
A14
ORIGINS AND EVOLUTIONARY GENOMICS OF THE 2009 SWINE-ORIGIN H1N1 INFLUENZA A EPIDEMIC81
Gavin J. D. Smith,82 Dhanasekaran Vijaykrishna,82 Justin Bahl,82 Samantha J. Lycett,83 Michael Worobey,84 Oliver G. Pybus,85 Siu Kit Ma,82 Chung Lam Cheung,82 Jayna Raghwani,83 Samir Bhatt,85 J. S. Malik Peiris,82 Yi Guan,82 and Andrew Rambaut83
In March and early April 2009, a new swine-origin influenza A (H1N1) virus (S-OIV) emerged in Mexico and the United States (CDC, 2009a). During the first few weeks of surveillance, the virus spread worldwide to 30 countries (as of May 11) by human-to-human transmission, causing the World Health Organization to raise its pandemic alert to level 5 of 6. This virus has the potential to develop into the first influenza pandemic of the twenty-first century. Here we use evolutionary analysis to estimate the time scale of the origins and the early development of the S-OIV epidemic. We show that it was derived from several viruses circulating in swine, and that the initial transmission to humans occurred several months before recognition of the outbreak. A phylogenetic estimate of the gaps in genetic surveillance indicates a long period of unsampled ancestry before the S-OIV outbreak, suggesting that the reassortment of swine lineages may have occurred years before emergence in humans, and that the multiple genetic ancestry of S-OIV is not indicative of an artificial origin. Furthermore, the unsampled history of the epidemic means that the nature and location of the genetically closest swine viruses reveal little about the immediate origin of the epidemic, despite the fact that we included a panel of closely related and previously unpublished swine influenza isolates. Our results highlight the need for systematic surveillance of influenza in swine, and provide evidence that the mixing of new genetic elements in swine can result in the emergence of viruses with pandemic potential in humans (CDC, 2009a).
Initial genetic characterization of the S-OIV outbreak by the United States Centers for Disease Control suggested swine as its probable source, on the basis of sequence similarity to previously reported swine influenza isolates (CDC, 2009a). Classical swine H1N1 viruses have circulated in pigs in North America and other regions for at least 80 years (CDC, 2009a). In 1998, a new triple- reassortant H3N2 virus—comprising genes from classical swine H1N1, North American avian, and human H3N2 (A/Sydney/5/97-like) influenza—was reported as the cause of outbreaks in North American swine, with subsequent establishment in pig populations (CDC, 2009a; Shortridge et al., 1977). Co-circulation and mixing of the triple-reassortant H3N2 with established swine lineages subsequently generated further H1N1 and H1N2 reassortant swine viruses (CDC, 2009a; Shortridge et al., 1977; Shope and Lewis, 1931) which have caused sporadic human infections in the United States since 2005 (Newman et al., 2008; Shinde et al., in press). Consequently, human infection with H1N1 swine influenza has been a nationally notifiable disease in the United States since 2007 (CDC, 2009a,b). In Europe, an avian H1N1 virus was introduced to pigs (‘avian-like’ swine H1N1) and first detected in Belgium in 1979 (Pensaert et al., 1981; CDC, 2009a). This lineage became established and gradually replaced classical swine H1N1 viruses, and also reassorted in pigs with human H3N2 viruses (A/PortChalmers/1/1973-like) (CDC, 2009a). It is noteworthy that, until now, there has been no evidence of Eurasian avian-like swine H1N1 circulating in North American pigs. In Asia, the classical swine influenza lineage circulates, in addition to other identified viruses, including human H3N2, Eurasian avian-like H1N1, and North American triple-reassortant H3N2 (Peiris et al., 2001; Jung and Song, 2007; CDC, 2009a; Shortridge, 1977).
Using comprehensive phylogenetic analyses, we have estimated a temporal reconstruction of the complex reassortment history of the S-OIV outbreak, summarized in Fig. A14-1 (Methods). Our analyses showed that each segment of the S-OIV genome was nested within a well-established swine influenza lineage (that is, a lineage circulating primarily in swine for >10 years before the current outbreak). The most parsimonious interpretation of these results is therefore that the progenitor of the S-OIV epidemic originated in pigs. Some transmission of swine influenza has, however, been observed in secondary hosts in North America, for example, in turkeys (CDC, 2009a). Although the precise evolutionary pathway of the genesis of S-OIV is greatly hindered by the lack of surveillance data (see later), we can conclude that the polymerase genes, plus HA, NP and NS, emerged from a triple-reassortant virus circulating in North American swine. The source triple-reassortant itself comprised genes derived from avian (PB2 and PA), human H3N2 (PB1) and classical swine (HA, NP and NS) lineages. In contrast, the NA and M gene segments have their origin in the Eurasian avian-like swine H1N1 lineage. Phylogenetic analyses from the early days of the outbreak, on the basis of the first publicly available sequences, quickly established this multiple genetic origin (Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, in
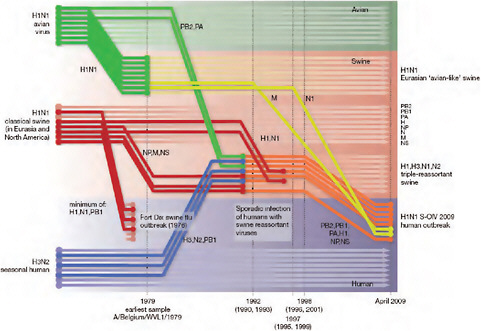
FIGURE A14-1 Reconstruction of the sequence of reassortment events leading up to the emergence of S-OIV. Shaded boxes represent host species; avian (green), swine (red) and human (grey). Coloured lines represent interspecies-transmission pathways of influenza genes. The eight genomic segments are represented as parallel lines in descending order of size. Dates marked with dashed vertical lines on ‘elbows’ indicate the mean time of divergence of the S-OIV genes from corresponding virus lineages. Reassortment events not involved with the emergence of human disease are omitted. Fort Dix refers to the last major outbreak of S-OIV in humans. The first triple-reassortant swine viruses were detected in 1998, but to improve clarity the origin of this lineage is placed earlier.
press; Trifonov et al., 2009; Garten et al., in press and http://influenza.bio.ed.ac.uk; CDC, 2009a; Shortridge et al., 1977).
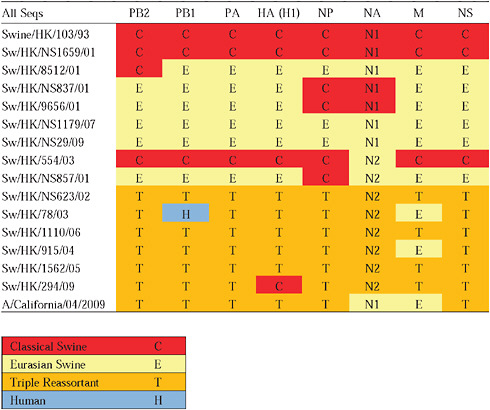
Given that S-OIV contains genes of Eurasian origin, we included in our phylogenetic analyses 15 newly sequenced swine influenza viruses from Hong Kong, sampled in the course of a surveillance program conducted since the early 1990s. The viruses were a mixture of seven H1N1 and eight H1N2 subtypes, and viruses belonging to the classical, Eurasian avian-like, and triple-reassortant swine lineages were all present. Both Eurasian and triple-reassortant strains were isolated in Hong Kong in 2009. Extensive reassortment among these three virus lineages was also observed from the Hong Kong surveillance data (Supplementary Table A14-3), with reassortment between Eurasian avian-like and triple-reassortant swine lineages occurring as early as 2003 (for example, Sw/HK/78/2003).
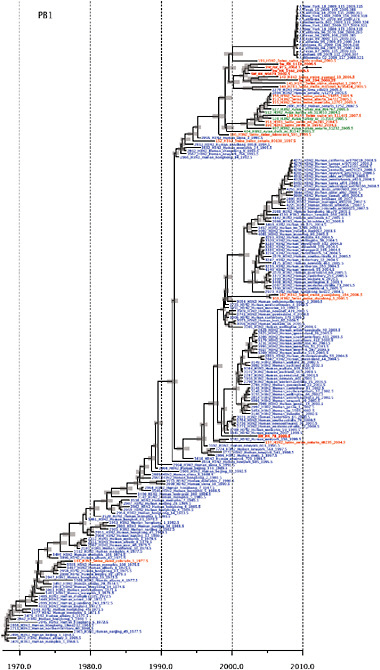
Notably, for the PB1,HA and M genes, some of these newly generated sequences are more similar to the S-OIV epidemic than any previously reported isolates (Supplementary Fig. A14-3). Notably, seven out of eight genomic segments found in a single 2004 isolate (Sw/HK/915/04 (H1N2)) were located in a sister lineage to the current outbreak. Not only does this suggest that the precursors of S-OIV were swine viruses, but also that they were geographically widely distributed. Crucially, however, the observation of a sister relationship between the current outbreak virus and Sw/HK/915/04 cannot be interpreted as evidence for a Eurasian origin of the outbreak, owing to the long branch of the phylogeny leading to the 2009 human strains (Fig. A14-2 and Table A14-1). This
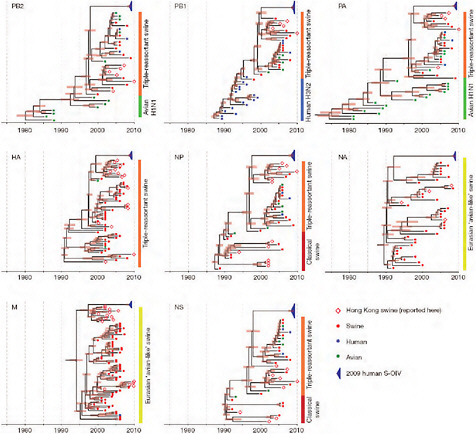
FIGURE A14-2 Genetic relationships and timing of S-OIV for each genomic segment. Symbols represent sampled viruses on a timescale of when they were sampled and coloured by host species (pigs, red; humans, blue; birds, green). Internal nodes are reconstructed common ancestors with 95% credible intervals on their date given by the red bars. The S-OIV outbreak strains are represented by a blue triangle, with the apex representing the common ancestor of these.
TABLE A14-1 Time of Most Recent Common Ancestors for the S-OIV Outbreak
|
Gene |
TMRCA of outbreak samples |
Duration of unsampled diversity (years) |
Mean evolutionary rate ×10−3 (subst. per site per year) |
|
HA |
28 Aug 2008 (1 Apr 2008, 2 Jan 2009) |
9.80 (8.41, 11.02) |
3.67 (3.41, 3.92) |
|
MP |
3 Aug 2008 (8 Dec 207, 5 Feb 2009) |
11.82 (10.17, 13.74) |
2.55 (2.19, 2.93) |
|
NA |
8 Aug 2008 (23 Feb 2008, 26 Dec 2008) |
17.15 (15.40, 18.88) |
3.65 (3.22, 4.12) |
|
NP |
27 Mar 2008 (15 Sep 2007, 19 Sep 2008) |
11.83 (10.53, 13.23) |
2.59 (2.34, 2.84) |
|
NS |
21 May 2008 (30 Sep 2007, 27 Nov 2008) |
11.47 (9.75, 13.21) |
2.62 (2.32, 2.92) |
|
PA |
7 Oct 2008 (1 Jun 2008, 1 Feb 2009) |
11.70 (10.25, 13.10) |
2.45 (2.20, 2.69) |
|
PB |
124 Oct 2008 (8 Jul 2008, 27 Jan 2009) |
9.24 (7.59, 10.48) |
2.34 (2.13, 2.53) |
|
PB |
29 Sep 2008 (12 Apr 2008, 9 Jan 2009) |
11.26 (9.93, 12.69) |
2.60 (2.29, 2.92) |
|
Genome* |
21 Jan 2009 (3 Aug 2008, 13 Mar 2009) |
N/A |
3.66 (0.61, 6.58) |
|
The values in parentheses represent the 95% credible intervals *This data set comprises complete or partial genomes of swine-origin influenza A (H1N1) virus outbreak isolates sampled predomin antly in the United States between March and May 2009. |
|||
branch must represent either an increased rate of evolution leading to the outbreak, or a long period during which the ancestors of the current epidemic went unsampled. To test these hypotheses, we regressed genetic divergence against sampling date for each gene, and found in favour of the latter: the evolutionary rate preceding the S-OIV epidemic is entirely typical for swine influenza (Supplementary Figs A14-4 and A14-5).
Therefore, to quantify the period of unsampled diversity, and to estimate the date of origin for the S-OIV outbreak, we performed a Bayesian molecular clock analysis for each gene (Methods). We also estimated the rate of evolution and time of the most recent common ancestor (TMRCA) of a set of genome sequences sampled from the S-OIV epidemic (between March and May 2009; isolates listed in Supplementary Table A14-5). We found that the common ancestor of the S-OIV outbreak and the closest related swine viruses existed between 9.2 and 17.2 years ago, depending on the genomic segment, hence the ancestors of the epidemic have been circulating undetected for about a decade. In contrast, the currently sampled S-OIV shared a common ancestor around January 2009 (no earlier than August 2008; Table A14-1). The long, unsampled history observed for every segment suggests that the reassortment of Eurasian and North American swine lineages may not have occurred recently, and it is possible that this single reassortant lineage has been cryptically circulating rather than two distinct lineages of swine flu. Thus, this genomic structure may have been circulating in pigs for several years before emergence in humans, and we urge caution in making inferences about human adaptation on the basis of the ancestry of the individual genes.
A search for amino acid residues in the S-OIV outbreak sequences that have been previously identified as phenotypic markers showed no evidence of v irulence-associated variation or adaptations to human hosts (CDC, 2009a; Shortridge et al., 1977; Shope and Lewis, 1931), consistent with the outbreak being of swine origin and causing relatively mild symptoms. Full molecular characterization of the human swine H1N1 viruses is provided in Supplementary Information.
We did detect a difference in the viral molecular evolution in the outbreak clade when compared to that observed in related swine influenza sequences: all S-OIV genes showed a comparatively higher non-synonymous to synonymous (dN/dS) substitution rate ratio (Supplementary Tables A14-2 and A14-3). This dN/dS ratio rise could be due to the increased detection of mildly deleterious mutations resulting from intensive epidemic surveillance; such mutations would more typically be eliminated and escape detection (CDC, 2009a). Alternatively, these mutations could be adaptations to the new host species.
Because this dN/dS ratio rise may affect our estimate of the TMRCA of the S-OIV outbreak strains (which was estimated using long-term rates of swine influenza evolution), we compared the mean dN/dS values of outbreak versus nonoutbreak data sets, thereby approximating the degree of excess of non- synonymous
mutations in the outbreak sequences (Methods). Once the dN/dS ratio rise is corrected for, the mean TMRCA of the S-OIV outbreak became 1 to 5 months more recent for each gene (Supplementary Tables A14-2 and A14-3). Furthermore, the adjusted TMRCA estimates are more uniform across genes, and are more similar to that obtained using internally calibrated S-OIV complete genomes (Table A14-1; a comparable estimate for the TMRCA of the HA gene only was recently reported [CDC, 2009a]). Irrespective of whether the dN/dS ratio rise is due to increased detection of deleterious mutations or to increased adaptive evolution, its presence may be a general feature of intensively sampled emerging epidemics, and should be accounted for in the evolutionary analysis of such events.
Movement of live pigs between Eurasia and North America seems to have facilitated the mixing of diverse swine influenza viruses, leading to the multiple reassortment events associated with the genesis of the S-OIV strain. Domestic pigs have been described as a hypothetical ‘mixing-vessel’, mediating by reassortment the emergence of new influenza viruses with avian or avian-like genes into the human population, and triggering a pandemic associated with antigenic shift (Shortridge et al., 1977). Previous research has suggested that occupational exposure to pigs increases the risk of swine influenza virus infection, and that swine workers should be considered in any surveillance programs (Shortridge et al., 1977).
The emergence of S-OIV provides further evidence of the role of domestic pigs in the ecosystem of influenza A. As reported recently, all three pandemics of the twentieth century seem to have been generated by a series of multiple reassort ment events in swine or humans, and to have emerged over a period of years before pandemic recognition (Shortridge et al., 1977). Our results show that the genesis of the S-OIV epidemic followed a similar evolutionary pathway: H1N1 viruses with human pandemic potential had been identified, transmission from swine to humans was known (Webby et al., 2000) and the disease had been made notifiable. Yet despite widespread influenza surveillance in humans, the lack of systematic swine surveillance allowed for the undetected persistence and evolution of this potentially pandemic strain for many years.
Methods Summary
We compared 15 newly sequenced Hong Kong swine influenza genomes and two genomes from the S-OIV outbreak with 796 genomes representing the spectrum of influenza A diversity (comprising 285 human, 100 swine and 411 avian isolates). Phylogenetic trees were constructed for each genomic segment independently (Supplementary Fig. A14-3). Next, for each genomic segment, viruses with known isolation dates that were genetically similar to the current outbreak were identified, and more detailed analysis using a Bayesian ‘relaxed molecular clock’ approach was performed (Drummond and Rambaut, 2007), thereby estimating rates of viral evolution and dates of divergence (Fig. A14-3). Finally, a similar
Bayesian molecular clock approach was applied to the 30 individual viruses isolated from the human outbreak since the end of March 2009 (Supplementary Table A14-5 and Supplementary Fig. A14-4). This analysis was performed assuming a model of exponential growth in the number of infections.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Received 24 May; accepted 4 June 2009.
Published online 11 June 2009.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Acknowledgements
We thank E. C. Holmes for comments and encouragement. We acknowledge support from The Royal Society of London (A. R. and O. G. P.), the National Institute of Allergy and Infectious Diseases (NIAID) (G. J. D. S. and M. W.), the Biotechnology and Biological Sciences Research Council (BBSRC) (S. J. L.), and the David and Lucile Packard Foundation (M. W.). A.R. works as a part of the Interdisciplinary Centre for Human and Avian Influenza Research (ICHAIR). This study was supported by the National Institutes of Health (NIAID contract HHSN266200700005C) and the Area of Excellence Scheme of the University Grants Committee (grant AoE/M-12/06) of the Hong Kong SAR Government.
Author Contributions
J. B., S. J. L., O. G. P., A. R., G. J. D. S., D. V. and M. W. conceived the study, performed analyses, co-wrote the paper, and all contributed equally to this work. J. S. M. P. co-wrote the paper, Y. G. conceived the study and co-wrote the paper, S. B. and J. R. performed analyses, S. K. M. conducted surveillance, and C. L. C. conducted sequencing. All authors commented on and edited the paper.
Author Information
Newly reported sequences have been deposited at GenBank under accession numbers GQ229259–GQ229378. Reprints and permissions information is available at www.nature.com/reprints. This paper is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence, and is freely available to all readers at www.nature.com/nature. Correspondence and requests for materials should be addressed to A. R. (a.rambaut@ed.ac.uk) or Y. G. (yguan@hku.hk).
Methods
Sequence Selection for Phylogenetic Analysis
We downloaded 3,986 complete influenza genomes of any subtype and sampling year (2,490 human, 185 swine and 1,311 avian) from the NCBI Influenza Virus Resource (Bao et al., 2008) on 29 April 2009. Each sequence set was given a unique ID of the form (ID number)_(Subtype)_(Host)_(isolate name), in which the isolate name is in lower case.
To reduce the number of very similar sequences, we listed all isolates in which the coding region in segment 1 (PB2) was at least one nucleotide different from the others. This left 1,759 human, 166 swine and 1,117 avian complete genome sets. Next we sampled the human, swine and avian sets, selecting one genome set per specific host (as defined in the isolate name, for example, chicken, duck), per specific location (for example, state or province), per year (although isolate name synonyms, for example, duck = dk, hongkong = hk were not accounted for). Two avian and four swine sequence sets were removed owing to bad sequences in one or more segments (for example, frameshifts), leaving 286 human (including S-OIVs), 100 swine and 411 avian sequences in the sampled subset. A further outbreak sequence set (A/Canada-ON/RV1527/2009), and the 15 new swine sequence sets were also added, making a total of 813 complete genome sets for analysis. For the more detailed, temporal analyses, all available S-OIV sequences were used.
The nucleotides in the coding regions of segments 1 (PB2), 2 (PB1), 3 (PA) and 5 (NP) were aligned using ClustalW (Thompson et al., 1994) followed by manual alignment to codon position. The full nucleotide sequences of segments 7 (M1 and M2) and 8 (NS1 and NS2) were also aligned using ClustalW, and the sequences were edited such that all of the codons in first open reading frame (ORF) were followed by the remaining codons in the second ORF (that is, nucleotides were not repeated between the two ORFs). The HA and NA genes (segments 4 and 6) were aligned to codon positions using Muscle (Edgar, 2004). Further H1, H3, N1 and N2 only alignments were also performed.
New Swine Influenza Sequences from Hong Kong
To evaluate the evolutionary history of swine/human influenza A H1N1 viruses, 15 viruses isolated from swine in Hong Kong during 1993 to 2009 were sequenced. Viral RNA was directly extracted from infected allantoic fluid or cell culture using QIAamp viral RNA minikit (Qiagen, Inc.). Complementary DNA was synthesized by reverse transcription reaction, and gene amplification by PCR was performed using specific primers for each gene segment. PCR products were purified with the QIAquick PCR purification kit (Qiagen Inc.) and sequenced by synthetic oligonucleotides. Reactions were performed using Big Dye- Terminator
v3.1 Cycle Sequencing Reaction Kit on an ABI PRISM 3730 DNA Analyser (Applied Biosystems) following the manufacturer’s instructions. All sequences were assembled and edited with Lasergene version 8.0 (DNASTAR). Full genome sequences of these viruses are available for download at GenBank under accession numbers GQ229259–GQ229378.
Molecular Evolution and Adaptation
We used the programs SLAC (Single-Likelihood Ancestor Counting) (Kosakovsky Pond and Frost, 2005) and SNAP (Synonymous Non- synonymous Analysis Program) (Korber, 2000) to compare the mean ratio of non- synonymous changes per non-synonymous site to synonymous changes per synonymous site (dN/dS) of outbreak versus non-outbreak sequences. SLAC calculates inferred ancestral sequences for each internal node in a phylogeny using a codon model (and disallowing stop codons), and then counts the synonymous and nonsynonymous mutations by comparing each codon to its immediate ancestor. SNAP counts the possible synonymous and non-synonymous codon changes across all pairs of sequences.
In brief, we calculated the effect of the excess of non-synonymous changes in the outbreak data as follows. Assume that S is the number of synonymous sites in a data set, N is the number of non-synonymous sites (typically ~3.5S for these data), and ω is the dN/dS ratio. If the proportional contribution to the overall rate from synonymous sites is s, then the proportional contribution to the overall rate from non-synonymous sites is equal to (N/S)(ω)s. N, S and ω are all readily estimated from the data. Assuming the same rate of synonymous substitution in both the outbreak and reference data sets, the relative rate expected in the outbreak sequences compared to the reference sequences is thus equal to
Phylogenetic Analyses
Phylogenetic trees were inferred using the neighbour joining distance method, with genetic distances calculated by maximum likelihood under the Hasegawa–Kishino–Yano (HKY) model with gamma-distributed rates among sites (HKY+Γ). Parameters of this model were estimated using maximum likelihood on an initial tree. Temporal phylogenies and rates of evolution were inferred using a relaxed molecular clock model that allows rates to vary among lineages within a Bayesian Markov chain Monte Carlo (MCMC) framework (Drummond and Rambaut, 2007).This was used to sample phylogenies and the dates of divergences between viruses from their joint posterior distribution, in which the sequences are constrained by their known date of sampling. A model comprising a codon-position specific HKY+ Γ substitution model was used. The
limited sampling timespan of the S-OIV samples required a simpler model to avoid over-parameterization, so a single HKY+Γ model over all sites was used. For the analyses using Bayesian MCMC sampling, in all cases chain lengths of at least 50 million steps were used with a 10% ‘burn-in’ removed. Furthermore, at least two independent runs of each were performed and compared to ensure adequate sampling.
References
Bao, Y. et al. The influenza virus resource at the national center for biotechnology information. J. Virol. 82, 596–601 (2008).
Brown, I. H. The epidemiology and evolution of influenza viruses in pigs. Vet. Microbiol. 74, 29–46 (2000).
Brown, I. H., Harris, P. A., McCauley, J. W. & Alexander, D. J. Multiple genetic reassortment of avian and human influenza A viruses in European pigs, resulting in the emergence of an H1N2 virus of novel genotype. J. Gen. Virol. 79, 2947–2955 (1998).
Centers for Disease Control and Prevention. Swine influenza A (H1N1) infection in two children—Southern California, March–April 2009. Morb. Mortal. Wkly Rep. 58, 400–402 (2009).
Centers for Disease Control and Prevention. Novel influenza A virus infections—2007 case definition. <http://www.cdc.gov/ncphi/disss/nndss/casedef/novel_influenzaA.htm. (24 May 2009).
Choi, Y. K. et al. H3N2 influenza virus transmission from swine to turkeys, United States. Emerg. Infect. Dis. 10, 2156–2160 (2004).
Drummond, A. J. & Rambaut. A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214(2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Fraser, C. et al. Pandemic potential of a strain of influenza A (H1N1): Early findings. Science doi:10.1126/science.1176062 (in the press).
Garten, R. J. et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) nfluenza viruses circulating in humans. Science doi:10.1126/science.1176225 (in the press).
Hatta, M., Gao, P., Halfmann, P.& Kawaoka, Y. Molecular basis of high virulence of Hong Kong H5N1 influenza A viruses. Science 7, 1840–1842 (2001).
Jung, K. & Song, D. S. Evidence of the cocirculation of influenza H1N1, H1N2 and H3N2 viruses in the pig population of Korea. Vet. Rec. 161, 104–105 (2007).
Korber, B. HIV Signature and Sequence Variation Analysis. Computational Analysis of HIV Molecular Sequences (eds Rodrigo, A. G. & Learn, G. H.) Ch. 4, 55–72 (Kluwer Academic Publishers, 2000).
Kosakovsky Pond, S. L. & Frost, S. D. W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (2005).
Le, Q. M., Sakai-Tagawa, Y., Ozawa, M., Ito, M. & Kawaoka, Y. Selection of H5N1 influenza virus PB2 during replication in humans. J. Virol. 83, 5278–5281 (2009).
Myers, K. P. et al. Are swine workers in the United States at increased risk of infection with zoonotic influenza virus? Clin. Infect. Dis. 42, 14–20 (2006).
Newman, A. P. et al. Human case of swine influenza A (H1N1) triple reassortant virus infection, Wisconsin. Emerg. Infect. Dis. 14, 1470–1472 (2008).
Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. doi:10.1056/NEJMoa0903810 (in the press).
Obenauer, J. C. et al. Large-scale sequence analysis of avian influenza isolates. Science 311, 1576–1580 (2006).
Peiris, J. S. M. et al. Cocirculation of avian H9N2 and contemporary “human” H3N2 influenza A viruses in pigs in southeastern China: potential for genetic reassortment? J. Virol. 75, 9679–9686 (2001).
Pensaert, M., Ottis, K., Vanderputte, J., Kaplan, M. M. & Buchmann, P. A. Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential for man. Bull. World Health Organ. 59, 75–78 (1981).
Pybus, O. G. et al. Phylogenetic estimation of deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 24, 845–852 (2007).
Shinde, V. et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. doi:10.1056/NEJMoa0903812 (in the press).
Shope, R. E. & Lewis, P. Swine influenza: experimental transmission and pathology. J. Exp. Med. 54, 349–359 (1931).
Shortridge, K. F., Webster, R. G., Butterfield, W. K. & Campbell, C. H. Persistence of Hong Kong influenza virus variants in pigs. Science 196, 1454–1455 (1977).
Smith, G. J. D. et al. Dating the emergence of pandemic influenza viruses. Proc. Natl Acad. Sci. USA. (in the press).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Trifonov, V., Khiabanian, H., Greenbaum, B. & Rabadan, R. The origin of the recent swine influenza A (H1N1) virus infecting humans. Euro Surveill. 14, 19193 (2009).
Webby, R. J. et al. Evolution of swine H3N2 influenza viruses in the United States. J. Virol. 74, 8243–8251 (2000).
Supplementary Information
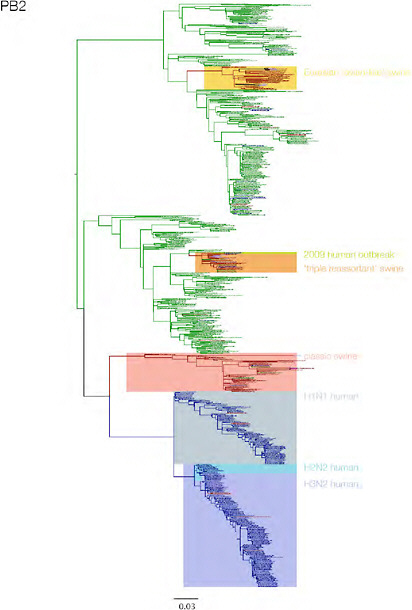
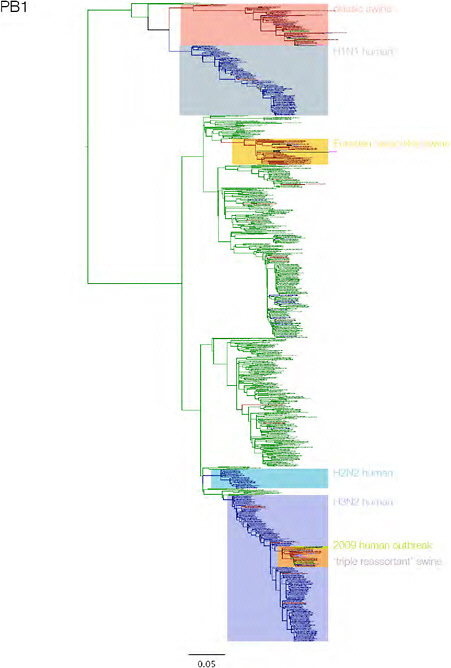
SUPPLEMENTARY FIGURE A14-3 Phylogenetic relationships of each gene segment (PB2, PB1, PA, HA, NP, NA, M & NS) of swine influenza A viruses indicating genetic components of the swine-origin influenza A (H1N1) virus. Clade labels indicate major swine lineages. Human viruses are coloured blue, swine viruses in red and avian viruses in green.
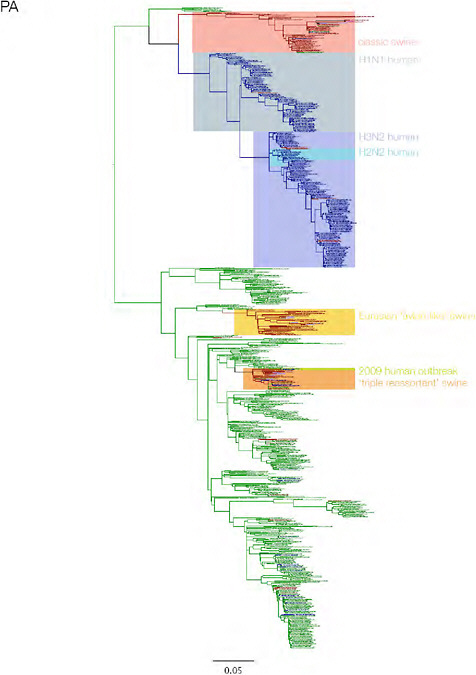
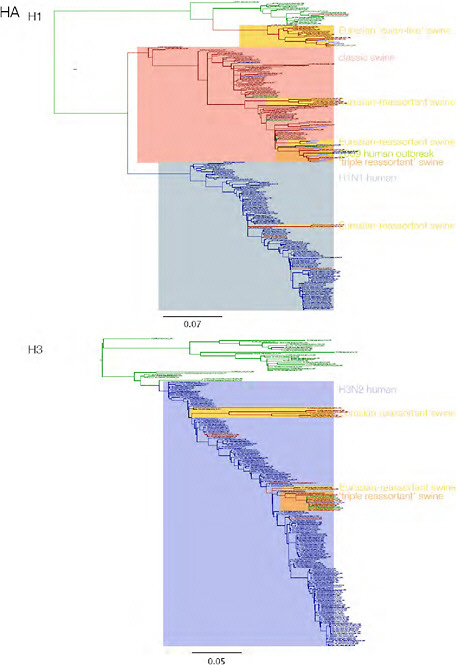
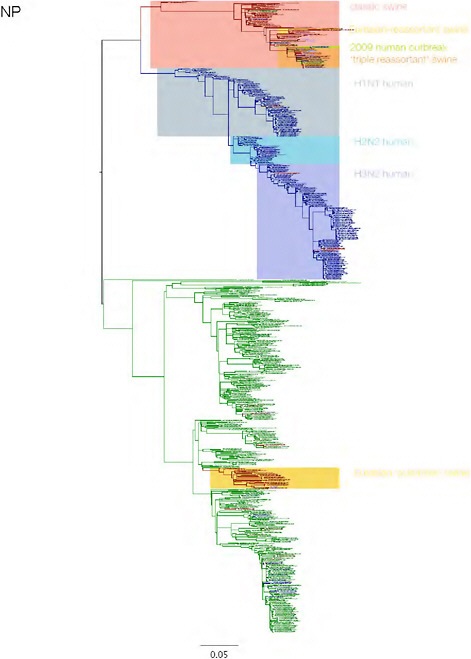
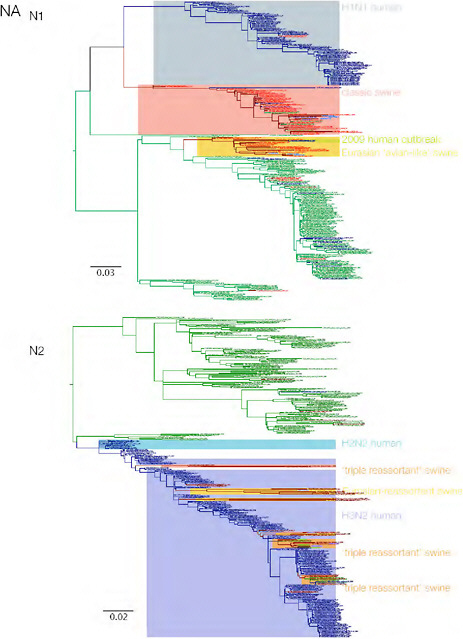
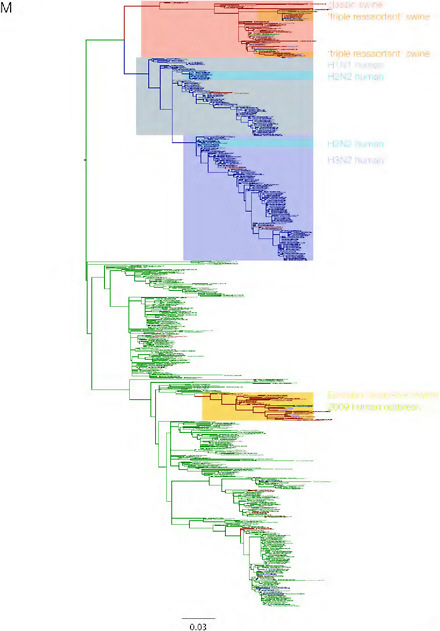
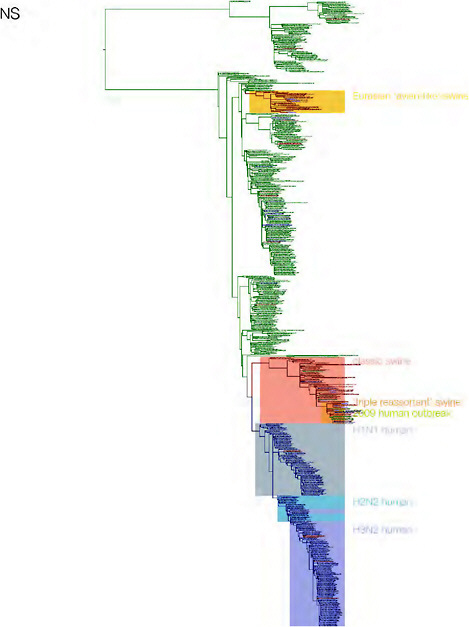
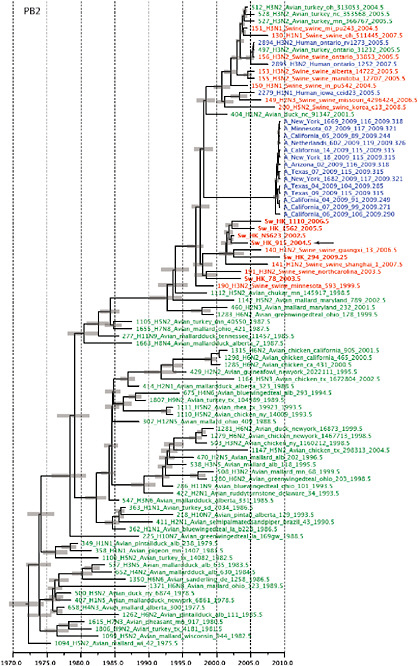
SUPPLEMENTARY FIGURE A14-4 Phylogenetic relationships scaled to time for each gene segment (PB2, PB1, PA, HA, NP, NA, M & NS) of the swine-origin influenza A (H1N1) virus as represented in Figure A14-2 of the main text but with full virus names and GenBank accession numbers. Internal nodes are reconstructed common ancestors with 95% credible intervals on their date given by the grey bars. Human viruses are coloured blue, swine viruses in red and avian viruses in green. Newly described Hong Kong sequences have bold labels. Sw/HK/915/04 (H1N2) is highlighted with an arrow when present (see main text). Timeline is in years.
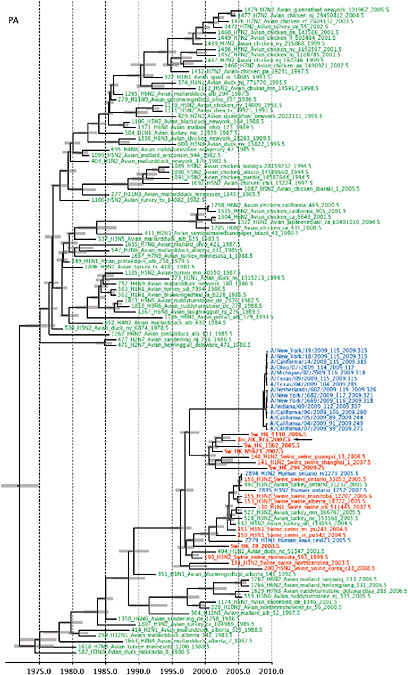
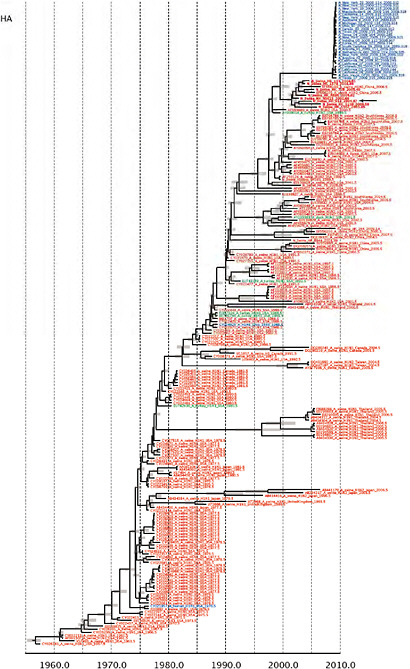
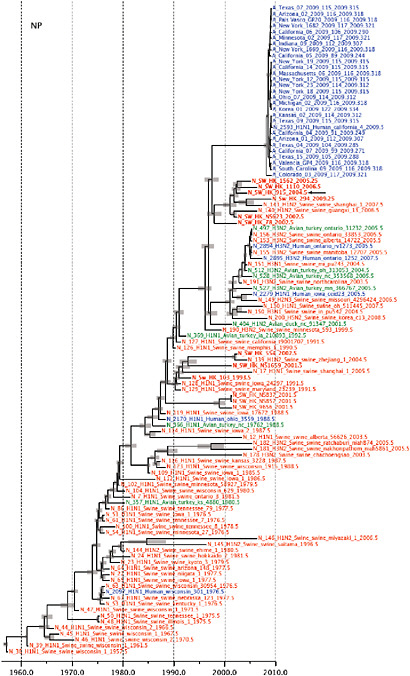
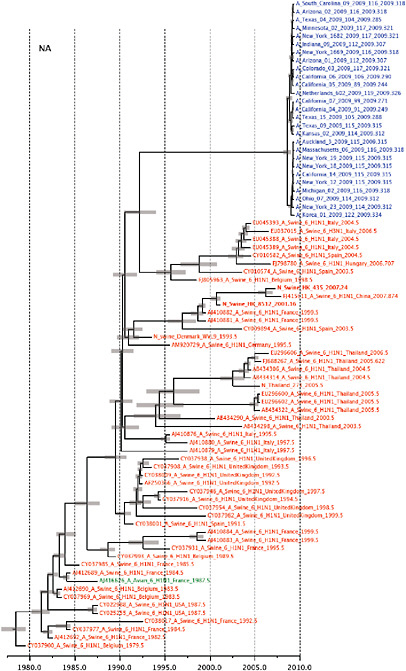

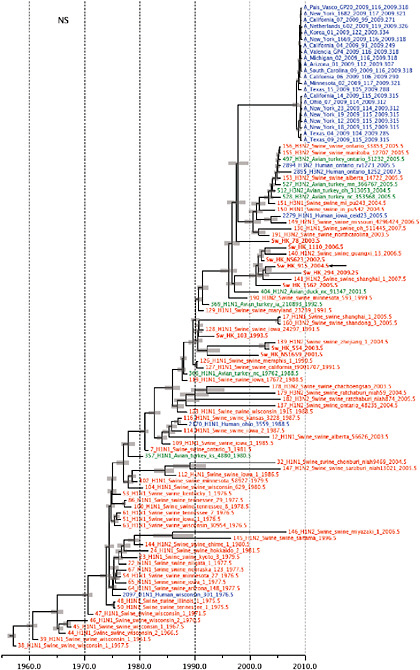
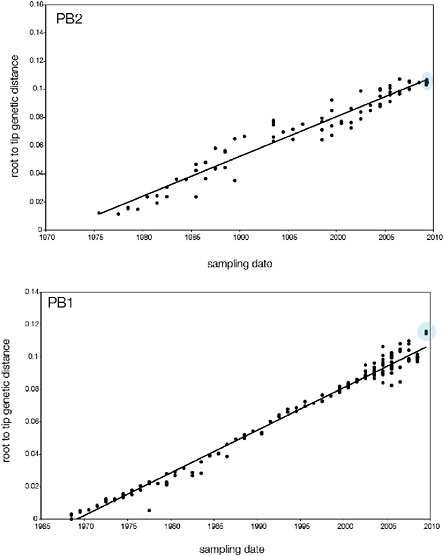
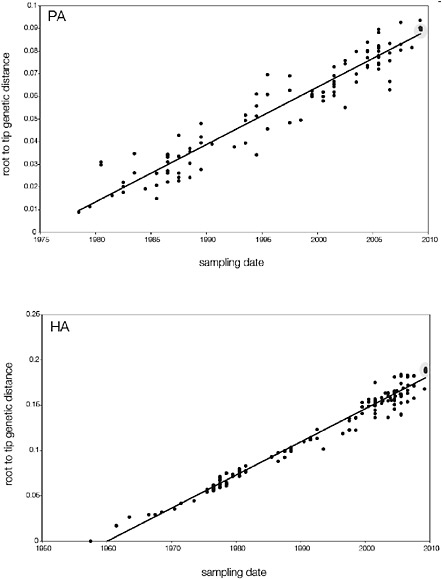
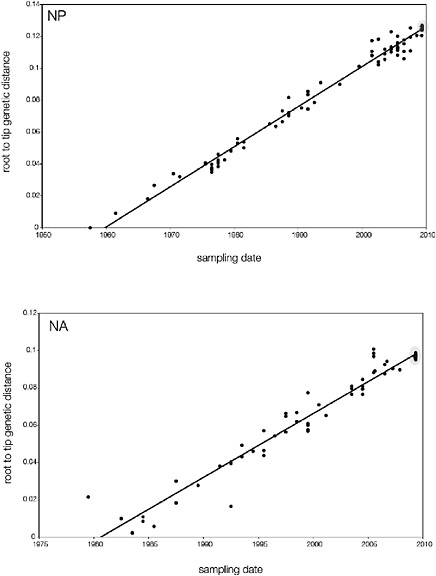
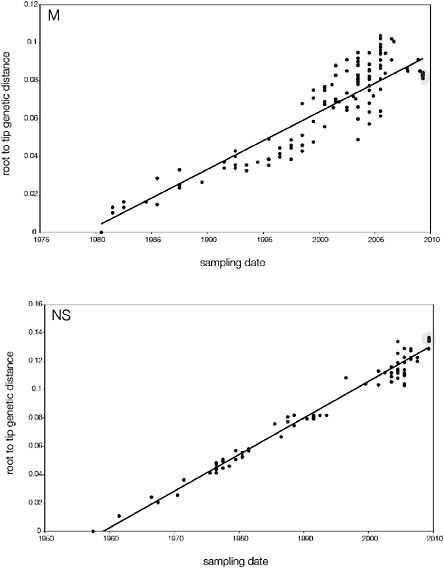
SUPPLEMENTARY FIGURE A14-5 For each gene segment (PB2, PB1, PA, HA, NP, NA, M & NS), we plot the isolation date of each influenza sequence against the genetic distance from that sequence to the root of the phylogeny. The linear regression gradient is therefore an estimate of the rate of sequence evolution and the x-intercept is an estimate of the TMRCA of the whole phylogeny. Phylogenies were estimated using neighbour-joining, with rooting chosen to maximise the regression fit. The chosen root was typically very close to the earliest sampled sequence. Residual analysis was performed to identify and remove significant outliers, which most likely result from isolation data annotation errors in the sequence database. For each gene, the degree of scatter about the linear regression reflects evolutionary rate heterogeneity among lineages, such that a “strict clock” corresponds to all the points falling exactly on the regression line. The 2009 outbreak sequences (highlighted in light blue) are entirely typically of the long termtrends in divergence, hence there is no evidence that the branch leading to the outbreak has evolved unusually rapidly or slowly. For further discussion of this methodology, see Drummond AJ, Pybus OG, Rambaut A. 2003. Inference of viral evolutionary rates from molecular sequences. Advances in Parasitology 54:331-358.
SUPPLEMENTARY TABLE A14-2 SLAC Results
|
gene |
outbreak dN/dSa |
reference dN/dS |
estimated relative rateb |
mean TMRCA |
mean TMRCA (adjusted) |
|
HA |
0.32 |
0.21 |
1.22 |
28-Aug-2008 |
11-Oct-2008 |
|
NA |
0.26 |
0.18 |
1.16 |
8-Aug-2008 |
12-Sep-2008 |
|
MP |
0.19 |
0.05 |
1.43 |
3-Aug-2008 |
26-Oct-2008 |
|
NP |
0.18 |
0.05 |
1.39 |
27-Mar-2008 |
19-Jul-2008 |
|
NS |
2.15 |
0.23 |
3.85 |
21-May-2008 |
6-Feb-2009 |
|
PA |
0.11 |
0.06 |
1.15 |
7-Oct-2008 |
6-Nov-2008 |
|
PB1 |
0.212 |
0.06 |
1.45 |
24-Oct-2008 |
22-Dec-2008 |
|
PB2 |
0.15 |
0.11 |
1.11 |
9-Sep-2008 |
4-Oct-2008 |
|
aCalculated using SLAC (S. Kosakovsky Pond, available at http://datamonkey.org) bSubstitution rate in outbreak clade relative to non-outbreak sequences based on excess nonsynonymous mutations inferred from the higher dN/dS ratio. |
|||||
SUPPLEMENTARY TABLE A14-3 SNAP Results
|
gene |
outbreak dN/dSa |
reference dN/dS |
estimated relative rateb |
mean TMRCA |
mean TMRCA (adjusted) |
|
HA |
0.24 |
0.09 |
1.41 |
28-Aug-2008 |
9-Nov-2008 |
|
NA |
0.32 |
0.1 |
1.59 |
8-Aug-2008 |
17-Nov-2008 |
|
MP |
0.3 |
0.03 |
1.82 |
3-Aug-2008 |
5-Dec-2008 |
|
NP |
0.14 |
0.02 |
1.39 |
27-Mar-2008 |
19-Jul-2008 |
|
NS |
0.31 |
0.13 |
1.43 |
21-May-2008 |
5-Sep-2008 |
|
PA |
0.18 |
0.05 |
1.43 |
7-Oct-2008 |
10-Dec-2008 |
|
PB1 |
0.16 |
0.03 |
1.45 |
24-Oct-2008 |
22-Dec-2008 |
|
PB2 |
0.13 |
0.07 |
1.15 |
9-Sep-2008 |
10-Oct-2008 |
|
aCalculated using SNAP (B. Korber, available at http://www.hiv.lanl.gov/content/sequemce/SNAP/SNAP.htm) bSubstitution rate in outbreak clade relative to non-outbreak sequences based on excess nonsynonymous mutations inferred from the higher dN/dS ratio. |
|||||
SUPPLEMENTARY TABLE A14-5 S-OIV Sequences Available on NCBI Influenza Virus Database at the Time of Analysis
|
|
Days1 |
PB22 |
PB1 |
PA |
HA |
NP |
NA |
MP |
NS |
|
A/Arizona/01/2009 |
112 |
|
|
|
GQ117067 |
GQ117063 |
GQ117064 |
GQ117066 |
GQ117065 |
|
A/Arizona/02/2009 |
116 |
GQ117076 |
GQ117075 |
|
GQ117079 |
GQ117074 |
GQ117077 |
GQ117078 |
|
|
A/Auckland/3/2009 |
115 |
|
|
|
|
FJ973552 |
FJ973553 |
|
|
|
A/California/04/2009 |
91 |
FJ966079 |
FJ966080 |
FJ966081 |
FJ966082 |
FJ966083 |
FJ966084 |
FJ966085 |
FJ966086 |
|
A/California/05/2009 |
89 |
FJ966955 |
FJ966952 |
FJ966957 |
FJ966952 |
FJ966953 |
FJ966956 |
FJ966954 |
|
|
A/California/06/2009 |
106 |
FJ966963 |
FJ966950 |
FJ966964 |
FJ966950 |
FJ966961 |
FJ971075 |
FJ966962 |
FJ971074 |
|
A/California/07/2009 |
99 |
FJ984387 |
FJ969531 |
FJ969529 |
FJ981613 |
|
FJ984386 |
FJ966975 |
|
|
A/California/14/2009 |
115 |
GQ117035 |
GQ117037 |
GQ117037 |
GQ117040 |
GQ117033 |
GQ117036 |
GQ117039 |
GQ117038 |
|
A/Colorado/03/2009 |
117 |
|
|
|
GQ117119 |
GQ117117 |
GQ117118 |
|
|
|
A/Indiana/09/2009 |
112 |
|
GQ117093 |
GQ117095 |
GQ117097 |
GQ117092 |
GQ117094 |
GQ117096 |
|
|
A/Kamsas/02/2009 |
114 |
|
|
|
GQ117059 |
GQ117057 |
Gq117058 |
|
|
|
A/Korea/01/2009 |
122 |
|
|
|
GQ131023 |
GQ131024 |
GQ132185 |
GQ131025 |
GQ131026 |
|
A/Massachusetts/06/2009 |
116 |
|
|
|
GQ117043 |
GQ117041 |
GQ117042 |
|
|
|
A/Michigan/02/2009 |
116 |
|
|
GQ117109 |
GQ117112 |
GQ117107 |
GQ117108 |
GQ117111 |
GQ117110 |
|
A/Minnesota/02/2009 |
117 |
GQ117070 |
GQ117069 |
|
|
GQ117068 |
GQ117071 |
GQ117073 |
GQ117072 |
|
A/Netherlands/602/2009 |
119 |
|
|
|
CY039527 |
|
CY039528 |
|
|
|
A/New York/1669/2009 |
116 |
CY039900 |
CY039899 |
CY039898 |
CY039893 |
CY039896 |
CY039895 |
CY039894 |
CY039897 |
|
A/New York/1682/2009 |
117 |
CY039908 |
CY039907 |
CY039906 |
CY039901 |
CY039904 |
CY039903 |
CY039902 |
CY039905 |
|
A/New York/12/2009 |
115 |
|
|
|
FJ984337 |
FJ984336 |
FJ984335 |
|
FJ984334 |
|
A/New York/18/2009 |
115 |
FJ984351 |
FJ984353 |
FJ984354 |
FJ984355 |
FJ984352 |
FJ984350 |
FJ984348 |
FJ984349 |
|
A/New York/19/2009 |
115 |
|
FJ984392 |
FJ984393 |
FJ984394 |
FJ984391 |
FJ984390 |
FJ984388 |
FJ984389 |
|
A/New York/23/2009 |
114 |
|
|
|
FJ984364 |
FJ984363 |
FJ984362 |
|
FJ984361 |
|
A/Ohio/07/2009 |
114 |
|
|
FJ984401 |
GQ117100 |
GQ117098 |
GQ117099 |
|
|
|
A/Paos Vasc0/GP20/2009 |
116 |
|
|
|
FJ985763 |
FJ985761 |
FJ985764 |
FJ985760 |
FJ985762 |
Supplementary Notes
Adaptation and Purifying Selection
We suggest in the main text that the increased presence of amino-acid polymorphisms may be present in the outbreak sequences relative to the swine influenza reference sequences due to the rapid growth in number of infections and the small window of time over which they were sampled. The fairly uniform distribution of nonsynonymous changes across the outbreak genomes (data not shown) and the relatively consistent dN/dS ratios across genes (Supplementary Tables A14-2 & A14-3) support this view. We predict that many of these mutations will be mildly deleterious and, given time, will be removed by purifying selection along an emergent ‘trunk’ lineage, and that the estimated evolutionary rate and gene-specific dN/dS pattern will approach that observed in the swine influenza data sets sampled over a time period of decades (see below). An interesting alternative is that the elevated rate in the outbreak sequences (and higher dN/dS ratio) is due to a burst of adaptive evolution in a new host, rather than a relaxation of purifying selection and that many of the ‘extra’ nonsynonymous mutations will become fixed in the population. If the current outbreak persists in the human population, it will be possible to distinguish these hypotheses.
Detailed Molecular Characterization
The presence of residue Asn 31 in the M2 protein invariably confers resistance to the adamantanes, a group of antiviral drugs used for treatment of human influenza (Scholtissek et al., 1998). Sequence analysis revealed that Asn 31 was present in all human isolates of the current outbreak, this mutation was also present in swine H1N1 and H1N2 viruses that were most closely related in the M gene. The widespread occurrence of the Asn 31 mutation in closely related viruses indicate that it may have descended from the swine lineage of amantadine-resistant M2 genes rather than independently acquired. It should be noted that majority of the seasonal H1N1 and H3N2 viruses are resistant to amantadine, except Brisbane-like H1N1 viruses (Barr et al., 2008). No mutations were observed in the NA gene that confers Oseltamivir resistance.
The molecular determinants of interspecies transmission of the A/California /04/2009-like virus to humans are unclear. In these viruses the amino acid residue at the receptor binding pocket of HA1– position Gln 226 and Gly 228 retain configurations (2,3-NeuAcGal linkages) predicted to have affinity for mammalian cell-surface receptors (Wiley et al., 1981; Rogers et al., 1983). Amino acid residues relevant to receptor binding were identical to those of classical swine H1N1 and North American H1N1 and recent H1N1 viruses in both the 130-loop and 190-helix. However, the amino acids at positions 133 and 135 are different from the current seasonal vaccine strain A/Brisbane/59/2007(H1N1)
used in both the northern and southern hemispheres, indicating that these viruses may show less cross-reaction to the current vaccine strain (Winter et al., 2009; Canton et al., 1982).
Mutations Glu627Lys and Asp701Asn of the PB2 gene that are thought to be associated with adaptation to mammals and increased virulence of influenza viruses in mice were not present (Hatta et al., 2001; Gabriel et al., 2008; Le et al., 2009; Steel et al., 2009; Obenauer et al., 2006; Jackson et al., 2008). The C terminal of the NS1 gene is truncated in the A/California/04/2009-like viruses and four residues of the PDZ ligand domain are not present (Obenauer et al., 2006).
Supplementary Methods
Viral RNA was directly extracted from infected allantoic fluid or cell culture using QIAamp viral RNA minikit (Qiagen, Inc., Valencia, Calif.). cDNA were synthesized by reverse transcription reaction and gene amplification by PCR were performed using specific primers for each gene segments. PCR products were purified with the QIAquick PCR purification kit (Qiagen Inc.) and sequenced by synthetic oligonucleotides. Reactions were performed using Big Dye- Terminator v3.1 Cycle Sequencing Reaction Kit on an ABI PRISM 3700 DNA Analyzer (Applied Biosystems) following the manufacturer’s instructions. All sequences were assembled and edited with Lasergene version 6.1 (DNASTAR, Madison, WI). Full genome sequences of these viruses are available for download at GISAID under the accession numbers (EPI177540 to EPI77658 and EPI177947).
References
Barr, I. G., Deng, Y. M., Iannello, P., Hurt, A. C. & Komadina, N. Adamantane resistance in influenza A (H1) viruses increased in 2007 in South East Asia but decreased in Australia and some other countries. Antiviral Research 80, 200–205 (2008).
Canton, A., Brownlee, G. G., Yewdell, J. W. & Gerhard, W. The antigenic structure of the influenza virus A/PR/*/34 hemagglutinin (H1 subtype). Cell 31, 417–27 (1982).
Gabriel, G. I., Herwig, A. & Klenk, H.–D. Interaction of polymerase subunit PB2 and NP with importin a1 is a determinant of host range of influenza A virus. PLoS Pathog. 4, e11 (2008).
Hatta, M., Gai, P., Halfmann, P. & Kawaoka, Y. Molecular basis of high virulence of Hong Kong H5N1 influenza A viruses. Science 7, 1840–1842 (2001)
Le, Q. M., Sakai-Tagawa, Y,, Ozawa, M., Ito, M. & Kawaoka, Y. Selection of H5N1 Influenza Virus PB2 during Replication in Humans. J. Virol. 83, 5278–5281 (2009).
Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA (2008) A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 105, 4381–4386 (2008).
Obenauer, J. C. et al. Large-Scale Sequence Analysis of Avian Influenza Isolates. Science 311, 1576–1580 (2006).
Rogers, G. N. et al. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304, 76–78 (1983).
Scholtissek, C., Quack, G., Klenk, H. D. & Webster, R. G. How to overcome resistance of influenza A viruses against adamantane derivatives. Antiviral Res. 37, 83-95 (1998).
Steel, J., Lowen, A. C., Mubareka. S. & Palese, P. Transmission of Influenza Virus in a Mammalian Host Is Increased by PB2 Amino Acids 627K or 627E/701N. PLoS Pathog. 5, e1000252 (2009).
Wiley, D. C., Wilson, I. A. & Skehel, J. J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Winter, G., Fields, S. & Brownlee G. G. Nucleotide sequence of the haemagglutinin gene of a human influenza virus H1 subtype. Nature 292, 72–75.







































