
5
Strengthening Evidence-Based Regulation
CHAPTER RECOMMENDATIONS
The previous chapters introduced the committee’s proposed biomarker evaluation framework and tested it using diverse case studies. While the committee’s evaluation framework will provide a more complete review of the evidence supporting different contexts of biomarker use, the effective implementation of the framework may require additional actions by the Food and Drug Administration (FDA) and other stakeholders. First, the committee concluded that the FDA’s current regulatory authority is not sufficient. Inadequate fulfillment of postmarketing studies and incomplete understanding of how consumers interpret food and dietary supplement claims prevent robust protection of public health. Therefore, the committee recommended that:
Recommendation 5:
|
5a. |
Congress should strengthen the FDA’s authority to request and enforce postmarket surveillance across drugs, devices, and biologics when approvals are initially based on putative surrogate endpoint data. |
|
5b. |
Congress should grant the FDA authority to request studies and sufficient authority to act on the results of studies on consumer understanding of claims on foods and supplements. |
To support this recommendation, the first section of this chapter summarizes the FDA’s role and outlines the FDA’s regulatory authority and
describes some of the limitations related to the FDA’s current regulatory capacities. Recommendation 5 has two parts due to the differing regulatory frameworks surrounding drugs, devices, and biologics as compared to foods and supplements. Its intent is parallel, nonetheless.
In addition to strengthened FDA regulatory capacity, the committee acknowledged that science-based decision making is reliant on the availability of scientific data. Although there are ongoing efforts to collect and analyze biomarker data, the committee concluded that these efforts are uneven and not optimally organized within the U.S. Department of Health and Human Services (HHS). Recognizing the value of a well-coordinated, comprehensive effort to collect and share biomarker information in advancing public health, the committee sought to improve ongoing biomarker data collection efforts. Improved FDA information infrastructure and surveillance systems may also enhance the agency’s ability to interpret biomarkers and their relation to public health. Based on these findings, the committee made the following recommendation:
Recommendation 6:
|
6a. |
The U.S. Department of Health and Human Services should facilitate a coordinated, department-wide effort to encourage the collection and sharing of data about biomarkers for all uses, including drugs, biologics, devices, and foods. |
|
6b. |
The FDA in coordination with other federal agencies should build needed data infrastructure and surveillance systems to handle the information necessary to gain sufficient understanding of the effects of biomarker use. |
The second part of this chapter reviews the FDA’s infrastructure capacity, and ongoing biomarker data collection efforts. Opportunities to facilitate data collection and sharing, such as precompetitive collaboration, will also be highlighted.
FDA REGULATORY AUTHORITY
Several federal agencies have responsibility for public health. In addition to the FDA and the other 10 agencies that comprise HHS, HHS also collaborates with units within the Departments of Defense, Veterans Affairs, Agriculture, and Education in carrying out its public health responsibilities. The FDA’s mission is as follows:
The FDA is responsible for protecting the public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, our nation’s food supply, cosmetics, and products that emit radiation. The FDA is also responsible for advancing
the public health by helping to speed innovations that make medicines and foods more effective, safer, and more affordable; and helping the public get the accurate, science-based information they need to use medicines and foods to improve their health. (FDA, 2008a)
The FDA’s task is large. The FDA regulates products that comprise about 25 percent of consumer spending in the United States, which comes to more than $1 trillion in spending (Subcommittee on Science and Technology, 2007). The FDA’s 2008 budget authority was $1.87 billion; with user fees added to this number, the FDA’s total 2008 budget was $2.42 billion (Office of Budget, 2009). As stated by Wood (2008) in his article Playing “Kick the FDA”—Risk-free to Players but Hazardous to Public Health,
Between 1988 and 2007, additional FDA responsibilities were imposed by 137 specific statutes, 18 statutes of general applicability, and 14 executive orders (Subcommittee on Science and Technology, 2007). At the same time, the FDA received a 2007 federal appropriation of only $1.57 billion—less than 75% of the budget for the school district in its home county in Maryland.
For another comparison, Coca-Cola’s advertising budget in 2008 was $3 billion (Coca-Cola, 2009). The money spent to promote one company’s products in one year is greater than the money spent to ensure the safety of products purchased with one out of every four consumer dollars in the United States.
Recommendation 7.1 from the Institute of Medicine (IOM) report The Future of Drug Safety stated that “to support improvements in drug safety and efficacy activities over a product’s lifecycle, the committee recommends that the Administration should request and Congress should approve substantially increased resources in both funds and personnel for the Food and Drug Administration” (IOM, 2007b). Food safety is also a challenge because responsibilities are spread over multiple agencies (IOM, 1998). IOM reports on food safety have also pointed out the need for sufficient funding to support a science-based food safety system (IOM, 1998). The call for adequate resources to protect food and drug safety has also been sounded by the FDA’s Science Board in its report FDA Science and Mission at Risk (Subcommittee on Science and Technology, 2007). The challenges facing the FDA as its duties expand and its resources shrink have also been noted by IOM committees and workshops (IOM, 2007a, 2007b) as well as other entities (GAO, 2009b; IOM, 2007a, 2007b; Wood, 2008).
With its large task and small budget, the FDA faces criticism from many directions: when there is an outbreak of illness caused by a foodborne pathogen, when there are pervasive safety problems in food plants,
when medical and food products imported from other countries are adulterated, when new life-saving drugs are not approved fast enough, and when unsafe drugs are taken by patients for years before their risks are recognized. Criticism comes from the public, industry, and government. Decisions from case law modify laws and regulations; these decisions do not always consider their impact beyond a particular case. Changing administrations and priorities within the executive branch also complicate the FDA’s ability to be successful in protecting public health.
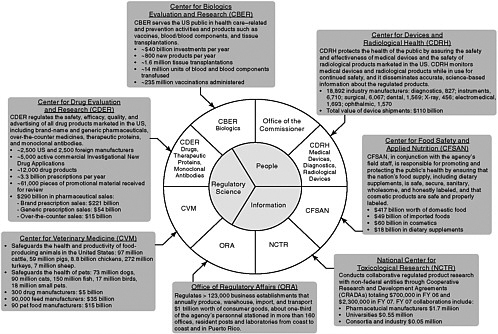
The FDA derives much of its regulatory authority from the Food, Drug, and Cosmetic Act (FDCA), which was originally passed in 1938 and has been amended over time (Box 5-1). From a relatively modest portfolio of activities in 1938, the responsibilities of the agency have continued to expand. In the past two decades, Congress has enacted more than 100 statutes that directly impact the FDA’s regulatory responsibilities—an average of 6 statutes per year, in addition to its core objectives. All of these statutes require some type of FDA action, such as the development or implementation of regulations or guidance documents, or the establishment of new regulatory programs. Although the FDA’s purview of responsibilities continue to expand, the FDA gained through appropriation only 646 employees, an increase of 9 percent, and lost more than $300 million to inflation (Subcommittee on Science and Technology, 2007). Figure 5-1 illustrates the scope of the FDA’s regulatory responsibilities, as outlined in 2006. In terms of dollars, the Center for Devices and Radiological Health (CDRH) regulated manufacturers with industry sales of $110 billion; the Center for Food Safety and Applied Nutrition (CFSAN) regulated $417 billion worth of domestic food, $49 billion in imported food, $60 billion in cosmetics, and $18 billion in dietary supplements; and the Center for Drug Evaluation and Research (CDER) regulated $275 billion in pharmaceutical sales (Subcommittee on Science and Technology, 2007).
The most recent amendment to the FDCA was the Food and Drug Administration Amendments Act of 2007 (FDAAA). The law expanded FDA authority and reauthorized the Prescription Drug User Fee Act (PDUFA), the Medical Device User Fee and Modernization Act, the Best Pharmaceuticals for Children Act, and the Pediatric Research Equity Act (FDA, 2009c). FDAAA requires more FDA involvement in ensuring that clinical trials are incorporated into ClinicalTrials.gov and provides the FDA with additional requirements, authorities, and resources related to pre- and postmarket drug safety, including the ability to require postmarketing studies, clinical trials, safety labeling changes, and Risk Evaluation and Mitigation Strategies (REMS). In addition, FDAAA requires new reporting of adverse events related to food (FDA, 2009b).
The traditional tools of regulatory agencies include regulation, approval or disapproval of applications, and enforcement (Hamburg and
|
BOX 5-1 Expanding FDA Responsibilities The modern regulatory functions of the Food and Drug Administration (FDA) began with the passage of the 1906 Pure Food and Drug Act, which prohibited interstate commerce in adulterated and misbranded food and drugs. The Food, Drug, and Cosmetic Act (FDCA), passed by Congress in 1938, overhauled the public health system by authorizing the FDA to require evidence of safety for new drugs, set standards for food, and conduct factory inspections. Since 1938, the FDA’s role has expanded enormously. Enactment of a series of statutes, beginning in the 1950s and continuing into the 1970s, provided the FDA with a much broader mandate. For example, the Kefauver-Harris Amendments of 1962 strengthened rules for drug safety and required manufacturers to demonstrate effectiveness of drugs. In 1976, the FDCA was amended to apply safety and effectiveness safeguards to new medical devices. Significant amendments to the FDCA since 1980 are included below:
SOURCES: FDA (2009e, 2009f); Subcommittee on Science and Technology (2007). |
Sharfstein, 2009). Prior to the 1970s, FDA functions were primarily related to law enforcement (e.g., issues of adulteration and misbranding). Current regulatory responsibilities are science based, as most of the FDA’s work has moved away from the court to regulatory decisions involving scientific competencies and technical knowledge. Across all centers, the core regulatory functions can be roughly divided into three categories: (1) premarket review, (2) marketed product adverse event surveillance and
|
BOX 5-2 Core FDA Regulatory Functions
SOURCE: Subcommittee on Science and Technology (2007). |
efficacy/safety assessment, and (3) ensuring marketed product safety and quality (see Box 5-2) (Subcommittee on Science and Technology, 2007).
Drugs and Biologics
Two centers, CDER and the Center for Biologics Evaluation and Research (CBER), are responsible for assessing the safety and effectiveness of drugs and biologics, respectively. CDER is the largest of the FDA’s five centers, and has responsibility for both prescription and over-the-counter drugs. CBER ensures the safety, purity, potency, and effectiveness of biological products, including vaccines, blood and blood products, cells, tissues, and gene therapies. The premarket review responsibilities of these
centers include reviewing and assessing sponsor applications and developing and/or implementing an FDA response for new, supplemental, or change-in-use products. Premarket applications related to these centers include Investigational New Drugs, New Drug Applications, and Biologic License Applications.
In addition to premarket review responsibilities, these centers are involved in adverse event surveillance and efficacy and safety assessment of marketed products. The FDA receives and analyzes reports of adverse events, participates in active surveillance and signal detection activities, takes action on safety and efficacy problems, and implements and evaluates risk communications. Particular concerns have been raised over the FDA’s drug safety system, and a large component of the FDA’s drug safety system consists of postmarketing surveillance activities. Previous reviews of the FDA, such as the IOM report The Future of Drug Safety (IOM, 2007b), suggest that the FDA has unclear and insufficient regulatory authorities related to enforcement. The following section outlines the limitations of current postmarking activities and the corresponding lack of FDA authority.
Postmarketing Surveillance in FDA Regulation of Drugs and Biologics
Phase I through III clinical trials are dedicated to demonstrating safety and effectiveness for FDA approval, and usually involve a few hundred to a few thousand individuals. However, many more individuals may ultimately receive the intervention post-FDA approval, and tracking clinical experience, through phase IV (postmarketing) studies is important for identifying relatively rare adverse events and determining effectiveness within different populations and circumstances. However, some evidence suggests that drug sponsors are not fulfilling their postmarketing obligations efficiently, and that the FDA lacks authority to hold drug sponsors to their commitments after drug approval.
Postmarketing surveillance may evaluate safety, efficacy, pharmacology, toxicology, and manufacturing controls, among other factors. The FDA requires drug applicants to conduct these studies in several situations. First, if a drug is approved under accelerated approval on the basis of a surrogate rather than clinical endpoint, then the FDA requires that postmarketing studies verify the safety and efficacy of the drug after it is on the market. If postmarketing studies do not substantiate clinical benefits, or raise safety concerns, the FDA may withdraw the drug. Second, in deferred pediatric studies, drugs approved in adults may be used in children with required postmarketing studies substantiating safety and efficacy in the pediatric populations. Third, when it is unethical to conduct clinical trials in humans, the FDA can approve drugs on
the basis of animal data, but also requires efficacy and safety data after approval. Finally, the FDA can request that the drug applicant conduct postmarketing studies prior to drug approval. Drug applicants agree to these commitments in writing, which the FDA then lists in its final drug approval letters.
The Food and Drug Administration Modernization Act of 1997 expanded the FDA’s authority to oversee postmarketing study commitments. The legislation requires drug sponsors to report on the status of certain postmarketing studies (via annual status reports) and establishes that some information contained in these reports is considered public information. The FDA requires annual status reports for postmarketing studies that address clinical safety, clinical efficacy, clinical pharmacology, and nonclinical pharmacology.1 In the annual status report, drug sponsors indicate the status of their postmarketing commitments, marking studies as pending (study has not started, but is not behind schedule), ongoing (ahead or on schedule), delayed, terminated (study ended before completion, but a final report has not been submitted to the FDA), or submitted.
There is concern that the current postmarketing surveillance system is inadequate in ensuring that drugs are safe and effective. The primary concern with the current system is that drug manufacturers are responsible for the collection, evaluation, and reporting of data from postmarketing studies of their own products. Statistics reveal that drug manufacturers are not efficiently fulfilling their postmarketing obligations; in 2004, fewer than half of promised postmarketing commitments had been initiated (Psaty et al., 2004). From 2004 to 2008, the number of open postmarketing commitments has remained relatively stable, at around 1,100–1,200, while the number of commitments met each year has also remained relatively stable, at around a much lower 100–160 each year.2
Drug manufacturers have little incentive to conduct timely postmarketing studies because these studies may reveal safety problems or other concerns that could result in more constrictive drug labeling, or withdrawal from the market, even though these studies may be a condition of drug approval. Drug manufacturers may also be tempted to conceal available data that suggest a drug has major risks. Examples of such concealment include unpublished data suggesting absence of benefit—or even risk of harm—of selective serotonin reuptake inhibitors in teens (Drummond, 2004), and data showing the interaction of cerivastatin with gemfibrozil and the risk of rhabdomyolysis. Although Bayer Corporation,
the manufacturer of cerivastatin, was aware of the risks of rhabdomyolysis as early as 4 months after the launch of the drug, the contraindication was not added to the package insert for more than 18 months3 (Fontanarosa et al., 2004; Psaty et al., 2004).
Beyond the disincentives of drug manufacturers to reveal possible drug risks postapproval, there are also concerns that the FDA is ill equipped to oversee postmarketing activities and lacks regulatory recourse to ensure drug manufacturer compliance. In 2006, HHS conducted a review of the FDA’s monitoring of its postmarketing study commitments, and came to two conclusions: first, the FDA cannot readily identify whether, or how timely, postmarketing study commitments are progressing toward completions; and second, that monitoring postmarketing study commitments are not a top priority at the FDA (Office of Inspector General, 2006). The report found that one-third of annual status reports were missing or incomplete and contained information with little utility, while the management information system supporting postmarketing activities was found to be ineffective. Compared to other priorities, postmarketing surveillance was also found to be of lower priority. Although PDUFA provided additional funding to the FDA to meet new time lines for drug approval, it prohibited the agency from using the user fees on postmarketing surveillance or other drug safety programs (Psaty et al., 2004), and FDA officials noted that PDUFA-associated activities (e.g., reviewing drug applications and documenting FDA/industry meetings) are of higher priority (Office of Inspector General, 2006). Furthermore, a Government Accountability Office (GAO) study found that there is a lack of criteria for determining which safety actions to take in response to postmarketing findings, and when they should occur (GAO, 2006).4 In reviewing FDA postmarketing surveillance activities, the IOM report The Future of Drug Safety showed that the FDA lacks clear, unambiguous authority to enforce drug sponsor compliance with regulatory requirements. The committee recommended that Congress ensure that the FDA has the ability to require postmarketing risk assessment and risk management programs, and is equipped with better enforcement tools to ensure drug sponsor compliance (IOM, 2007b).
In September 2007 FDAAA was enacted. Among its provisions were new authorities to require postmarket studies and clinical trials, safety labeling changes, and REMS. As a result of FDAAA, between March 25, 2008, and September 14, 2009, CDER and CBER issued 74 letters with postmarketing requirements to assess safety issues for drugs and biologics (FDA, 2009a). Whereas these kinds of studies would have had to have been undertaken voluntarily, they are now required with enforceable time lines. In addition, in 2008, the FDA introduced the Sentinel Initiative, a national integrated electronic database to detect adverse events of drugs and other medical products. It is hoped that the system will eventually monitor as many as 100 million individuals, and will be built from participating electronic health records and claims databases (IOM, 2009; Platt et al., 2009). The Sentinel System will be a distributed network, where all clinical data remains within the source systems’ databases, with centralized software to query approved network questions.
Devices
The FDA Center for Devices and Radiological Health is responsible for regulating medical devices as well as radiation-emitting electronic products. CDRH categorizes devices into three classes, depending on this risks they pose. Class I devices, which include items such as tongue depressors, toothbrushes, and bedpans, have the lowest regulation and do not require review by the FDA prior to marketing. Class II devices face an intermediate level of regulation, including a clearance process that usually does not require submission of clinical data to the FDA. Class III devices, including implants and other high-risk devices, are the most regulated device category and require submission of clinical evidence of safety and effectiveness to secure FDA approval prior to marketing. Regulation of medical devices tends to lag behind the regulation of pharmaceuticals (IOM, 2005). In addition to establishing the three categories of devices, The Medical Device Amendments Act of 19765 also gave the FDA authority to create a system for reporting adverse events associated with devices. In 1984 the FDA issued regulations requiring manufacturers and importers of devices to report information indicating that a device might have caused or contributed to a death or serious injury.
The Safe Medical Devices Act of 1990 added requirements that hospitals and other facilities report to the FDA and manufacturers any events indicating that a device caused or contributed to an event. Additionally, the legislation established new requirements for manufacturers to track specific types of high-risk medical devices and gave the FDA authority
to order recalls of devices in some circumstances. The 1990 legislation further provided that the FDA direct manufacturers to conduct additional information collection activities for certain implants and other devices with the potential to cause serious harm. While this Act increased the scope of device regulation, it also enabled certain medical devices for small user populations without requiring substantial clinical evidence of effectiveness through a Humanitarian Device Exemption.
The 1997 FDA Modernization Act reversed some provisions of the 1990 legislation, such as eliminating certain requirements for adverse event reporting, and ended provisions for mandatory postmarketing surveillance. The legislation prioritized FDA resources to higher risk devices and authorized a new adverse event reporting system based on a sample of hospitals and other user facilities. In addition to providing a system of user fees for FDA premarket reviews, the Medical Device User Fee and Modernization Act of 2002 authorized additional appropriations for postmarketing surveillance, but Congress did not appropriate the funds (reviewed by IOM, 2005).
Postmarketing Surveillance and Premarket Approval in FDA Regulation of Devices
Several factors indicate that the postmarketing surveillance activities for devices are inadequate. For the Safe Medical Devices for Children report (IOM, 2005), the IOM committee was asked to assess if the postmarket surveillance of medical devices provides adequate safeguards for pediatric populations. The committee found shortfalls in FDA performance that were, by and large, not limited to children. The committee found that the agency lacked effective procedures for monitoring the status of required postmarket studies, and there was a lack of public information regarding these studies. The committee recommended that the FDA establish a reliable system to track postmarket studies, and that this information should be publicly available (IOM, 2005). In 2009, the GAO added the FDA’s oversight of medical products, including devices, to its list of high-risk areas warranting attention by Congress and the executive branch. In regard to postmarket surveillance of medical devices, the GAO reported that the number of adverse event reports associated with medical devices increased substantially from 2000 to 2006, but concluded that there are shortcomings in FDA’s regulatory authority related to postmarket surveillance. According to the FDA, the volume of adverse event reports exceeds the agency’s ability to consistently enter or review the reports in a routine manner. FDA officials told the GAO in 2008 that it had a number of strategies to prioritize the reviews of adverse event reports, but cannot review all of the reports received (GAO, 2009c).
In addition to limitations in postmarketing surveillance, the strength of evidence supporting premarketing device approvals has been questioned. Two recent reviews (Dhruva et al., 2009; Kramer et al., 2009) assessed the strength of studies used to support the premarket approval of cardiovascular devices. The first, sponsored by the FDA, found that more than 40 percent of studies used to approve cardiovascular devices lacked high quality data about either the treatment or safety goals of the study, and 25 percent of trials failed to adequately follow the outcomes of a sufficient number of patients (Kramer et al., 2009). The second study (Dhruva et al., 2009) found that 33 of 123 studies (27 percent) used to support the FDA approval of cardiovascular devices were randomized and 17 of 123 were blinded; 65 percent of premarket approvals were based on a single study. This review found that 187 of 213 (88 percent) primary endpoints were surrogate measures and 122 of 157 (78 percent) had a discrepancy between the number of patients enrolled in the study and the number analyzed. In recognition of concerns over the quality of studies used in device approval, the FDA is in the process of developing guidelines to set tougher scientific standards (Meier, 2009). Most likely, the FDA will urge device makers to use more sharply defined targets to measure success of clinical trials and more closely follow patients enrolled in trials to determine whether these targets are met.
Foods
Chapter 2 outlines the legal basis for health claims and describes the different types of claims (see Table 5-1). In addition, Chapter 2 discusses how the FDA has used biomarkers as surrogate endpoints in the evaluation of authorized and qualified health claims, and the current “surrogate endpoints of disease risk” used by CFSAN. The following section describes the FDA’s regulatory authority related to foods, while Box 5-3 discusses FDA regulatory authority related to dietary supplements.6
Legislative mandates and legal action have influenced the way foods are regulated in the United States. Notably, the Nutrition Labeling and Education Act of 1990 (NLEA) first set the circumstances under which nutrition and health claims could be used. NLEA was designed to give consumers more scientifically valid information about foods they eat, and the statute directed the FDA to issue regulations providing for the use of
TABLE 5-1 Types of Claims
|
Type of Claim |
Description |
|
Health claims based on significant scientific agreement (SSA), or authorized health claims |
Health claims based on a high level of confidence in the validity of the relationship between the substance and the disease or health-related condition. Authorized health claim language must meet all regulatory requirements (e.g., development of hypertension or high blood pressure depends on many factors. [This product] can be part of a low-sodium, low-salt diet that might reduce the risk of hypertension or high blood pressure). Authorized health claims require Food and Drug Administration (FDA) review and approval. |
|
Health claims based on authoritative statements |
Claims based on authoritative statements about the substance/disease relationship by other scientific bodies of the U.S. government or the National Academy of Sciences.a Authoritative statements can be used without approval by an FDA review process. |
|
Qualified health claims |
Claims that do not meet the SSA standard can be allowed on the basis of lower evidence if the claim is accompanied by qualifying language. Qualified health claims require FDA review and approval. |
statements that describe the relationship between a substance and disease in labeling of foods, including dietary supplements, after the statements have been reviewed and authorized by the FDA (CFSAN, 2009). Based on NLEA, the FDA authorizes health claims on the basis of significant scientific agreement (SSA), or:
based on the totality of publicly available scientific evidence (including evidence from well-designed studies conducted in a manner which is consistent with generally recognized scientific procedures and principles), that there is significant scientific agreement among experts qualified by scientific training and experience to evaluate such claims, that the claim is supported by such evidence.7
Legal action challenged the SSA standard and resulted in a process to allow claims with lesser scientific evidence, with qualifying language (qualified health claims) (Schneeman, 2007; Taylor and Wilkening, 2008).
Consumer Understanding and FDA Regulation in Food
In order to ensure the safety of nutrition-related uses of biomarkers, more research is needed on strategies for effective communication to consumers about health-related information. In the next paragraphs, research on communication for conveyance of health information related to foods is presented. Although the FDA has stipulated the numerous types of claims that manufacturers can include in product labeling, it can be difficult for consumers to assess the scientific merit of these
|
BOX 5-3 Dietary Supplements Adding to the complexity of the FDA’s regulatory authority, the FDA regulates dietary supplements under a different set of regulations than those covering conventional foods. Dietary supplements are defined in the Dietary Supplement Health and Education Act of 1994 (DSHEA) as food products, that among other things, are intended to supplement the diet and contain one or more of the following “dietary ingredients”: vitamins, minerals, herbs or other botanicals, amino acids, or other dietary supplements for use by humans to supplement the diet by increasing the total dietary intake, or concentrates, metabolites, constituents, extracts, or combinations for these ingredients (FDA, 2009d). Prior to 1994, dietary supplements were subject to the same regulatory requirements as other foods. However, DSHEA created a new regulatory framework for the safety and some aspects of labeling of dietary supplements. Under DSHEA, dietary supplements are broadly presumed to be safe, and the FDA does not have the authority to require dietary supplements to be approved for safety and efficacy before they enter the market (GAO, 2009a). Although the FDA must be notified of new dietary ingredients and the manufacturer must provide evidence that the new dietary ingredient is reasonably expected to be safe, the starting assumption is that dietary ingredients are safe (Yetley, 2007). The manufacturer is responsible for ensuring that any claims made about dietary supplements are substantiated by adequate evidence to show that they are not false or misleading (FDA, 2009d). In contrast to authorized and qualified health claims for food, the FDA permits statements of nutritional support to be made in dietary supplement labeling without a scientific review of the evidence.a These claims are permitted under the following circumstances: (1) if the manufacturer has substantiation that a statement is truthful and not misleading; (2) if the labeling includes prominently displayed text that “This statement has not been evaluated by the Food and Drug Administration. This product is not intended to diagnose, treat, cure, or prevent any disease”; (3) the manufacturer notifies the FDA no later than 30 days after first marketing the dietary supplement with the statement.b In 2000, the FDA published final regulations on the types of unacceptable structure/function claims in the labeling of dietary supplements.c Because of the regulatory framework for dietary supplements, the FDA has little information about the safety and effectiveness of dietary supplements. In 2007, the FDA issued a final rule establishing regulations to require good manufacturing practices to improve oversight of dietary supplements (FDA, 2007). However, a GAO report on dietary supplements reported that “consumers remain vulnerable to risks posed by potentially unsafe products” and found that the FDA’s ability to |
|
identify dietary supplement safety concerns is hindered by a lack of information (GAO, 2009a). With this limited information, a primary method for the FDA to identify safety concerns is postmarketing surveillance. In 2006, the Dietary Supplement and Nonprescription Drug Consumer Protection Actd amended the FDCAe to require dietary supplement companies that receive serious adverse event reports to submit information about the event to the FDA, effective December 22, 2007. Since the mandatory reporting requirement went into effect, the FDA has seen a three-fold increase in the number of total adverse events reported: from January through October 2008, the FDA received 948 adverse event reports, compared with 298 reports received over the same period in 2007. Of the 948 adverse event reports, 596 reports were mandatory reports of serious adverse events submitted by industry. However, the FDA recently estimated that the actual total number of adverse events (including mild, moderate, and serious adverse events) related to dietary supplements is more than 50,000, suggesting underreporting, limiting the amount of information the FDA receives (GAO, 2009a). Once the FDA has identified a potential safety problem, the agency has several options, including issuing warning letters and consumer alerts, working with a company on a voluntary product recall, and banning an ingredient, among other options. However, the FDA’s authority to respond is limited. According to the recent GAO report, the limitations are two-fold: first, the FDA dedicates relatively few resources to dietary supplement oversight activities; and second, under the significant or unreasonable risk standard, the FDA has difficulty establishing adulteration for dietary supplement products (GAO, 2009a). The FDA’s experience with ephedra demonstrates how regulatory authority influences the agency’s response to reports of safety problems. Based on numerous reports of possible adverse effects associated with the use of these products, the FDA began a process of investigating the safety of ephedra, collating and evaluating the evidence while ephedra remained on the market. Although the agency first convened an advisory committee in 1995 to investigate the adverse effects, the published final regulations banning the use of ephedra did not occur until 2004 (Yetley, 2007). Agency officials and other stakeholders attribute the difficulty of banning dietary supplements on the FDA’s requirement to establish adulteration under the significant or unreasonable standard, and limited data on the safety of dietary supplements compounds the problem (GAO, 2009a). |
claims in practice. Studies have demonstrated the lack of consumer understanding of the differing types of claims, leading to confusion, and in some cases, a perception that claims of lower scientific evidence are more valid than claims based on SSA. To enhance communication, more research is needed on methods of improving consumer understanding, which is a necessary prerequisite to ensuring that effective science-based strategies are used to improve consumer interpretations of health claims. Consideration should also be given to appropriate implementation of these strategies in population subgroups with differing cultural and educational backgrounds.
Several consumer studies have highlighted the difficulty of determining which strategies are most effective in influencing consumer behavior. In a 2-year effort to provide information in grocery stores on healthier food choices, Levy and colleagues (1985) found that a number of other factors were more influential on consumer purchasing behavior than the interventions tested. More influential factors included the city tested, socioeconomic status, product price, in-store purchasing trends, and seasonal trends. Levy et al. (1992) also discovered that consumers do not necessarily prefer the most effective nutrition-label formats. Mazis and Raymond (1997) found that consumers have more accurate interpretations of health claims when nutritional labels are also present, but Roe et al. (1999) found that consumers are less likely to look at Nutrition Facts panels when health claims are present on the front of food packages.
CFSAN has had a consumer studies staff for many years. This group conducts studies on consumer understanding of nutrition labeling, including health claims. There is a strong need for this type of research to be continued. These studies have informed the content of the Nutrition Facts panel through consumer studies evaluating effectiveness of various label types and consumer preferences (Heimbach and Stokes, 1982; Levy et al., 1996; Lewis and Yetley, 1992).
Additional research was conducted on consumer understanding of health claims after the introduction of qualified health claims in the marketplace. Some research showed that consumers preferred simple, succinct claim language (Williams, 2005), and other studies showed that the length and wording of claims made it difficult for consumers to identify the type of claim or strength of evidence supporting claims (Hooker and Teratanavat, 2008; Kapsak et al., 2008). The International Food Information Council Foundation (IFICF) conducted a study on consumer understanding of health claims and found that consumers rate the scientific evidence and other attributes of a product containing an authorized (SSA-level) claim similar to products containing a structure/function claim or dietary guidance statement for which FDA authorization is not required (IFICF,
2005). Research also indicates that consumers have difficulty understanding the “qualifying language” that is intended to help consumers distinguish among the four levels of scientific evidence in authorized and qualified health claims.
Both the IFICF and CFSAN reached several conclusions regarding consumer understanding of qualified health claims: first, consumers had difficulty distinguishing among the differing evidentiary levels for claims, especially with language-only claims (as opposed to graphic representations; see Figure 5-2) (CFSAN, 2005; IFICF, 2005). The IFICF study also found that words such as “promising,” “inconclusive,” and “may” were perceived to mean different things to different consumers, which altered their perception of the health claim (IFICF, 2005). The FDA study revealed similar perception biases: “[e]ven when qualified health claims were understood as intended, qualifying statements had unexpected effects on consumers’ judgments about the health benefits and overall healthfulness of the product bearing the claim. Sometimes these qualified health claims led to more positive product perceptions” (CFSAN, 2005). Alarmingly, the FDA study also found that B grades were understood by consumers to convey greater scientific certainty than authorized health claims, or those that meet the SSA standard (in the CFSAN study, an A letter grade wasn’t included for SSA health claims; instead, the substance/disease relationship was stated) (CFSAN, 2005). Further studies have suggested the need to study how consumers perceive health claims, including how the information is presented, to foster better understanding (Borra, 2006; Hooker and Teratanavat, 2008; Kapsak et al., 2008; Mazis and Raymond, 1997; Williams, 2005). Since conventional foods can make a number of claims without premarket review by the FDA (such as structure/function claims and dietary guidance statements; reviewed in Chapter 2), evidence that consumers have difficulty assessing the scientific merit of claims suggests that the multitude of claims, with differing levels of scientific support, may not adequately protect public health.
Enforcement of health claims The FDA is responsible for enforcing the correct use of food label claims; however, the general enforcement capacity of the FDA has been questioned. Prescription for Harm: The Decline in FDA Enforcement Activity (2006), a report requested by Rep. Henry Waxman (D-CA), found that FDA enforcement declined by more than 50 percent from 2000 to 2005. According to the report, there has been a decline in the overall number of FDA enforcement actions, including fewer warning letters and seizures. Likewise, the Center for Science in the Public Interest (CSPI) began a litigation initiative in 2005 to stop deceptive labeling, fraudulent advertising, and use of dangerous food additives, saying that these actions were necessary because the FDA and

FIGURE 5-2 Comparison of Food and Drug Administration (FDA) health-related food label statements: (A) an authorized health claim for the relationship of calcium and osteoporosis—authorized health claims require strong evidence and FDA review; (B) a nutrient content claim—these require substantiating data to be kept by the company and FDA notification but do not require FDA review; (C) a structure–function claim—these require substantiating data to be kept by the company and FDA notification but do not require FDA review; and (D) a dietary guidance statement—these are categorized separately from health claims because they make statements about healthy diet in general rather than about a specific substance in the product on which the statement appears. While claim A is based on significant scientific agreement (SSA), B, C, and D do not need to reach SSA-level evidence. Studies have indicated that consumers have difficulty understanding, or are unaware of, the levels of evidences associated with each type of label claim.
SOURCE: CFSAN (2007).
the Federal Trade Commission “have done a poor job enforcing the law in these areas” (CSPI, 2005). For example, CSPI threatened the Quaker Oats Company with a lawsuit over food labeling and advertising that “exaggerated the health benefits of eating oatmeal” (CSPI, 2007). In exchange for no longer describing oatmeal as a “unique whole grain food” that “actively finds the excess cholesterol” and a graph that overstates the cholesterol-lowering ability of oatmeal, CSPI dropped the lawsuit against the Quaker Oats Company.
However, recent enforcement actions by the FDA may indicate heightened enforcement of food labeling, including health claims. Cheerios has a cereal box that suggests “you can lower your cholesterol four percent in six weeks” (Grocery-aisle gotchas, 2009). This box refers consumers to a General Mills study that found that 3 cups of Cheerios (as opposed to 3 cups of cornflakes) with 1.5 cups of milk a day lowered cholesterol levels by 3.8 percent and low-density lipoprotein levels by 4.2 percent in six weeks (Johnston et al., 1998). Wording such as “lowers cholesterol” appears to trigger drug status, especially considering the SSA model health claim language approved by the FDA for oat bran and heart disease: “soluble fiber from foods such as oat bran in Brand Name Cereal, as part of a diet low in saturated fat and cholesterol, may reduce the risk of heart disease.”8
On May 5, 2009, the FDA sent General Mills Inc. a warning letter over the Cheerios labeling. The FDA argued that the labeling saying “you can lower your cholesterol four percent in six weeks” violated the FDCA and applicable regulations. The letter indicated that Cheerios was being marketed as an unapproved drug and was misbranded (FDA, 2009b). A General Mills spokesperson indicated that the claim has been used for more than 2 years (Corbett Dooren, 2009). The director of CFSAN indicated that the agency is ready to send out more warning letters if it finds more violators, noting food companies have had a tendency to cross the line into the drug category by making specific health claims on packaging (Corbett Dooren, 2009).
In March 2010, the FDA notified 17 food manufacturers that the labeling for 22 of their food products violated the FDCA. The violations cited include unauthorized health claims, unauthorized nutrient content claims, and the unauthorized use of terms, such as “healthy,” which have strict regulatory definitions (FDA, 2010a). In an open letter to industry, the FDA commissioner Margaret Hamburg noted that the warning letters “cover a range of concerns about how false or misleading labels can undermine the intention of Congress to provide consumers with labeling information that enables consumers to make informed and healthy food
choices” (Hamburg, 2010). Hamburg noted that the FDA should provide as clear and consistent guidance as possible about food labeling claims and nutrition information to help consumers construct healthy diets, and that these warning letters will give the food industry further clarification about what is expected of them as they review their current labeling.
FEDERAL AGENCIES AND DATA COLLECTION
Numerous agencies within HHS are involved in efforts to collect information on medical or lifestyle interventions, observational data on health, and biomedical discovery efforts. Some of the agencies involved in data collection include the NIH, the FDA, the Centers for Disease Control and Prevention (CDC), the Centers for Medicare & Medicaid Services (CMS), and the Agency for Healthcare Research and Quality (AHRQ). In addition to the federal government, many other stakeholders are involved in the collection of data, including academia, industry, and nonprofit organizations. Government agencies and other stakeholders collect diverse information depending on the focus and needs of the organization. For example, as the steward of medical and behavioral research for the nation, the NIH conducts and supports basic and translational research. The CDC collects data to monitor health, detect and investigate health problems, to enhance prevention, and to develop and advocate sound public health policies, among its other responsibilities. In fulfilling its mission to improve the quality, safety, efficiency, and effectiveness of health care for all Americans, AHRQ conducts research that helps people make more informed decisions and improve the quality of health care services. As the nation’s largest and most representative claims database, CMS collects administrative claims data, including information on diagnoses, treatment, as well as outcomes. The FDA collects information to support regulatory decision making, including the evaluation of safety and efficacy of products and ensuring that products are honestly, accurately and represented to the public.
Biomarkers are an important focus of research and data collection. Both the federal government and other stakeholders have recognized the potential role of biomarkers in the development of medical interventions, selection of populations for therapy, assessment of safety and efficacy of interventions, in clinical decision making, and in surveillance activities. Efforts to collect biomarker data are currently underway, and involve collaborations among government, industry, academic, and philanthropic stakeholders. The following section outlines ongoing efforts to collect information about biomarkers and discusses HHS’ important role in facilitating and coordinating these efforts.
Collective Efforts to Collect and Share Biomarker Data
Many of the efforts to collect and share biomarker data result from precompetitive collaborations. As described by the IOM Cancer Biomarkers report (2007a), the challenge and expense of biomarker discovery and development may make it impossible for a company or organization to undertake the work single-handedly. Because biomarkers have the potential to facilitate research activities for multiple stakeholders, the sharing of precompetitive data and cooperation could be important to accelerating discovery and development. By pooling skills, technologies, and other resources, precompetitive collaborations may be able to leverage the strengths of different partners, leading to greater efficiency and effectiveness (IOM, 2007a). Government, industry, academia, and nonprofit organizations may potentially play roles in the sharing of precompetitive data to advance biomarker research.
The Biomarkers Consortium
For example, the Biomarkers Consortium is a public–private biomedical research partnership administered by the Foundation for the National Institutes of Health (FNIH).9 The Consortium “endeavors to develop, validate, and qualify biological markers (biomarkers) to speed the development of medicines and therapies for detection, prevention, diagnosis, and treatment of disease and improve patient care” (FNIH, 2007). The Consortium is focused on identifying “high-impact biomarker opportunities” that address significant unmet medical needs, promise immediate practical impact on outcomes such as development of treatments and patient care, and can be accomplished within practical limits on time frames and cost. It has four disease/therapeutic focus areas: cancer, immunity and inflammation, metabolic disorders, and neuroscience. The founding partners of The Biomarkers Consortium include the NIH, the FDA, the Pharmaceutical Research and Manufacturers of America, and FNIH. Other partners include CMS and the Biotechnology Industry Organization.
The Biomarkers Consortium Executive Committee, composed of the founding partners and other stakeholders, has created four disease-specific Steering Committees to identify, prioritize, and refine biomarker-related project concepts and projects within their focus areas. Each Steering Committee is led by two cochairs appointed by the Executive Committee, and has broad membership, including relevant experts from key Consortium partners (NIH, FDA, CMS, industry, academia, and the advocacy community). Once the steering committee has a high-priority concept selected and refined (generally 3–4 pages in length), it will assemble a volunteer “project team” of 8–20 people with relevant expertise, including representatives from NIH, FDA, and industry. This team then develops a detailed project design and protocol (generally 30–100 pages), including a time line with expected milestones, under the direction of a staff scientific program manager. That document is then reviewed and approved by the steering committee and executive committee prior to launching a formal solicitation for funding. Most projects are launched within 4–6 months, with four to five funders supporting the study.10
One of the Consortium’s projects includes an evaluation of the performance of adiponectin as a biomarker predictive of glycemic efficacy. Using a statistical analysis of combined data from multiple phase II clinical trials performed by four pharmaceutical companies, the project assessed the relationship between adiponectin and glucose lowering in response to PPAR (peroxisome proliferator-activated receptors) agonists. The analysis suggested that in type 2 diabetes mellitus patients, adiponectin level is a robust predictor of glycemic response to PPAR agonists, but not to non-PPAR drugs (Wagner et al., 2009). In addition, this project established important precedents for biomarker data-sharing principles among stakeholders in the Biomarkers Consortium and demonstrated the benefits of cross-company collaboration.
Under the auspices of the FNIH Biomarkers Consortium, several pharmaceutical and biotechnology industries collaborated with the National Cancer Institute (NCI), the FDA, and academic investigators to further the use of biomarkers in breast cancer treatment. The I-SPY2 (Investigation of Serial Studies to Predict Your therapeutic response with imaging and molecular analysis) trial aims to simultaneously and serially test several targeted treatments and biomarker tests to more rapidly assess which biomarkers best predict a therapeutic response (Barker et al., 2009).
The Critical Path Institute
The Critical Path Institute (C-Path) provides an important venue for the collection of biomarker data. C-Path, an independent, publicly-funded institute, brings together scientists from the FDA, academia, and industry to accomplish goals outlined in the Critical Path Initiative.11 According to C-Path, its work can be viewed as a series of projects funded by grants and performed by collaborations and consortia; however, much of the organization’s work falls under the goal of creating methods to enable personalized medicine that improves public health, including the creation of tools, such as biomarkers, and methods qualified by the FDA for use in medical product development (C-Path, 2010a).
As mentioned in Chapter 2, C-Path’s Predictive Safety Testing Consortium (PSTC) is a public–private partnership that brings together pharmaceutical companies to share and validate each other’s safety testing methods (C-Path, 2008) Through the PSTC, consortium members are sharing new preclinical biomarker tests for examination and cross-validation by other consortium members. As a result of the work of the PSTC, seven biomarkers of drug-induced nephrotoxicity in rats were validated and qualified by the FDA and the EMEA (European Medicines Agency) (FDA, 2008b).
C-Path has also initiated the Coalition Against Major Diseases (CAMD). CAMD’s focus is to develop new tools and methods leading to better treatments for neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease. The coalition will define clinical data standards and establish a pooled database of control arms of pharmaceutical clinical trials in order to develop quantitative disease progression models. Additionally, the coalition will attempt to incorporate imaging, biochemical, and molecular biomarkers that have the greatest potential to identify patient populations that are most likely to benefit from new therapies (C-Path, 2010b).
CEO Roundtable on Cancer’s Life Science Consortium
The CEO Roundtable on Cancer’s Life Science Consortium has been working to establish a new precompetitive environment to facilitate the development and use of biomarkers in cancer drug development (Curt, 2009). In recognition of the lack of standardization and qualification of biomarkers (Curt, 2009; IOM, 2007a), the Life Science Consortium envisions a new precompetitive environment that enables drug companies to
|
11 |
As mentioned in Chapter 1, the Critical Path Initiative is the FDA’s national strategy to drive innovation in the scientific processes through which FDA-regulated products are developed, evaluated, manufactured, and used (FDA, 2010b). |
present their biomarker programs for cancer drug development, under confidentiality, to the NCI (Curt, 2009). This precompetitive safe harbor allows the NCI to gain a unique perspective, unobservable to its individual industry partners, to identify areas of overlap and redundancy as well as gaps. By selecting the most promising partners for further biomarker development and then sharing the validated markers with the academic and industry communities at large, the NCI provides a neutral platform that can enable cancer drug development across companies and academia because the risks are shared and collaboration replaces competition. This new approach has already come to fruition. The NCI identified a promising assay for measuring the activity of poly (ADP-ribose) polymerase inhibitors and worked to further develop and validate the assay, which has now been used in a phase 0 human trial (Kinders et al., 2008; Kummar et al., 2009; Yang et al., 2009).
Oncology Biomarker Qualification Initiative
The Oncology Biomarker Qualification Initiative (OBQI) is an agreement among the FDA, the NCI, and CMS to collaborate on improving the development of cancer therapies and the outcomes for cancer patients through biomarker development and evaluation (NCI, 2006). According to the memorandum of understanding for the OBQI collaboration, extensive cross-sector and multi-disciplinary efforts are needed to understand and develop the clinical utility of a new generation of biomarker technologies. The three agencies agreed to collaborate through working groups and steering committees to develop strategic plans, set priorities, and leverage resources and expertise from multiple sources, including the private sector (FDA, 2009g). Goals of the OBQI include the development of biomarker technologies and validation protocols to improve the detection, diagnosis, treatment, and prevention of cancer; development of guidance for the use of biomarkers to facilitate cancer drug development; and the ability to make informed decisions about reimbursement of new or existing treatment regimens based on biomarker-guided knowledge (Barker, 2006). Cancer imaging, molecular assays and targeted therapies, biomarker-driven clinical trial designs, and data mining are initial priorities of the OBQI.
Role of HHS in Biomarker Data Collection
These ongoing efforts to collect and share information about biomarkers will provide necessary information for the effective application of the committee’s proposed evaluation framework. Recognizing the value of a well-coordinated, comprehensive effort to collect and share biomarker
information in advancing public health, the committee sought to improve ongoing biomarker data collection efforts. HHS can play an important role in ensuring that these efforts are optimally organized and coordinated. If biomarker discovery and development are uneven, HHS may be able to focus attention on underdeveloped areas, such as biomarkers for food and nutrition.
Unlike biomarker discovery efforts for drug and diagnostic development, relatively little research regarding biomarkers, or clinical outcomes, has been conducted for nutritional uses. One of the main reasons for this difference is that the regulatory setting for nutrition, such as foods and supplements, is quite different than for drugs and even devices. For example, drugs are generally considered unsafe and ineffective until clinical trials proving otherwise are conducted. With most foods, they are “generally recognized as safe” and can be introduced on the market without review of the safety evidence by FDA scientists. When a food manufacturer wishes to make an authorized or qualified nutritional claim about a food, the resulting health claim is not exclusive to the manufacturer, but broadly applied to the food substance across a range of food products. Other manufacturers may use an authorized health claim on their products, which decreases the incentives to collect biomarker or clinical outcome data on the food substance in their food products.
In addition, HHS coordination may ensure that biomarker data collection efforts are effective and that they leverage the stakeholders’ strengths and capacities. For example, NIH has played a critical role in advancing biomarker discovery, development, and qualification (see Box 5-4). In addition, HHS can facilitate precompetitive collaborations that may encourage multiple stakeholders to share data, including industry. Such precompetitive collaborations are already underway, and include the Biomarkers Consortium, C-Path, and the OBQI. By coordinating biomarker efforts, HHS can ensure that biomarkers are effectively utilized across all contexts of use, including drugs, biologics, devices, and foods.
TRACKING THE EFFECTS OF BIOMARKER USE AT THE FDA
Within the regulatory environment, ensuring high-quality data collection is paramount to evidence-based regulation. Although the FDA has to make decisions in the presence of uncertainty, it is critical that regulatory decision making incorporates sound scientific information (Yetley, 2007). Reliance on scientific data for regulatory decision making provides legitimacy for agency actions and strengthens public trust in the FDA: “Establishing the FDA as a public health agency requires a culture that encourages scientific exchange and respects alternative viewpoints along the path of decision making. It also requires that the agency define and
|
BOX 5-4 Role of NIH in Biomarker Data Collection The NIH has played an instrumental role in the development and qualification of biomarkers for all purposes. NIH has initiated a number of efforts aimed at improving collaboration between stakeholders and increasing the amount of publicly available information on promising biomarkers. These efforts include workshops on the state of the science for various biomarkers, the Biomarkers Consortium, and the Oncology Biomarker Qualification Initiative. NIH-led workshops on use of biomarkers for purposes with regulatory impact have been held via the Office of Dietary Supplements, NHLBI, and others. In 1999, the NIH and the FDA held a workshop on “Biomarkers and Surrogate Endpoints: Advancing Clinical Research and Applications” (Abstracts of the NIH-FDA conference, 1998). Topics ranged from definitions to needs and applications in disease areas from cardiovascular to psychiatric conditions. While this report does not describe the contributions of NIH and its separate institutes and offices in detail, these cannot be underestimated. The expertise, leadership, and resources of the NIH enable much rigorous science, interagency and inter-sector collaboration, and the public availability of biomarker data that would otherwise not occur. The NIH may also help play a role in prioritizing the development of biomarkers in underdeveloped areas, such as food and nutrition. |
protect integrity in its basic processes” (Hamburg and Sharfstein, 2009). According to a 1998 report from the U.S. House of Representatives Committee on Science, a necessary step toward evidence-based decision making is ensuring access to sound scientific data. The report recommends that sufficient resources are committed to science that informs policy decisions so that research, whenever possible, precedes policy decisions (Committee on Science, 1998). However, there are concerns that the FDA’s science capacity is at risk, threatening the agency’s ability to meet current and emerging regulatory responsibilities (Subcommittee on Science and Technology, 2007).
The Subcommittee on Science and Technology concluded that: “science at the FDA is in a precarious position: the Agency suffers from serious scientific deficiencies and is not positioned to meet current or emerging regulatory responsibilities” (Subcommittee on Science and Technology, 2007). According to the subcommittee, three areas requiring improvement include strengthening mission-supportive scientific research programs, excellent staff with appropriate scientific expertise, and an information infrastructure and processing capability to ensure the FDA has access to the best data and information necessary to support regulatory science.
The subcommittee found significant deficiencies in the ability of FDA
regulatory programs to assess and use information. Although the FDA is dependent on accurate and timely information to deliver its regulatory mission, the information crisis is putting their mission at risk. The subcommittee found that there is evidence of important, but slow, progress to improve information sciences and technology at the FDA over the past few years, yet significant gaps remain. In particular, the subcommittee concluded that the FDA cannot fulfill its surveillance mission because of inadequate staff and IT resources to implement cutting-edge approaches to modeling, risk assessment, and data analysis (Subcommittee on Science and Technology, 2007). The FDA is in the process of implementing a number of initiatives to improve its capacity to collect and interpret surveillance data. The following section describes these efforts and other efforts undertaken that may be important resources to the FDA as it collects outcome data on FDA-regulated products.
Efforts to Collect Information on Outcomes
The committee recommends that the FDA ensures that appropriate data infrastructure and surveillance systems are in place to gain sufficient understanding of the effects of biomarker use. There are a number of ongoing efforts to collect information on outcomes related to FDA-regulated products. These include the Sentinel Initiative, MedWatchPlus, and the Observational Medical Outcomes Partnership. In addition, the International Serious Adverse Events Consortium, the Cardiac Safety Research Consortium, and ClinicalTrials.gov may provide important information on outcomes. Information on outcomes will need to be linked to biomarkers, so that the FDA can gain sufficient understanding of the use of biomarkers in regulatory decision making.
Sentinel Initiative
As mentioned previously, the Sentinel Initiative aims to develop and implement a proactive system to track reports of adverse events linked to the use of the FDA’s regulated products (FDA, 2010c). It is hoped that the Sentinel Initiative will be a national electronic system that will transform FDA’s ability to track the safety of drugs, biologics, medical devices—and ultimately all FDA-regulated products once they reach the market. The Sentinel Initiative will be developed and implemented in stages. Currently, the FDA is working on the mini-Sentinel program, or developing a Sentinel prototype. Two aspects of this prototype include developing a coordinating center for a distributed system and evaluating emerging methods in safety science (Platt, 2010). Mini-Sentinel will include drugs, biologics, and devices; data sources include administrative
claims databases, outpatient and impatient electronic medical records, and registries.
MedWatchPlus
The FDA is currently developing MedWatchPlus, an electronic system for receiving, processing, storing, and analyzing adverse event reports and other safety-related information for all FDA-regulated products. This system will combine the FDA’s various safety reporting processes and systems and will provide a single point of entry for reporters. Additionally, the FDA and the NIH are collaborating to develop a “rational questionnaire” to ensure that submitting adverse events and problem reports are easier, more complete, and more consistent (FDA, 2009h). FAERS (FDA Adverse Event Reporting System) will be FDA’s new repository with enhanced analytic methods to enable staff to efficiently analyze thousands of safety reports and identify potential safety problems.
Observational Medical Outcomes Partnership
The Observational Medical Outcomes Partnership (OMOP) is a public–private partnership designed to help improve the monitoring of drugs for safety. The OMOP is funded and managed through the FNIH and draws on the expertise of the FDA, other federal agencies, the pharmaceutical industry, and non-profit organizations. The partnership is conducting a 2-year initiative to research methods that are feasible and useful to analyze existing healthcare databases to identify and evaluate safety and benefit issues of drugs already on the market (FNIH, 2010). In particular, the partnership is evaluating whether multi-source observational data can improve the ability to assess drug safety and benefits (Ryan, 2010).
International SAE Consortium
The International Serious Adverse Events Consortium (iSAEC) is a nonprofit organization comprised of pharmaceutical companies, the Wellcome Trust, and academic institutions that receives scientific and strategic input from the FDA and international regulatory bodies. This consortium attempts to identify DNA variants that may be useful in predicting the risk of drug-related serious adverse events (iSAEC, 2010). The iSAEC phase 1 objectives include creation of a publicly available knowledge base of cross drug safety pharmacogenomics markers for predicting key serious adverse events and supporting the execution of the Critical Path Initiative (Holden, 2010).
Cardiac Safety Research Consortium
The Cardiac Safety Research Consortium (CSRC) is a public–private partnership of the Critical Path Initiative that focuses on cardiac safety and new medicine product development. Duke University’s Clinical Research Institute manages the CSRC, which involves industry, academics, and regulators. The CSRC has developed a model for precompetitive data sharing in which electronic ECG submissions to the FDA are made available for research by the consortium, with an initial focus on QT interval issues. Additional areas of focus include using the ECG library to qualify new ECG biomarkers for cardiac risk and developing additional research and regulatory evaluation tools to facilitate clinical decision making and future medical product development (CSRC, 2010).
Clinicaltrials.gov
ClinicalTrials.gov was created in 1997 after passage of the Food and Drug Administration Modernization Act (FDAMA). As a result of FDAMA and FDAAA, Congress has required that the FDA implement registration prior to recruitment of all clinical trials that fall under the regulatory authority of the FDA and, within 1 year of completion, the reporting of results in a database. These databases have been developed by the National Library of Medicine (NLM) at the NIH in collaboration with the FDA. Drug, device, and biologic trials are included in the legislative mandates; nutritional and supplement studies are not mandated for registration or reporting, nor are observational studies covered. Nonetheless, the databases accept and encourage registration of observational studies, and about 15 percent of the registry consists of observational studies. An unknown proportion of studies are nutritional, behavioral, or health services; again, these are voluntary. Overseas trials are registered if the sponsor intends to register the drug in the United States or has U.S. study sites.
More than 80,000 trials are registered and about 500 results are available. Clinical trial results for trials initiated after September 2007 are to be provided even if the trial findings remain unpublished, as occurs in about 30 percent of trials; such reporting remains uneven. NIH and other publicly funded trials are also required to be registered and reported, but there is some confusion about the requirements. Noncompliance carries substantial penalties, so registration levels are high, with a possible exception in the device area. Individuals or researchers can use search terms that permit rapid and effective identification and aggregation. The database also links to PubMed and publications. This database has been used to scan all potential informative trials and is used in meta-analyses, which could inform qualification and validation/interpretation.
This database is built on a robust platform with public access and many links to related sources. Although the database was created and is maintained with the input of the FDA, the capability and staff necessary to administer the database reside at the NLM. Legislation is required to make changes in this reporting system, and a directive is needed regarding the implementation within the legislation. Congress has been aggressive in seeking transparency of results and their use. A major caveat is that the reporting is by investigators and sponsors, and the NLM has responsibilities for archiving, not validating, reports. Therefore, no interpretation of individual study findings is provided at the site, although the FDAAA requires an examination of whether this information can be included in an unbiased way.
Reaction to ClinicalTrials.gov has been mixed. Companies feared that disclosure of clinical trial results would put them at a competitive disadvantage and impact the viability of the pharmaceutical development enterprise (Drazen and Wood, 2005). For a period of time, some companies failed to include meaningful data in their registry entries. However, efforts on the part of the medical research community resulted in improved data submissions (Drazen and Wood, 2005, 2006). Editors of medical journals supported the database, eventually requiring authors to have registered clinical trials in the database or be barred from publication in many medical journals (Drazen and Wood, 2005). It is not yet clear how beneficial the database will be for patients and the public, due to challenges of implementation and the short time that the database has been available (Hirsch, 2008; Zarin et al., 2007).
REFERENCES
Abstracts of the NIH-FDA conference “Biomarkers and Surrogate Endpoints: Advancing Clinical Research and Applications.” 1998. Disease Markers 14(4):187-334.
Barker, A. 2006. Oncology Biomarker Qualification Initiative: NCI-FDA-CMS collaboration to speed development of cancer therapies. Presentation to the National Cancer Advisory Board June 14, 2006. Bethesda, MD.
Barker, A., C. C. Siggman, J. K. Kelloff, N. M. Hylton, D. A. Berry, and L. J. Esserman. 2009. I-SPY 2: An Adaptive Breast Cancer Trial Design in the Setting of Neoadjuvant Chemotherapy. Clinical Pharmacology and Therapeutics 86(1):97–100.
Borra, S. 2006. Consumer perspectives on food labels. American Journal of Clinical Nutrition 83(5):1235S–1235S.
C-Path (Critical Path Institute). 2008. Predictive Safety Testing Consortium (PSTC). http://www.c-path.org/pstc.cfm (accessed September 28, 2009).
C-Path. 2010a. Sharing knowledge that leads to innovation. http://www.c-path.org/programs.cfm (accessed March 13, 2010).
C-Path. 2010b. Coalition Against Major Diseases. http://www.c-path.org/CAMD.cfm (accessed March 13, 2010).
CFSAN (Center for Food Safety and Applied Nutrition). 2005. Questions and answers: Qualified health claims in food labeling. http://www.cfsan.fda.gov/~dms/qhc-qa.html (accessed March 17, 2009).
CFSAN. 2007. Experimental study of health claims on food packages: Preliminary topline frequency report. http://www.cfsan.fda.gov/~comm/crnutri4.html (accessed March 26, 2009).
CFSAN. 2008. A food labeling guide. http://www.cfsan.fda.gov/~dms/2lg-8.html#health (accessed March 26, 2009).
CFSAN. 2009. Guidance for industry: Evidence-based review system for the scientific evaluation of health claims. http://www.cfsan.fda.gov/~dms/hclmgui6.html (accessed February 25, 2009).
Coca-Cola. 2009. 2008 Annual report on form 10-K. http://www.thecoca-colacompany.com/investors/form_10K_2008.html (accessed September 30, 2009).
Committee on Science. 1998. Unlocking our future: Toward a new national science policy. Washington, DC: Committee on Science, U.S. House of Representatives.
Corbett Dooren, J. 2009. Cheerios’ health claims break rules, FDA says. The Wall Street Journal, May 13.
CSPI (Center for Science in the Public Interest). 2005. Food watchdog group announces litigation initiative. http://www.cspinet.org/new/200505031.html (accessed March 26, 2009).
CSPI. 2007. Quaker agrees to tone down exaggerated health claims on oatmeal. http://cspinet.org/new/200704171.html (accessed March 26, 2009).
CSRC (Cardiac Safety Research Consortium). 2010. Cardiac safety: Background. http://www.cardiac-safety.org/background (accessed March 14, 2010).
Curt, G. 2009. Step change in safe harbors: Public–private partnerships. The Oncologist 14(4):308–310.
Dhruva, S. S., L. A. Bero, and R. F. Redberg. 2009. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. Journal of the American Medical Association 302(24):2679–2685.
Drazen, J. M., and A. J. J. Wood. 2005. Trial registration report card. New England Journal of Medicine 353(26):2809–2811.
Drazen, J. M., and A. J. J. Wood. 2006. Clinical trials report card. New England Journal of Medicine 354(13):1428–1429.
Drummond, R. 2004. Trial registration: A great idea switches from ignored to irresistible. Journal of the American Medical Association 292(11):1359–1362.
FDA (Food and Drug Administration). 2007. Dietary supplement current good manufacturing practices (CGMPs) and interim final rule (IFL) facts. http://www.fda.gov/Food/DietarySupplements/GuidanceComplianceRegulatoryInformation/RegulationsLaws/ucm110858.htm (accessed March 2, 2010).
FDA. 2008a. FDA’s mission statement. http://www.fda.gov/opacom/morechoices/mission .html (accessed December 10, 2008).
FDA. 2008b. FDA, European Medicines Agency to consider additional test results when assessing new drug safety. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116911.htm (accessed November 23, 2009).
FDA. 2009a. FDAAA implementation—Highlights two years after enactment. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FoodandDrugAdministrationAmendmentsActof2007/ucm184271.htm (accessed November 20, 2009).
FDA. 2009b. FDAAA implementation—Highlights two years after enactment. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FoodandDrugAdministrationAmendmentsActof2007/ucm184271.htm (accessed November 17, 2009).
FDA. 2009c. Food and Drug Administration Amendments Act (FDAAA) of 2007. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FoodandDrugAdministrationAmendmentsActof2007/default.htm (accessed November 17, 2009).
FDA. 2009d. Overview of dietary supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/ucm110417.htm#what (accessed November 6, 2009).
FDA. 2009e. Regulatory information: Legislation. http://www.fda.gov/RegulatoryInformation/Legislation/default.htm (accessed November 5, 2009).
FDA. 2009f. Significant amendments to the FD&C Act. http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/default.htm (accessed November 17, 2009).
FDA. 2009g. Memorandum of understanding between the Food and Drug Administration and the National Cancer Institute and the Center for Medicare and Medicaid Services for the FDA/ NCI/CMS Oncology Biomarker Qualification Initiative. http://www.fda.gov/AboutFDA/PartnershipsCollaborations/MemorandaofUnderstandingMOUs/DomesticMOUs/ucm115681.htm (accessed March 14, 2010).
FDA. 2009h. MedWatchPlus/FAERS. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/SpotlightonCPIProjects/ucm083295.htm (accessed March 14, 2010).
FDA. 2010a. FDA calls on food companies to correct labeling violations; FDA commissioner issues an open letter to the industry. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm202814.htm (accessed March 13, 2010).
FDA. 2010b. FDA’s Critical Path Initiative: Transforming the way FDA-regulated products are developed, evaluated, manufactured and used. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/ucm076689.htm (accessed March 13, 2010).
FDA. 2010c. FDA’s Sentinel Initiative. http://www.fda.gov/Safety/FDAsSentinelInitiative/default.htm (accessed March 14, 2010).
FNIH (Foundation for the National Institutes of Health). 2007. The Biomarkers Consortium. http://www.biomarkersconsortium.org/ (accessed March 13, 2010).
FNIH. 2009. Foundation for the National Institutes of Health. http://www.fnih.org/ (accessed November 17, 2009).
FNIH. 2010. Observational Medical Outcomes Partnership. http://omop.fnih.org/node/22 (accessed March 15, 2010).
Fontanarosa, P. B., D. Rennie, and C. D. DeAngelis. 2004. Postmarketing surveillance—Lack of vigilance, lack of trust. Journal of the American Medical Association 292(21):2647– 2650.
GAO (Government Accountability Office). 2006. Drug safety: Improvement needed in FDA’s postmarket decision-making and oversight process. Washington, DC: GAO.
GAO. 2009a. Dietary supplements: FDA should take further actions to improve oversight and consumer understanding. Washington, DC: GAO.
GAO. 2009b. Food and Drug Administration: FDA faces challenges meeting its growing medical product responsibilities and should develop complete estimates of its resource needs. Report No. GAO-09-581. Washington, DC: GAO.
GAO. 2009c. Medical devices: Shortcomings in FDA’s premarket review, postmarket surveillance, and inspections of device manufacturing establishments. http://www.gao.gov/new.items/d09370t.pdf (accessed November 22, 2009).
GAO. 2009d. New drug approval: FDA needs to enhance its oversight of drugs approved on the basis of surrogate endpoints. Washington, DC: GAO.
Grocery-aisle gotchas. 2009. Consumer Reports on Health 21(2):8–9.
Hamburg, M. A. 2010. Open letter to industry from Dr. Hamburg. http://www.fda.gov/Food/LabelingNutrition/ucm202733.htm (accessed March 13, 2010).
Hamburg, M. A., and J. M. Sharfstein. 2009. The FDA as a public health agency. The New England Journal of Medicine 360(24):2493–2495.
Heimbach, J. T., and R. C. Stokes. 1982. Nutrition labeling and public health: Survey of American Institute of Nutrition members, food industry, and consumers. American Journal of Clinical Nutrition 36(4):700–708.
Hirsch, L. 2008. Trial registration and results disclosure: Impact of US legislation on sponsors, investigators, and medical journal editors. Current Medical Research and Opinion 24(6):1683–1689.
Holden, A. International SAE Consortium, LTD. 2010. An international industrial biomedical consortium researching the genetic basis of drug related serious adverse events. Presentation at Second Annual Sentinel Initiative Workshop. Washington, DC: The Brookings Institution.
Hooker, N. H., and R. Teratanavat. 2008. Dissecting qualified health claims: Evidence from experimental studies. Critical Reviews in Food Science and Nutrition 48(2):160–176.
IFICF (International Food Information Council Foundation). 2005. Qualified health claims consumer research project executive summary. http://www.ific.org/research/qualhealth claimsres.cfm (accessed March 17, 2009).
IFT (Institute of Food Technologists). 2005. Functional foods: Opportunities and challenges. Chicago, IL: Institute of Food Technologists.
IOM (Institute of Medicine). 1998. Ensuring safe food: From production to consumption. Washington, DC: National Academy Press.
IOM. 2005. Safe medical devices for children. Washington, DC: The National Academies Press.
IOM. 2007a. Cancer biomarkers: The promises and challenges of improving detection and treatment. Washington, DC: The National Academies Press.
IOM. 2007b. The future of drug safety: Promoting the health of the public. Washington, DC: The National Academies Press.
IOM. 2009. Beyond the HIPAA privacy rule: Enhancing privacy, improving health through research. Washington, DC: The National Academies Press.
iSAEC (International Serious Adverse Events Consortium). 2010. International SAE Consortium. http://www.saeconsortium.org/ (accessed March 14, 2010).
Johnston, L., H. Reiss Reynolds, D. B. Hunninghake, K. Schultz, and B. Westereng. 1998. Cholesterol-lowering benefits of a whole grain oat ready-to-eat cereal. Nutrition in Clinical Care 1(1):6–12.
Kapsak, W. R., D. Schmidt, N. M. Childs, J. Meunier, and C. White. 2008. Consumer perceptions of graded, graphic and text label presentations for qualified health claims. Critical Reviews in Food Science and Nutrition 48(3):248–256.
Kinders, R. J., M. Hollingshead, S. Khin, L. Rubinstein, J. E. Tomaszewski, J. H. Doroshow, R. E. Parchment, and National Cancer Institute Phase 0 Clinical Trials Team. 2008. Pre-clinical modeling of a phase 0 clinical trial: Qualification of a pharmacodynamic assay of poly(ADP-ribose)polymerase in tumor biopsies of mouse xenografts. Clinical Cancer Research 14(21):6877–6885.
Kramer, D. B., E. Mallis, B. D. Zuckerman, B. A. Zimmerman, and W. H. Maisel. 2009. Premarket clinical evaluation of novel cardiovascular devices: Quality analysis of premarket clinical studies submitted to the Food and Drug Administration 2000-2007. American Journal of Therapeutics 17(1)2–7.
Kummar, S., R. Kinders, M. E. Gutierrez, L. Rubinstein, R. E. Parchment, L. R. Philips, J. Ji, A. Monks, J. A. Low, A. Chen, A. J. Murgo, J. Collins, S. M. Steinberg, H. Eliopoulos, V. L. Giranda, G. Gordon, L. Helman, R. Wiltrout, J. E. Tomaszewski, and J. H. Doroshow. 2009. Phase 0 clinical trial of the poly(ADP-ribose)polymerase inhibitor ABT-888 in patients with advanced malignancies. Journal of Clinical Oncology 27(16):2705–2711.
Levy, A. S., O. Mathews, M. Stephenson, J. E. Tenney, and R. E. Schucker. 1985. The impact of a nutrition information program on food purchases. Journal of Public Policy & Marketing 4(1):1–13.
Levy, A. S., S. B. Fein, and R. E. Schucker. 1992. More effective nutrition label formats are not necessarily preferred. Journal of the American Dietetic Association 92(10):1230–1234.
Levy, A. S., S. B. Fein, and R. E. Schucker. 1996. Performance characteristics of seven nutrition label formats. Journal of Public Policy & Marketing 15(1):1–15.
Lewis, C. J., and E. A. Yetley. 1992. Focus group sessions on formats on nutrition labels. Journal of the American Dietetic Association 92(1):62–66.
Mazis, M. B., and M. A. Raymond. 1997. Consumer perceptions of health claims in advertisements and on food labels. Journal of Consumer Affairs 31(1):10–26.
Meier, B. 2009. F.D.A. to seek new standards on human test data. The New York Times, December 30, 2009.
NCI (National Cancer Institute). 2006. New federal health initiative to improve cancer therapy. Patients will benefit from rapid development and delivery of new cancer treatments. http://www.cancer.gov/newscenter/pressreleases/OBQI (accessed November 17, 2009).
Office of Budget. 2009. Fiscal Year 2010 Budget in Brief. http://www.hhs.gov/asrt/ob/docbudget/2010budgetinbriefa.html (accessed September 30, 2009).
Office of Inspector General. 2006. FDA’s monitoring of postmarketing study commitments. Washington, DC: Department of Health and Human Services.
Platt, R. 2010. FDA’s Mini-Sentinel Program. Presentation at the Second Annual Sentinel Initiative Workshop. Washington, DC: Brookings Institution.
Platt, R. M. Wilson, A. Chan, J. S. Benner, J. Marchibroda, and M. McClellan. 2009. The new Sentinel Network—Improving the evidence of medical-product safety. New England Journal of Medicine 361(645-647).
Prescription for harm: The decline in FDA enforcement activity. 2006. Washington, DC: U.S. House of Representatives Committee on Government Reform—Minority Staff Special Investigations Division.
Psaty, B. M., C. D. Furberg, W. A. Ray, and N. S. Weiss. 2004. Potential for conflict of interest in the evaluation of suspected adverse drug reactions: Use of cerivastatin and risk of rhabdomyolysis. Journal of the American Medical Association 292(21):2622–2631.
Roe, B., A. S. Levy, and B. M. Derby. 1999. The impact of health claims on consumer search and product evaluation outcomes: Results from FDA experimental data. Journal of Public Policy & Marketing 18(1):89–105.
Ryan, P. 2010. Observational Medical Outcomes Partnership: Overview and lessons learned. Presentation at the Sentinel Initiative Public Workshop, Washington, DC: The Brookings Institution.
Schneeman, B. 2007. FDA’s review of scientific evidence for health claims. Journal of Nutrition 137(2):493–494.
Subcommittee on Science and Technology. 2007. FDA science and mission at risk. FDA Science Board: Rockville, MD.
Taylor, C. L., and V. L. Wilkening. 2008. How the nutrition food label was developed, part 2: The purpose and promise of nutrition claims. Journal of the American Dietetic Association 108(4):618–623.
Wagner, J. A., E. C. Wright, M. M. Ennis, M. Prince, J. Kochan, D.J.R. Nunez, B. Schneider, M.-D. Wang, Y. Chen, S. Ghosh, B.J. Musser, and M.T. Vassileva. 2009. Utility of adinopectin as a biomarker predictive of glycemic efficacy is demonstrated by collaborative pooling of data from clinical trials conducted by multiple sponsors. Clinical Pharmacology & Therapeutics 86(6):619-625.
Williams, P. 2005. Consumer understanding and use of health claims for foods. Nutrition Reviews 63(7):256–264.
Wood, A. J. 2008. Playing “kick the FDA”—Risk-free to players but hazardous to public health. New England Journal of Medicine 358(17):1774–1775.
Yang, S. X., S. Kummar, S. M. Steinberg, A. J. Murgo, M. Gutierrez, L. Rubinstein, D. Nguyen, G. Kaur, A. P. Chen, V. L. Giranda, J. E. Tomaszewski, J. H. Doroshow, and The National Cancer Institute Phase 0 Working Group. 2009. Immunohistochemical detection of poly(ADP-ribose)polymerase inhibition by ABT-888 in patients with refractory solid tumors and lymphomas. Cancer Biology and Therapy 8(21)2004–2009.
Yetley, E. A. 2007. Science in the regulatory setting: A challenging but incompatible mix? Novartis Foundation Symposium 282:59–68; discussion 69–76, 212–218.
Zarin, D. A., N. C. Ide, T. Tse, W. R. Harlan, J. C. West, and D. A. Lindberg. 2007. Issues in the registration of clinical trials. Journal of the American Medical Association 297(19):2112– 2120.






































