black, H. Shinefield, J.I. Ward, E.J. Chang, R.T. Chen, D. Shatin, J. Hill, T. Lieu, and J.M. Ogren. 2001. Population-based study of rotavirus vaccination and intussusception. Pediatr Infect Dis J 20:410-416.
Mariner, W. 1992. Legislative report: The National Vaccine Injury Compensation Program. Health Aff (Spring):255-265.
Moylett, E.H., and I.C. Hanson. 2004. Mechanistic actions of the risks and adverse events associated with vaccine administration. J Allergy Clin Immun 114(5):1010-1020.
Murphy, T.V., P.M. Gargiullo, M.S. Massoidi, D.B. Nelson, A.O. Jumaan, C.A. Okoro, L.R. Zanardi, S. Setia, E. Fair, C.W. LeBaron, M. Wharton, J.R. Livingood, and the Rotavirus Intussusception Investigation Team. 2001. Intussusception among infants given an oral rotavirus vaccine. NEJM 344:564-572.
NCSL (National Conference of State Legislators). 2009. States with Religious and Philosophical Exemptions from School Immunization Requirements. Available: http://www.ncsl.org/Default.aspx?TabId=14376 [accessed November 20, 2009].
NIAID (National Institute of Allergy and Infectious Diseases). 1998. Task Force on Safer Childhood Vaccines: Final Report and Recommendations. Bethesda, MD: National Institutes of Health.
NVAC (National Vaccine Advisory Committee). 2009a (February). The National Vaccine Program 2008 State of the Program Report. Available: http://www.hhs.gov/nvpo/nvac/2008StateoftheProgramReportFeb09.pdf [accessed March 2009].
NVAC. 2009b (June). Recommendations on the Centers for Disease Control and Prevention Immunization Safety Office Draft 5-Year Scientific Agenda. Available: http://www.hhs.gov/nvpo/nvac/NVACRecommendationsISOScientificAgendaFinal.pdf [accessed September 9, 2009].
ONCHIT (Office of the National Coordinator for Health Information Technology). 2009 (June 16). Meaningful Use: A Definition, Recommendations from the Meaningful Use Workgroup to the Health IT Policy Committee. Available: http://healthit.hhs.gov/portal/server.pt/gateway/PTARGS_0_10741_872720_0_0_18/Meaningful%20Use%20Preamble.pdf [accessed December 9, 2009].
Rennels, M.B. 2000. The rotavirus vaccine story: A clinical investigator’s view. Pediatrics 106(1):123-125.
Rennels, M.B., U.D. Parashar, R.C Holman, C.T. Le, H-C. Chang, and R.I. Glass. 1998. Lack of an apparent association between intussusception and wild or vaccine rotavirus infection. Pediatr Infect Dis J 17:924-925.
Salmon, D.A., L.H. Moulton, and N.A. Halsey. 2004. Enhancing public confidence in vaccines through independent oversight of postlicensure vaccine safety. AJPH 94(6):947-950.
Shefer, A. 2008 (May). The Childhood Immunization Program. Available: http://www.cdc.gov/omhd/CAMICC/LectureSeries2008/PDFs/Shefer.pdf [accessed January 2009].
Slade, B.A., L. Leidel, C. Vellozzi, E.J. Woo, W. Hua, A. Sutherland, H.S. Izurieta, R. Ball, N. Miller, M.M. Braun, L.E. Markowitz, and J. Iskander. 2009. Postlicensure safety surveillance for quadrivalent human papillomavirus recombinant vaccine. JAMA 302(7):750-757.
Vaccine Healthcare Centers Network. 2009. About the VHC. Available: http://www.vhcinfo.org/subpage.asp?page=about [accessed January 2009].
Varricchio, F., J. Iskander, F. Destefano, R. Ball, R. Pless, M.M. Braun, and R.T. Chen. 2004. Understanding vaccine safety information from the Vaccine Adverse Event Reporting System. Pediatr Infect Dis J 23(4):287-294.
Verstraeten, T., A.L. Baughman, B. Cadwell, L. Zanardi, P. Haber, R.T. Chen, and the Vaccine Adverse Event Reporting System Team. 2001. Enhancing vaccine safety surveillance: A capture-recapture analysis of intussusception after rotavirus vaccination. Am J Epidemiol 154(11):1006-1012.
Zanardi, L.R., P. Haber, G.T. Mootrey, M.T. Niu, M. Wharton, and the VAERS Working Group. 2001. Intussusception among recipients of rotavirus vaccine: Reports to the vaccine adverse event reporting system. Pediatrics 107(6):e97.




























