
7
Medical Devices: Research and Development for Rare Diseases
The Vertical Expandable Prosthetic Titanium Rib (VEPTR), a device that has saved the lives of 300 infants and young children who otherwise would have died from lack of breath [thoracic insufficiency syndrome], has been approved by the U.S. Food and Drug Administration (FDA)… . The titanium rib is curved like a ribcage and has holes that allow the surgeons to expand the device in outpatient surgery every six months. The rib is implanted in infants as young as 6 months and in teenagers until skeletal maturity, typically age 14 in girls and age 16 in boys… . “It took 13 years to gain FDA approval because it took a long time to accumulate a lot of patients with rare diseases” Dr. [Robert] Campbell [the inventor] said.
UTHSCSA, 2004
For rare diseases, efforts to accelerate research and product development clearly focus on drugs and biological products. Devices and the need for devices are much less frequently mentioned in articles or conversations. When devices for rare conditions are discussed, it is generally in connection with pediatric populations.
To acknowledge the emphasis on drugs for rare diseases is not to imply that devices are not important for many people with rare medical conditions. Some people depend critically on devices targeted at distinctive features of their condition, for example, children who have received the implanted titanium rib described above. No pharmaceutical or biological product can provide the mechanical support afforded by this implant. Ge-
netic tests that are necessary for the diagnosis and treatment of certain rare conditions are, in certain cases, regulated as medical devices. In addition, people with rare conditions benefit from a large number of medical devices that are used generally in connection with complex surgery, anesthesia, respiratory support, nonsurgical cardiac procedures, administration of certain medications, diagnostic and therapeutic imaging of various kinds, laboratory testing, and other services.
Clinical studies of the titanium rib were supported under the orphan products grants program described in Chapter 3. Earlier, the National Organization for Rare Disorders provided a seed grant from its donated research funds. The two companies that were involved in manufacturing the device for research use participated out of interest in children’s health rather than expectations of profit (Campbell, 2007). After years of investigation and adaptation, the device was approved by the Food and Drug Administration (FDA) in 2004 through a Humanitarian Device Exemption (HDE). This process was established in the Safe Medical Devices Act of 1990 (P.L. 101-629) to provide incentives for the development of medical devices for small populations. Although medical devices for small populations are grouped under the label orphan products in the grants program created by the Orphan Drug Act and are within the charge of the Office of Orphan Product Development (OOPD), the term orphan medical device does not appear in legislative or regulatory language.
Regulatory requirements and product development pathways differ significantly for medical devices compared to drugs and biologics. Thus, this report devotes a separate chapter to medical device development, regulation, and reimbursement.
This chapter begins with a brief overview of important differences between devices and drugs. It then reviews device regulation and reimbursement with an emphasis on the HDE process and other policies or procedures that are potentially most relevant to complex, high-risk devices intended for small populations. This discussion is followed by an overview of the research and development process for complex devices and a discussion of barriers and opportunities for the development of devices for small populations. As this chapter highlights, the stringency of government regulation of devices is related to the risk presented by the device.
DIFFERENCES BETWEEN MEDICAL DEVICES AND DRUGS
Compared to pharmaceuticals, medical devices are an extremely diverse group of products. Some are as simple as adhesive bandages, tongue depressors, and plastic tubing. Others are complex, for example, various implanted cardiac and neurological devices, stair-walking wheelchairs, robotic surgical systems, and magnetic resonance imaging devices. In contrast
to single-molecule drugs, many complex devices involve a number of components that, together, form a system.1 Table 7-1 summarizes several additional differences between devices and drugs as they relate, in particular, to implants and other complex medical devices.
In addition to the cost-related differences noted in the table, companies that develop medical devices also have to consider other costs that may be only minor considerations for most pharmaceutical companies. One category of such costs involves the support and servicing of complex devices once they are released into the market. Depending on the device, highly skilled company personnel may provide training to physicians, clinical staff, and patients (and their families) on the proper use of the device. Service technicians often must be available promptly in case device-related problems arise. Companies must also consider potential obligations to patients if a decision is made at some point to discontinue the device.2
As is true of its products, the medical device industry is likewise quite variable. Some companies are large and have diverse product lines and substantial resources to devote to product development and interactions with government regulators. Compared to the drug industry, a larger proportion of device firms are small, focused on single products and narrow market segments, and limited in their resources (see Gelijns et al., 2006; Linehan et al., 2007).
Entrepreneurs at small start-up companies develop many innovative medical devices, including devices that address needs of small patient populations. Company motivations for taking the start-up path to market vary. In some cases, those involved may see the approach as a focused way to address an unmet need and contribute to society without having to navigate the decision-making processes of a large, complex company. In some cases, the projected business opportunity is too small or too risky to be worth attention from an existing company but is still attractive enough to attract venture capitalists or a small group of entrepreneurs. In exchange for partial ownership of the start-up company, angel investors and venture
TABLE 7-1 Complex Medical Devices Tend to Differ from Drugs
|
Complex Medical Device |
Small-Molecule Compound |
|
Physical, engineering-based object (or set of components) |
Chemical formulation |
|
Direct mechanism of action and, usually, readily apparent, near-term response |
Indirect biochemical mechanism of action via blood, other body fluids, or tissue diffusion |
|
Site- or organ-specific therapy |
Usually systemic treatment |
|
Patient responses to therapy generally similar and not dependent on dose response |
Patient responses variable (benefits and adverse effects) and dose dependent |
|
High initial product costs amortized over service life |
Costs for product accumulate over the course of treatment |
|
Application often requires professional expertise (e.g., surgical implantation); patient use might involve complex instructions |
Application or use is often simple and patient controlled (e.g., taking a pill) |
|
Continuing product refinement and short product life cycle that may improve effectiveness and reduce costs |
Product (basic molecule) not modified, long product life cycle |
|
Moderate to high development cost |
High development cost |
|
Few basic patents, many incremental patents and products |
Basic patent, fewer incremental patents or products |
|
SOURCES: Adapted from Linehan et al., 2007; Citron, 2008; see also Feigal et al., 2003. |
|
capitalists often provide the financing needed to bring nascent innovations to the market. In addition to infusions of capital, venture capitalists who have worked with other new companies may provide management expertise and strategic advice to guide the managers of a start-up company.
As discussed further below, the processes of device development and refinement also differ in significant ways from the processes that characterize the development of drugs and biologics (see generally Linehan et al., 2007; Pietzsch, 2009; Zenios et al., 2010). Because medical device companies are often engaged in a continuous process of product refinement and innovation, patents and similar protections may be less important as a source of competitive advantage for device companies than they are for drug companies. As discussed in Chapter 3, once a new drug is approved by FDA, a pharmaceutical company will have marketplace exclusivity for a specific formulation for a period of time and may also receive patent-term restoration that extends the remaining patent life of the drug. In contrast,
several device companies may compete simultaneously in the marketplace with devices for the same indication that differ in only limited respects. This might be because the devices are not patented or because manufacturers have been able to design around the patents that protect a particular competitor’s devices. Consequently, although FDA-approved devices are eligible for patent-term restoration, patents may not be as useful in protecting devices from competition as they are for pharmaceutical products. Even in instances when patents could provide an element of protection from market competition, the patent holder may elect to license its patents to one or more competitors in exchange for royalties or to cross-license patents in order to acquire access to patents held by a competitor. Nevertheless, medical device companies are aggressive in defending their intellectual property from infringements by competitors (Budd and Liebman, 2009).
REGULATION OF MEDICAL DEVICES
Basic Framework of Medical Device Regulation
Although the Federal Food, Drug, and Cosmetic Act of 1938 mentioned therapeutic medical devices, devices were a relatively inconsequential component of FDA’s jurisdiction. The statute specified that devices be adequately labeled and provide adequate instructions for use but did not give FDA premarket regulatory authority over devices. In the 1970s, following widely publicized problems with the Dalkon Shield (an intrauterine contraceptive device) (Hubacher, 2002), Congress turned to the regulation of medical devices with the Medical Device Amendments of 1976 (P.L. 94-295). The legislation created the basic framework for device regulation. As defined by statute (21 USC 321(h)), a device is
an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
-
recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them,
-
intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
-
intended to affect the structure or any function of the body of man or other animals, and
which does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes.
Within FDA, the Center for Devices and Radiological Health (CDRH) regulates most medical devices. The Center for Biologics Evaluation and Research regulates devices related to blood and cellular products such as blood collection, screening, and processing devices. The OOPD has roles in designation of devices eligible for HDE approval and in awarding product development grants, which are available for device as well as drug development.
Device Classification and Regulation
A fundamental element of the 1976 law was a risk-related device classification scheme that forms the basis for risk-related regulatory requirements. To simplify, the law designated devices of lowest risk and relatively little complexity as Class I; devices of moderate risk and greater complexity as Class II; and devices that support or sustain life or otherwise present a high risk to the patient as Class III.3
In general, Class I and II devices have substantially equivalent predecessor or “predicate” devices that are already on the market. Some new devices may be classified automatically as Class III devices because they have no such predicate device. FDA may reclassify such devices as Class II devices based on an analysis of the risk they present. For example, such a reclassification was requested for the first device available to screen newborn infants for inherited abnormalities of amino acids and deficiencies in certain enzymes (Lloyd, 2004).4
Regardless of its complexity, any device can present potential harms to patients if it is misused, mislabeled or poorly labeled, badly designed, poorly manufactured, or misrepresented. Thus, the regulatory framework created by Congress covers all classes of devices and extends, in some cases, to requirements for sponsors to conduct postmarket studies to collect data about safety and effectiveness after a device is approved for marketing.
For Class I devices, manufacturers generally must register with FDA and follow FDA’s quality system regulations, including adherence to good manufacturing practices. These devices are usually not subject to premarket notification or review.
Manufacturers of Class II devices usually must get FDA clearance of a “510(k) notification” (named for the relevant section of the law) to legally market these devices. The process requires the submission of considerable technical information and sometimes animal study data related to safety and performance characteristics of the new device, in order to demonstrate its “substantial equivalence” to the predicate device. The 510(k) notifica-
tion typically does not require clinical data unless the technology for a new device differs from that of the predicate device and clinical data are necessary to evaluate the potential impact of this difference on safe and effective performance. Clinical data are included in approximately 10 percent of 510(k) notifications (Tillman and Gardner, 2004; Rosecrans, 2010). Both a CDRH working group and an Institute of Medicine (IOM) committee (as requested by FDA) are evaluating aspects of the 510(k) process. The CDRH group issued its preliminary report in August 2010 (CDRH, 2010a).
For Class III devices, which account for a small proportion of all legally marketed medical devices, manufacturers must submit premarket approval (PMA) applications and provide data from clinical trials to demonstrate reasonable assurance that a device is safe and effective (what this report terms efficacy) for the intended use in the intended patient population. Examples of Class III devices include implanted devices such as the titanium rib, some diagnostic test kits, and certain surgical sealants. Securing FDA approval of such a device is usually complex, costly, and time-consuming, taking on the order of several years. The cost will vary depending on the complexity of the device and the kinds of nonclinical and clinical data that the sponsor must submit to demonstrate safety and efficacy.
This report focuses on complex devices intended specifically to treat complex rare conditions. Most will be Class III devices and thus will require formal authorization by FDA, usually through the PMA process or, in some cases, the 510(k) process.
For qualifying devices intended for a small population, approval can also come through the HDE process described below. The committee is not aware of any analysis that attempts to catalog devices that have been cleared under the 510(k) process or approved under a PMA specifically for the treatment of rare conditions defined according to the Orphan Drug Act (i.e., conditions affecting fewer than 200,000 individuals). It has found examples of such devices. For example, for the rare eye condition keratoconus, CDRH has cleared devices under the 510(k) process (K992466 and K024164) and also approved a different type of device for the condition under an HDE (H040002). At least one HDE-approved device, Bioglue, was subsequently approved for broader indications under a PMA (P010003).
For devices that are designated as “significant risk devices” because they have the potential to cause serious harm to research participants, manufacturers must secure an Investigational Device Exemption (IDE) before they can conduct clinical studies in humans with the devices.5 Similar to the Investigational New Drug application, an IDE application must include
data about preclinical studies and any available clinical information. It must also provide a description of the proposed research and analysis strategy. An IDE may prompt extensive discussions and negotiations between the manufacturer and FDA to arrive at agreement on a research plan that will provide data of acceptable quality to support FDA approval of the device. As described in one review of the process, the “first and arguably most important step in this process is the pre-IDE meeting, in which the company, often accompanied by the lead clinical investigator(s), meets with FDA/ CDRH to present data about the device, its clinical development program, and its intended use after approval” (Kaplan et al., 2004, p. 3071).
As is the case for pharmaceuticals, FDA may approve medical devices with requirements for postmarketing studies, including clinical studies. For example, when CDRH approved a transcatheter pulmonary valve system under an HDE, it required two postapproval clinical studies (Tillman, 2010). CDRH now tracks the status of postapproval studies required after January 1, 2005, and posts tracking information on a public web page.
Diagnostic Devices, In Vitro Devices, and Genetic Tests
FDA regulates a range of diagnostic devices under the procedures described above. Diagnostic devices include such diverse items as blood pressure cuffs, vision evaluation instruments, cardiac monitors, and sophisticated imaging equipment. Based on their complexity, diagnostic devices are generally assigned to one of the three classes discussed above and regulated accordingly. CDRH has approved HDEs for three diagnostic testing devices.6
Diagnostic devices also include an array of products known as in vitro diagnostic devices, which “are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions” (21 CFR 809.3(a)). FDA regulates in vitro diagnostic devices that are developed and sold by device manufacturers as test kits. In vitro diagnostic devices include genetic and other tests that are important in diagnosing many rare diseases.7
In addition to using in vitro diagnostic test kits to perform diagnostic testing, some clinical laboratories develop their own in-house assays, known as laboratory-developed tests. These laboratory-developed tests are currently regulated under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) and state laws (Maloney, 2010). With rare exceptions, laboratory-developed tests usually are not regulated by FDA.8 Recently, however, FDA announced its intention to regulate all laboratory-developed tests as medical devices, as discussed below.
FDA regulations do, however, require that a clinical laboratory that develops a test using an analyte-specific reagent must disclose its regulatory status and must add a statement on test reports that the test has not been cleared or approved by FDA (21 CFR 809.30(e)). Analyte-specific reagents (which include polyclonal and monoclonal antibodies, specific receptor proteins, nucleic acid sequences, and similar reagents) are the building blocks that clinical laboratories use to develop in-house assays. Also, although laboratory-developed tests themselves are not usually regulated by FDA, analyte-specific reagents are regulated as “restricted devices.” Manufacturers cannot make any claim of clinical or diagnostic effectiveness for an analyte-specific reagent and can only describe what substance it will identify. If a manufacturer combines analyte-specific reagents into a kit, or otherwise offers them for sale together, then the product must be approved as a medical device.
Because most genetic tests are available only as laboratory-developed tests, they are not regulated by FDA (Huang and Javitt, 2008). In a report on the regulation of genetic tests prepared by the Secretary’s Advisory Committee on Genetics, Health, and Society, the group identified shortcomings in several areas, including regulations governing clinical laboratory quality and “oversight of the clinical validity of genetic tests” (SACGHS, 2008, p. 191).
In the past several years, various groups have recommended that FDA should regulate either all genetic tests or all laboratory-developed tests under the medical device authorities (Mansfield and Tezak, 2010). In June 2010, CDRH announced a public meeting and requested comments on issues related to the regulation of laboratory-developed tests (75 Fed. Reg. 34463). Although the agency has indicated that it plans to regulate some of these tests as medical devices, the specifics and priorities have yet to be decided. In noting the challenges of encouraging innovation while assuring
the safety and efficacy of laboratory-developed tests, the CDRH announcement specifically cited tests for rare conditions.
Another area of regulatory complexity is co-development of a drug and a companion diagnostic. An example is a diagnostic test kit to assess whether a breast cancer patient has a gene mutation that is targeted by the drug trastuzumab (Herceptin). FDA held up approval of the drug until an approved in vitro diagnostic could be substituted for the laboratory-developed test that was initially used in clinical trials. It approved both the drug and the diagnostic in 1998 (Madsen, 2004). After a 2005 concept paper on the topic generated considerable criticism (see, e.g., PMC, 2009), the FDA Commissioner indicated that a new draft guidance document would be published in 2010 and would reflect public comments and scientific and other developments (Hamburg, 2009; Ray, 2010). In the meantime, FDA has been applying a case-by-case approach to regulation of companion diagnostics (Carver, 2010).
Combination Products
Some medical products combine a medical device and a drug or biologic. Examples include the drug-eluting coronary stent (which adds a drug coating to a metal stent in order to reduce the risk of reocclusion of the coronary artery) and the fentanyl patch (which delivers the drug through the skin). Combinations can take several different forms. For a product such as the drug-eluting stent, the device and drug components are truly combined into a single entity. Two items that are physically distinct but packaged together also qualify as a combination product. The category can also cover a product such as a drug that is packaged separately but is labeled as being for use only with a specific device or type of device (such as a specific diagnostic test).9
At least one combination product has been approved by CDRH through the HDE process (OP-1 Putty under H020008).10 As discussed below, the
different incentives for the development of orphan drugs and for the development of devices for small populations theoretically could complicate collaboration on combination products for small populations.
Alternate Approval Route for Medical Devices for Small Populations
As is true for companies that manufacture drugs and biologics, device companies naturally seek business opportunities in markets of sufficient size and profitability to warrant the investment risk. Particularly if FDA requires extensive clinical data for approval of the device, companies may be discouraged from pursuing devices for small markets by the expense and practical challenges of conducting acceptable trials to demonstrate safety and effectiveness.
The Safe Medical Devices Act of 1990 authorized the Humanitarian Device Exemption to encourage the development and introduction of complex device technologies to meet the needs of small patient populations. Although neither the text nor the title of the 1990 law uses the term “rare disease” or “orphan product,” the purpose is broadly similar to the purpose of the Orphan Drug Act. The specifics vary in part because the details of device regulation differ and in part because the incentives (particularly market exclusivity) that were viewed as important for drug manufacturers were viewed as less meaningful for device manufacturers.
An HDE application is the same as a PMA application except that it need not include evidence of effectiveness, a characteristic that also distinguishes the requirements for an HDE from the requirements for FDA approval of an orphan drug. The HDE application must, however, “contain sufficient information for FDA to determine that the device does not pose an unreasonable or significant risk of illness or injury, and that the probable benefit to health outweighs the risk of injury or illness from its use” (CDRH, 2009, unpaged).
To be eligible for an HDE, a manufacturer must first request that the device be designated by the OOPD as a Humanitarian Use Device (HUD). A HUD is a “medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the United States per year” (21 CFR 814.102(a)(5)). (If a device is for diagnostic purposes, the documentation in an HDE application must demonstrate that fewer than 4,000 patients per year would be subjected to diagnosis by the device in the United States.) The statutory language has caused some confusion about whether it refers to incidence or
prevalence, but FDA provided the following interpretation in the preamble to the HDE regulations issued in June 1996 (61 Fed. Reg. at 33233):
The agency believes that defining the criteria on a per year basis is consistent with the intent of section 520(m) of the act …, whereas a point prevalence definition would be considerably more restrictive and provide less of an incentive for the development of such devices. In response to comments, FDA also has added “or is manifested” to the definition of a HUD in order to establish that HUD designation may be appropriate in cases where more than 4,000 people have the disease but fewer than 4,000 manifest the condition.
CDRH now interprets the 4,000-individual restriction to allow a company to ship up to 4,000 devices a year (or a higher number if the data show that patients need more than one device within a year). The shipment limit means that substantially expanded use of a device either within the approved indication or off-label is controlled in a way that does not apply for orphan drugs. Sponsors must report data to CDRH on a periodic basis to support the continued appropriateness of the HUD designation. The agency may ask companies to withdraw an HDE if evidence indicates that the population criterion is no longer met. (In contrast, a designated orphan drug may be approved with exclusivity even if the affected population has, during the time between designation and approval, exceeded 200,000.)
CDRH will not approve an HDE if a comparable device has been cleared or approved for the same indication through either the 510(k) notification process or the PMA approval process under the procedures described in the preceding section.11 It will, however, consider an HDE application if a comparable device has been approved under another HDE or if a comparable device is being studied under an IDE (CDRH, 2009).
Comparison of HDE and Orphan Drug Incentives
Table 7-2 summarizes several ways in which the provisions for HDEs differ from the incentives for the development of orphan drugs. In contrast to the orphan drug policy, the HDE policy has no provisions for market exclusivity. This difference reflects the process of ongoing device refinement described earlier and the less significant role of patent or patent-like protection in the medical device industry. Also in contrast to orphan drugs, Con-
TABLE 7-2 Incentive Comparison: Drugs or Biologics Versus Devices
|
Incentive |
Orphan Drug or Biologic |
Humanitarian Use Device |
|
Product development assistance |
Tax credit for qualified clinical testing expenses |
|
|
|
FDA orphan products grants |
FDA orphan products grants |
|
Market exclusivity |
7 years |
No equivalent |
|
Pricing discretion |
Sponsor sets selling price |
Limited to cost recovery Profits allowed for products for pediatric populations up to a specified annual limit |
|
Requirement for demonstration of safety or effectiveness (efficacy) |
Clinical evidence of safety and effectiveness (efficacy) similar to nonorphan products |
Evidence of safety and data showing that probable benefit exceeds risk |
|
Population size constraint |
Fewer than 200,000 people with the condition in the U.S. |
Fewer than 4,000 people per year in the U.S. (i.e., 4,000 devices shipped per year, unless a patient uses more than one device a year) |
|
Waiver of fees |
NDA submission fees waived for sponsors of orphan drugs |
Submission fees waived for sponsors of HDEs |
|
Other |
|
An HDE cannot be granted if a comparable device for the same intended use is available under usual approval procedures. More than one HDE can exist for same intended use |
gress did not authorize tax credits for clinical research for an HDE, which perhaps reflects the lack of a requirement for clinical evidence of efficacy.
For device manufacturers, the lack of a requirement for clinical evidence of a device’s effectiveness (efficacy) can be viewed as an incentive because clinical trials to support effectiveness claims are expensive and can take years to complete. In general, the costs of clinical trials that are usually needed to support a PMA make small markets unattractive or infeasible, particularly for start-ups and small device companies.
Another HDE incentive (and one that parallels that for orphan drugs) is the waiver of the filing fees normally required under the Medical Device User Fee Act (more than $200,000 per PMA application for FY 2010). In addition, the time period specified for regulatory review of an application
is shorter for HDEs (including the time for the HUD designation step) than for regular premarket approval applications.
Like developers of orphan drugs, developers of devices are also eligible for orphan products grants. Seven devices for which grants were awarded have subsequently been approved, five through the HDE process and two through the PMA process (Linda C. Ulrich, M.D., Medical Officer, FDA Office of Orphan Product Development, April 26, 2010, personal communication).
At the same time that the HDE route to approval has some advantages compared to the PMA process, it also comes with a critical restriction in addition to the 4,000-unit limit per year. Specifically, sponsors are not permitted to make a profit on the sale of the HUD if the device is sold for more than $250. They can recover certain costs, for example, those related to research and development, manufacturing, and distribution.12 The sponsor must provide supporting financial documentation to FDA about the price it proposes to charge. Another complication is that even though the sponsor can charge for the device, the HDE device might not be purchased for use in clinical practice—usually within a hospital—if adequate third-party reimbursement is not available to cover an institution’s cost to purchase, as discussed in a later section of this chapter.
One unique feature of the HDE policy is the requirement that use of an HDE device requires approval by an institutional review board (IRB) at the institution where the device is to be used. A clinician can usually request IRB approval in advance for several patients so that emergency procedures do not need to be invoked.
The primary responsibility of IRBs is to protect human research participants through review of proposed research. Their role in approving the use of an HDE device is an anomaly and a potential source of confusion because the purpose in question is not research on the device but use of the device in clinical care. The task of securing IRB approval is often a difficult, costly, and sequential (institution-by-institution) task for the HDE sponsor. In addition, marketing of the device to individual centers (which must seek IRB approval) may be more difficult in the absence of the usual FDA premarket approval. The requirement for IRB approval thus is potentially another factor that may discourage companies from developing products for small markets under HDE procedures.
The requirement for IRB approval and monitoring also creates complexities for IRBs. IRBs are charged with the review of research involving human subjects, but an HDE application does not relate to the conduct of research. An HDE holder may, however, conduct research on the device without an IDE if the study involves the approved indications, but IRB approval is still required. This aspect of HDE policy may add to confusion for IRBs and sponsors. A survey of IRB chairs in 2008 (with an 18 percent response rate) found that half reported an HDE review within the preceding 5 years and that many were confused about the process (Gordon, 2008).
In 2008, FDA issued draft guidance for HDE holders, IRBs, clinical investigators, and FDA staff in the form of questions and answers. In 2010, it issued a final guidance document (CDRH-CBER, 2010). Although this guidance is helpful, the process and the guidance are still confusing for device companies and IRBs. For example, the guidance states that the local IRB can defer to another IRB but does not explain whether it transfers all of its obligations to the other IRB (including continuing review) or just the initial review. It would have been useful if the guidance had included a sample letter for such a deferral. To cite another shortcoming of the guidance, it states that IRBs will receive reports of adverse events from the FDA Medical Device Reporting system, but it does not explain what IRBs should do with the information.
As noted above, the different incentives for orphan drugs and HDE devices could potentially create difficulties for an innovative combination product in some situations. If the testing of safety and effectiveness for an innovative drug required simultaneous use of the innovative device, that clinical testing should provide evidence to support both approval of the drug and clearance or approval of the device. For example, one company is simultaneously testing an intrathecal drug delivery device with a drug for Hunter syndrome; if testing shows safety and efficacy, the results should support regular FDA clearance or approval of the device (see description at http://clinicaltrials.gov/ct2/show/NCT00920647). (An intravenous formulation of the drug has orphan drug approval.)
If, however, a device were to be developed separately, for example, as an improved method to deliver an already approved orphan drug, it is possible that the different incentives for drugs and devices for small populations could be mismatched in a way that could discourage device companies from collaborating with drug companies on this type of combination product. (This issue could also arise as the development and regulation of companion diagnostic tests evolves, for example, when the companion diagnostic to predict patient responsiveness to a drug for a rare condition is not tested as part of the clinical trial of the drug itself [Swanson, 2009].) The committee did not find information on combination products that were discouraged or impeded because of the differences between the incentives
for orphan drug development and the HDE incentives, but it is theoretically possible and may require FDA response in the future.
HDE Approvals
The regulations implementing the HDE process were issued in June 1996 and became effective in October 1996. Between 1996 and the end of 2009, the OOPD received 232 requests for a HUD designation and granted 146 of these (Lewis, 2010). The first HDE was approved in February 1997. As of April 2010, CDRH had approved 50 therapeutic and diagnostic devices through the HDE process. Three HDEs have been withdrawn by the sponsors after FDA indicated that the patient population served by the device had grown to exceed the limits for HDE devices.13 Approximately two-thirds of HDE devices are implants, and HDEs have been most commonly approved for vascular, cardiac, neurological, and pediatric indications (Bernad, 2009).
Some HDEs are approved for use with patients who have a rare disease as such. For example, the titanium rib is approved for use with Jeune’s syndrome or other rare rib cage conditions. Mostly, however, HDE approvals have cited indications that involve a very severely ill subgroup of a larger patient population or a subgroup that has not benefited from usual therapy. (Orphan drug designations and approvals may likewise specify medically relevant subgroups.) This is illustrated in Box 7-1.
Incentives for the Development of Pediatric Medical Devices
Because children are generally a healthy population, companies often do not find it commercially feasible or attractive to develop devices specific to pediatric diseases or to develop smaller versions of adult devices for relatively small numbers of children who might benefit from them.14 For devices with indications for pediatric use, Congress acted in 2007 to modify
|
BOX 7-1 Examples of Devices Approved Under the Humanitarian Device Exemption The Spiration IBV is indicated to control prolonged air leaks of the lung, or significant air leaks that are likely to become prolonged air leaks, following lobectomy, segmentectomy, or lung volume reduction surgery. An air leak present on postoperative day 7 is considered prolonged unless present only during forced exhalation or cough. An air leak present on day 5 should be considered for treatment if it is (1) continuous, (2) present during normal inhalation phase of inspiration, or (3) present upon normal expiration and accompanied by subcutaneous emphysema or respiratory compromise. Approved October 24, 2008 (H060002). TAS Ecarin Clotting Time Test is used to determine the anticoagulant effect of recombinant hirudin (r-hirudin) during cardiopulmonary bypass in patients who have heparin-induced thrombocytopenia. Approved May 11, 2000 (H990012). Epicel (cultured epidermal autografts) is for use with patients who have deep dermal or full-thickness burns comprising a total body surface area of greater than or equal to 30 percent. It may be used in conjunction with split-thickness autografts or alone in patients for whom split-thickness autografts may not be an option due to the severity and extent of their burns. Approved October 25, 2007 (H990002). DeBakey VAD Child Left Ventricular Assist System is to provide temporary left-side mechanical circulatory support as a bridge to cardiac transplantation for pediatric patients (5-16 years old, with BSA ≥ 0.7 m2 and <1.5 m2) who are in NYHA Class IV end-stage heart failure, are refractory to medical therapy, and are (listed) candidates for cardiac transplantation. Approved February 25, 2004 (H030003). Abiocor Implantable Replacement Heart is indicated for use in severe biventricular end-stage heart disease patients who are not cardiac transplant candidates and who are less than 75 years old, require multiple inotropic support, are not treatable by left ventricular assist device (LVAD) destination therapy, and are not weanable from biventricular support if on such support. Approved September 5, 2006 (H040006). Activa Dystonia Therapy is for unilateral or bilateral stimulation of the internal globus pallidus (GPi) or the subthalamic nucleus (STN) to aid in the management of chronic, intractable (drug-refractory) primary dystonia, including generalized and/or segmental dystonia, hemidystonia, and cervical dystonia (torticollis) in patients 7 years of age or older. Approved April 15, 2003 (H020007). SOURCE: FDA listing of CDRH Humanitarian Device Exemptions. HDE approval numbers are in parentheses. |
the incentives associated with an HDE approval (P.L. 110-85). Notably, it removed the general restriction on profits. It also directed the Government Accountability Office (GAO) to assess by 2012 the effects of removing that restriction.
The law requires FDA to specify an annual distribution limit on the number of devices that can be sold at a profit (up to a maximum of 4,000). As described in draft agency guidance, the Pediatric Medical Device Safety and Improvement Act of 2007, this number “is determined by estimating the number of individuals (pediatric and adult patients) affected by the disease or condition and likely to use the device each year multiplied by the number of devices reasonably necessary to treat each individual. If the number calculated is less than or equal to 4,000, then this number is the ADN [annual distribution number]. If the number calculated is more than 4,000, then the ADN is 4,000 because in no case can the ADN exceed 4,000 devices. See section 520(m)(6)(A)(ii) of the Act.”
As of January 2010, CDRH has approved one HDE under the pediatric provisions. In the approval order for the Medtronic Melody transcatheter pulmonary valve (H080002), CDRH specified an annual distribution number of 2,996, which includes use with both children and adults,. The order includes no explanation of the number, but on its website, Medtronic, the device manufacturer, states that approximately 34,000 children are born each year with congenital heart disease, of which 20 percent are born with a malformation affecting blood flow between the heart and lungs (Medtronic, 2010). A subset of these infants will have a prosthetic conduit surgically implanted, and some of these devices will malfunction, which will require new surgery. The device is intended to extend the life of the malfunctioning conduit without open heart surgery.
Congress also created a grants program to promote the development of pediatric medical devices. OOPD announced the first awards to three pediatric device consortia in 2009 (OOPD, 2009). Box 7-2 describes the expectations for these consortia. One provision, which is similar to a recommendation in this report, specifies that consortia coordinate with companies and FDA to take approval or clearance processes and requirements into account.
Custom Devices
Perhaps the ultimate in devices for small populations is the custom device, which is not subject to 510(k) or PMA requirements. As described by a former FDA Commissioner, a custom medical device is an “example of individualized therapy … that is requested of the device manufacturer by a physician for a specific patient. A custom device is a one-of-a-kind device designed for an immediate need and for which the need is not likely
|
BOX 7-2 Expectations for Consortia to Stimulate Pediatric Device Development A consortium receiving a grant or contract under Section 305 will facilitate the development, production, and distribution of medical devices by
Each consortium will coordinate with the FDA Commissioner and device companies to facilitate applications for approval or clearance of devices labeled for pediatric use. Each consortium will coordinate with the NIH [National Institutes of Health]. SOURCE: From Pediatric Medical Device Safety and Improvement Act of 2007 (P.L. 110-85, section 305). |
to reoccur. In essence, a physician and a manufacturer collaborate to design a device for a specific circumstance. For such devices, it would be virtually impossible to conduct a clinical trial, and—assuming the device does meet all the criteria defining it as a custom device—the device would be exempt from premarket approval” (Henney, 2000).
FDA regulations specify several criteria that custom devices must meet. These include that the device (1) necessarily deviates from generally available devices or from a PMA requirement in order to comply with the order of an individual physician; (2) is not generally available to other physicians; (3) is not generally available in finished form for purchase or dispensing; and (4) is not offered for commercial distribution through labeling or advertising (21 CFR 812.3(b)). The agency has narrowly interpreted the custom device exemption and has brought enforcement actions against certain manufacturers who have sought to rely upon it.15
Although the committee did not examine the approval and use of custom devices or investigate possible concerns about these devices, the question arose whether a change in the custom device exemption might assist patients with rare conditions. Specifically, could protections for patients still be maintained if FDA were permitted to authorize the approval of a specific custom device for a very small group of patients (e.g., 5 or 10) who had the same rare problem? If the assessment of unmet needs recommended at the end of this chapter includes needs for custom devices, the assessment could help in determining whether allowing slightly broader approval of custom devices could benefit patients with very rare conditions.
COVERAGE AND REIMBURSEMENT FOR HDE MEDICAL DEVICES
Despite the restriction on profits for devices that have been approved through the HDE process, manufacturers may still set substantial prices for such devices based on the costs that they may legally recover under the law. Thus, in considering whether to pursue development of a device that would fit HDE requirements, manufacturers will also consider whether public and private health plans are likely to cover the device and what they might pay.
Most of the devices that have HDE approval are complex devices that are implanted or otherwise applied in an inpatient hospital setting.16 For care under Medicare, this means that coverage and payment for a device will be subject to the provisions of the Part A program. As described in Chapter 6, Medicare pays hospitals a bundled or per-case payment for institutional services provided in the course of treatment for a particular diagnosis with payment varying depending on the severity of the diagnosis and other factors. For medical device manufacturers, the key relevant feature of the payment method is that the diagnosis-related group (DRG) payment to a hospital will not necessarily be adjusted to reflect any higher costs should a newly approved HDE or other device be used with a particular patient.
To recognize the added costs of desirable new technologies, Medicare can authorize a temporary “add-on” payment to a DRG if three conditions are met. The technology must be new (generally meaning that it was approved by FDA within the preceding 2 to 3 years); it must not be adequately covered by the existing DRG payment; and it must offer a substantial clinical benefit over existing options. This last requirement may be difficult for manufacturers of HDE devices because approval of the device does not require clinical evidence of effectiveness. Absent the availability of an add-on payment for new-technology HDE devices, a hospital might not make
the device available for use if the device was substantially more expensive than existing technology. (Use of an HDE device still requires IRB approval as described above.)
Moreover, by statute, Medicare generally limits payment to items or services that are “reasonable and necessary” for the diagnosis and treatment of illness or injury or to improve the functioning of a malformed body member” (42 USC 1395y). This has generally been interpreted to mean that a service or item must be safe and effective, medically necessary and appropriate, and not experimental in order to qualify for reimbursement.
The Centers for Medicare and Medicaid Services (CMS) have approved add-on payments for some devices that have HDE approval. One is the artificial implantable heart described earlier (AbioMed, 2005). Another is the device for treatment of pulmonary air leaks mentioned in Box 7-1 (Spiration, 2009). Overall, of the seven applications for add-on payments approved between 2001 and 2008, six were for products classified as medical devices (Clyde et al., 2008).
As this report was being completed, CMS and FDA announced a memorandum of understanding to share information and expertise related to the review and use of FDA-regulated devices and other products (75 Fed. Reg. 48699). The agencies are also considering a process of parallel review that would reduce the lag between FDA marketing authorization decisions and CMS national coverage determinations (75 Fed. Reg. 57045).
The committee did not examine the coverage and reimbursement policies of state Medicaid programs or private health plans, but it did find illustrative examples of variation in health plan policies. Some private health plans have authorized coverage for specific uses of an HDE and rejected it for others. For example, Aetna will cover certain uses of total artificial heart devices and left ventricular assist devices, but it considers other uses experimental and investigational (Aetna, 2010). At least one health plan has posted a general policy on coverage that states Humanitarian Use Devices are subject to individual review and prior approval (Wellmark Blue Cross Blue Shield, 2009).
MEDICAL DEVICE RESEARCH AND DEVELOPMENT
In order to lay the foundation for the committee’s recommendations for encouraging the development of medical devices for rare diseases, it is useful to review briefly some features of medical device innovation and development. For example, breakthrough implantable devices were made possible, in part, by scientific and engineering advances in areas outside biomedicine. Creative device ideas have often originated with physicians in the clinic who are trying to address specific problems they encounter or to help a specific patient with the tools at hand. The life cycle of devices
includes iterative improvements over time, often involving collaborations between engineering and other disciplines.
Emergence of Complex Medical Devices
I cannot believe that six whole months have soared by since I was given a new lease on life. A tiny device called an ICD [implantable cardioverter defibrillator] was surgically implanted beneath a patch of muscle tissue in my chest … and now I have a small metal box in my chest that I affectionately refer to as the iFib … [which] is both a pacemaker and defibrillator all boxed up in a compact little package. It is about the same size as an iPod Nano, but it can’t play music. All it does is guard against sudden death. Let’s see an iPod do that!
Sands, 2010
This man, who has lived for decades with muscular dystrophy, has been assisted by a variety of medical devices. As is the case with many devices used for patients with rare conditions, none were devised specifically for patients with muscular dystrophy but all have helped him survive. The most sophisticated of his devices, the implantable cardioverter defibrillator (ICD), is used with patients who have a number of different conditions that put them at risk of sudden cardiac death. Its development and subsequent refinement were made possible by a number of scientific and engineering advances.
Although medical devices have a long history in the form of basic surgical instruments, braces, medical thermometers, and similar relatively simple objects, the development of technologically sophisticated, complex devices advanced significantly in the 1950s and early 1960s, based in part on technological innovations in other arenas. Notably, the transistor, invented in 1947 at Bell Labs, provided the foundation for solid-state electronics, which in turn made possible the miniaturization of electronic devices and improved capabilities. In 1957, surgeons used the world’s first transistorized therapeutic medical device, the external cardiac pacemaker, to maintain an appropriate heart rate and adequate cardiac output following open-heart surgery on a young boy (MMF, 2007).
Other innovation within and outside the medical device industry—led to the availability of durable and biologically compatible materials for use in orthopedic, neurological, cardiac, and other implants. Advances in mechanical valve materials and designs and newly available heart-lung machines made replacement heart valves feasible in the 1960s.
Innovation Process for Complex Medical Devices
Although moving from idea to marketing typically takes many years for both drugs and complex medical devices, the nature of medical device
innovation and product development and the underlying technical expertise differ in some significant ways for devices. In simple terms, the innovation pathway for drugs is a laboratory-based discovery process that is led by biomedical scientists, chemists, and pharmacologists. Clinicians assume a primary role toward the end of the process, that is, when drugs undergo clinical testing in humans, which regulations require for all drugs. In contrast, device innovation and development has been primarily an engineering process that combines technical expertise from multiple disciplines. Clinicians may be involved from the outset and may continue to be involved in ongoing refinement once a device is authorized for marketing.
As is true for engineered products generally, the process of device development is iterative and circular. Figure 7-1 depicts the device development life cycle for more complex devices as beginning with a basic concept to address an unmet need, followed by initial prototype development and test-
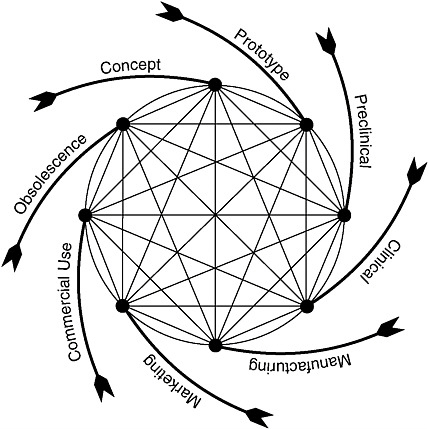
FIGURE 7-1 Total product life cycle for medical devices.
SOURCE: Feigal, 2002.
ing.17 This latter phase includes consultation with FDA about the evaluative methods and information needed to support FDA decisions to authorize the marketing of the device. After marketing authorization, modifications to the device typically continue for a variety of reasons, sometimes as design enhancements or sometimes in response to safety issues discovered once the device is on the market.
Figure 7-1 draws attention to a key aspect of medical devices, specifically, an “end-of-life” phase. In some cases, a device is supplanted by a radically different product that effectively makes obsolete, or reduces reliance on, the current product. For example, the development of implanted cardiac devices made obsolete the early external devices that often tethered individuals to power sources. In other cases, a device product is altered to make it smaller, safer, more effective, more convenient, or otherwise different in ways that make the older versions less desirable or less cost-effective. In contrast, small-molecule drugs may stay on the market unaltered for decades (except perhaps for additional formulations or methods of administration, e.g., a pill, a time-release capsule, or a liquid).
In addition, in contrast to pharmaceutical development, the process of developing a medical device often is not based on scientific discovery per se. Rather, the process involves the use of existing technological building blocks that are assembled into a “device” that satisfies certain desired performance characteristics related to a clinical need. If an initial approach proves unsatisfactory or clearly has features that can be improved, engineers may create a new design, reconfigure the existing design components, or even invent a new component, for example, one using a novel biomaterial that delivers the performance desired. The titanium rib illustrates a new conceptualization by a physician who had an engineering background. In some cases, an insurmountable performance roadblock is encountered and further development of the device is suspended.
Another distinguishing aspect of device development involves the roles played by clinicians in the innovation process for the most complex and technologically sophisticated therapeutic devices (Citron, 2008). In addition to identifying unmet needs, physicians are sometimes inventors who see the “flash of light” of a new idea and who even take an active role in pursuing it, as did the physician inventor of the titanium rib. Likewise, a clinician
conceived the fundamental idea for the ICD (Cannom and Prystowsky, 2004).
At other times, physicians provide vital clinical insight regarding the suitability of a proposed technology for their patients, and they also may propose improvements and enhancements for new or developing products. For devices such as the titanium rib or the cardiac pacemaker, they devise surgical techniques necessary for safe implantation. Clinicians also participate in clinical trials to support regulatory submissions, and based on research and clinical practice, they may identify clinical and technical problems and suggest refinements to improve performance. Clinicians involved in product development and testing may also teach their colleagues how to use a new technology correctly. In short, expert physicians are often an integral part of the research and development continuum, not just customers for the end product.
This involvement may add to the challenges of identifying and managing conflicts of interest, particularly if clinicians have an equity interest or other financial stake in the product. Likewise, when clinicians who consult with companies on product refinements also have a role in the choice of implants or other devices used during orthopedic and other surgeries, the potential for the financial interest to bias judgments is a concern. The identification of physician relationships with industry18 and the management of conflicts of interest19 have drawn increasing attention in recent years.
The development of deep-brain stimulation provides an example of academic inspiration in the device industry. French professors discovered that they could reduce the effects of movement disorders through neurostimulation using an existing device for an off-label (not FDA approved) indication (Linehan et al., 2007). Although Medtronic, the manufacturer of the device, was not involved in or even initially aware of the early results, later collaboration between the researchers and the company provided a starting point for work that led to FDA approval of the modified device for new uses. The company also learned, largely through the initial investigations of other neurosurgeons, that the device could treat additional neurological
disorders. Deep-brain stimulators were eventually approved by FDA for several new indications, each involving relatively minor technical changes to the device. The implant received PMA approval for essential tremor in 1997 and advanced Parkinson disease in 2002 and then was granted HDE approval for dystonia in 2003 and chronic, severe, treatment-resistant obsessive compulsive disorder in 2009.
University researchers have been actively involved in developing the technologies used in the genetic and drug discovery research described in earlier chapters. The American Institute for Medical and Biological Engineering cites the development of genomic sequencing and microarrays in its “hall of fame” (AIMBE, 2005). The bioengineering program at Stanford University, summarized in Box 7-3, offers one example of the intersection of device engineering and scientific advances in biological sciences.
Although some technological advances in medical devices have taken advantage of government-supported research and development, direct government investment in accelerating medical device research and development was initially limited. One exception is the total artificial heart
|
BOX 7-3 Stanford University Biodesign Program About 10 years ago, faculty at Stanford University developed a systematic approach to solving significant medical problems in which invention and innovation were a team activity and were part of a process. The impetus for this initiative, the Stanford Biodesign Program, was the realization that innovations in the medical area involved many technical and scientific disciplines and these disciplines need to collaborate and inform each other. Biodesign is associated with Stanford’s Bio-X program, which promotes interdisciplinary research in biology and medicine. As described on that program’s website, “Ideas and methods embodied in engineering, computer science, physics, chemistry, and other fields are being brought to bear upon important challenges in bioscience. In turn, bioscience creates new opportunities in other fields. Significant discoveries and creative inventions are accelerated through formation of new collaborative teams” (http://biox.stanford.edu/about/index.html). The Biodesign Program creates multidisciplinary collaborative teams composed of graduate students from engineering, medicine, and business. These teams follow a three-stage process or method to create cost-effective, state-of-the-art medical devices for the benefit of patients, industry, and society. The method includes three stages: need identification; concept development; and business or project planning. The program has been emulated both domestically and internationally. Stanford faculty have published a textbook Biodesign (Zenios et al., 2010) that describes the process. |
program at the National Heart, Lung, and Blood Institute (NHLBI), which began in 1964 (IOM, 1991). Although the program’s focus shifted to ventricular assist devices (which have a larger target population than the artificial heart per se), the initial investments provided important knowledge for the development of less ambitious but clinically relevant cardiac support systems. The report of an expert panel convened by NHLBI noted that it “is not probable that development of assist devices would have occurred without the government support that is now being increasingly assumed by industry as clinically effective devices move towards marketing approval” (NHLBI, 2000, p. 3).
During the 1970s, the engineering community sought to increase the visibility of biomedical engineering and to educate the National Institutes of Health (NIH) about the nature and value of research in bioengineering and bioimaging (Hendee et al., 2002). In 2000, after a number of unsuccessful proposals in the 1990s, Congress created the National Institute of Biomedical Imaging and Bioengineering (NIBIB, 2009). NIBIB seeks to advance basic research and improve patient care by integrating the physical and engineering sciences with the life sciences. Relevant disciplines extend beyond biological sciences and various engineering disciplines to include (among others) the information sciences, physics, chemistry, mathematics, materials science, and computer science. The range of NIBIB interests covers, among other areas, biomaterials, bioinformatics, structural biology, drug and gene delivery systems and devices, tissue engineering, microbiomechanics, nanotechnology, sensors, surgical instruments, diverse kinds of imaging, and rehabilitation technology. Some areas of device innovations that illustrate the interaction between innovations in engineering and biological sciences are summarized in Box 7-4. In addition, the discussion in Chapter 4 of discovery research and diagnostic developments identifies other areas in which scientific and technological advances in biomedicine will likely shape innovation in diagnostic devices.
Clinical Studies for Medical Devices
As discussed earlier, FDA clears the majority of medical devices for marketing without requiring formal clinical studies. For a small proportion of devices—in particular, implanted devices or other high-risk devices—FDA requires data from clinical studies. For significant-risk devices, FDA requires formal approval of a request to begin clinical studies.
Under its regulations governing clinical studies to support PMAs, FDA states that the agency relies only on “valid scientific evidence to determine whether there is reasonable assurance that the device is safe and effective” (21 CFR 860.7(c)). The same document goes on to describe valid scientific evidence as
|
BOX 7-4 Innovations in Engineering and Biological Sciences and Medical Device Innovation Replacement organs: Using man-made scaffolds and other biomedical techniques, tissue engineering and regenerative medicine discoveries will permit scientists to “grow” organs in the laboratory to replace patients’ failed organs. Tissue-engineered urinary bladders have already been implanted in patients. Proof-of-principle laboratory demonstration of a total beating heart (rat) has also been achieved. Such technologies have the potential to overcome the shortage of available donor transplants and to offer patients a biological tissue solution rather than an electromechanical therapy. Drug delivery: Oral delivery of drugs has significant limitations, including patient compliance, first-pass inactivation by the liver, systemic rather than site-specific effects, and undesirable variability of blood levels between doses. Early generations of implanted drug delivery systems for chronic diseases or symptoms such as spasticity and intractable pain have demonstrated capabilities that address many of the limitations cited for oral delivery. Future generations are expected to provide sensor-based closed-loop operation, delivering drugs only when needed and at appropriate dosages. Delivery will be either site-specific, treating only affected tissue, or systemic. Of particular interest is the development of a fully implanted artificial pancreas that will deliver appropriate amounts of insulin in response to continuous monitoring of diabetic patient’s blood glucose levels. Implanted diagnostics: The concept of an implanted laboratory-on-a-chip has the possibility of revolutionizing how disease is diagnosed and preventing certain diseases or crisis episodes. Biosensors, as described for implanted insulin pumps above, serve as an example of how this might work. Implanted “chips” that contain an array of sensors will continually monitor a patient’s condition and, if warranted, communicate to the patient’s health care provider that attention is needed. Prototypic versions of rudimentary diagnostic systems have already demonstrated the ability to reduce serious events related to heart failure and also to reduce the number of visits to the emergency room. Miniaturization: A collection of enabling technologies, some emerging from the field of nanotechnology, will expand possibilities of minimally invasive surgery, produce novel bio-interactive coatings, and reduce the size and expand the service life of implants, making them more suitable for pediatric patients. SOURCES: AdvaMed, 2004; Braunschweig, 2007; El-Khatib et al., 2010; Trafton, 2010. |
evidence from well-controlled investigations, partially controlled studies, studies and objective trials without matched controls, well-documented case histories conducted by qualified experts, and reports of significant human experience with a marketed device, from which it can fairly and responsibly be concluded by qualified experts that there is reasonable as-
surance of the safety and effectiveness of a device under its conditions of use. The evidence required may vary according to the characteristics of the device, its conditions of use, the existence and adequacy of warnings and other restrictions, and the extent of experience with its use. Isolated case reports, random experience, reports lacking sufficient details to permit scientific evaluation, and unsubstantiated opinions are not regarded as valid scientific evidence to show safety or effectiveness.
The requirements for evidence to support FDA approval of a PMA explicitly provide for more variability (linked to the particular characteristics and uses of a device) than is found in the corresponding expectations outlined in Chapter 3 for the approval of drugs. A recent study examined FDA summaries of the evidence used to support FDA approvals for 78 “high-risk cardiovascular devices” (Dhruva et al., 2009). The analysts reported that nearly two-thirds (65 percent) of the applications were approved based on a single study, that 27 percent of the 123 submitted studies were randomized, that 88 percent of the primary endpoints used were surrogate measures, that 52 percent of the endpoints were compared with controls, and that 31 percent of these controls were historical. Moreover, as described earlier, FDA does not require the same level of evidence for a device approved through an HDE as it does for those approved through a PMA.
For medical devices that require clinical evaluation, a pilot or feasibility study usually involves an initial clinical evaluation of the safety of a prototype device in individuals with the condition for which the device is designed. Such a study may suggest modifications to the prototype device to improve its performance. For a device that requires complex surgery for its implantation or that otherwise is technically demanding to use, the pilot phase may also provide an important period for learning about the process and skills required for the safe and effective clinical use of the device. Experience gained from pilot studies also contributes to the design of pivotal studies, which usually recruit larger numbers of research participants and may involve multiple study sites and centers. If surgical procedures are involved, the process may also require training of investigators at sites not involved in the pilot study.
As is also the case for orphan drugs, the accumulation of sufficient participants may take years for a device that is intended for a small population of patients. For example, clinical testing for the titanium rib (cited at the start of this chapter) occurred over 14 years, a long period that reflected in part the challenges of working with a rare condition and in part the request by FDA for long-term information on the device, which requires repeated adjustment as a child grows (Campbell, 2004).
In addition, many devices present special challenges for the design of clinical trials. Especially for surgically implanted devices, the classic ran-
domized, double-blind comparative study is often not feasible or ethical. A few trials of surgical implant procedures have included sham surgeries for comparison in single-blind studies, but the surgical team obviously had to be aware of which procedure was used (see, e.g., Moseley et al., 2002). Such trials are controversial (see, e.g., Miller, 2003; Mehta et al., 2007). For some electronic devices such as cardiac or neurological stimulators, clinical studies have sometimes used a design that involves implantation of the device in a study population and then comparing a subset of the group in which the device is kept switched on with another subset in which it is switched off for a predetermined period of time (see, e.g., Greenberg et al., 2006).
Staff at CDRH are planning two projects that should provide a better understanding of issues in the design of clinical studies for devices (Linda C. Ulrich, M.D., Medical Officer, FDA Office of Orphan Product Development, April 26, 2010, personal communication). One project is an analysis of the clinical safety and efficacy data submitted in support of PMAs. The other project is preparation of a guidance document on clinical trial design for device trials.
In addition, reflecting the characteristics of device trials, CDRH has developed guidance on the use of Bayesian statistics with clinical trials of medical devices, including situations involving confirmatory trials, device modifications, incomplete data, and opportunities for adaptive design strategies. (FDA released draft guidance in 2006 and final guidance in 2010 [CDRH-OSB, 2010].) FDA development and education efforts in this area date back well over a decade (Campbell, 2008, 2009). Although the Bayesian approach requires companies to have expert statistical advice and to engage in early consultation with FDA, it has the potential to reduce the costs of trials. As of 2009, at least 20 PMAs or PMA supplements using Bayesian analysis were under review (Campbell, 2009). As part of the guidance and education programs on small clinical trials that NIH and FDA are conducting, a commentary examining applications of Bayesian statistics to device trials involving small populations could be useful. In general, recent years have seen a growing appreciation of the special challenges of device trials, the importance of innovative trial and statistical methods, and the opportunities for FDA-industry interaction to improve trial design and analysis techniques and their use (Campbell, 2008).
DEVICE INNOVATION AND THE HDE OPTION
Although individuals and companies may pioneer devices for rare diseases for purely altruistic reasons, such altruism is uncommon because device innovation entails significant costs, protracted time for research and development, and commitment to ongoing support and administrative costs once a device is approved. For complex “new to the world” devices, the
research and development costs can run into the tens of millions of dollars, although details of these costs are not readily available. The time lines to produce practical, safe, and reliable implanted devices can be very long, measured in years and even decades.
An HDE approval reduces the time to market because demonstration of effectiveness is not required. Companies can also recover certain costs, for example, research and development costs. Nevertheless, without a reasonable opportunity to make a profit, as provided by the Orphan Drug Act, the costs and investment risks to bring a new technology forward for small markets are substantial and are likely not to appeal to many companies and investors.
These reservations may be moderated if development of a device for a small population is considered a stepping stone for a future application that may serve a larger market. In situations where the technology is truly novel, the HDE can offer a company the opportunity to learn more about it while continuing its development for a broader use. For example, the company Spiration, Inc., received HDE approval for a device to control prolonged air leaks in the lung following lung surgery (H060002). The company is also conducting clinical testing of the device to treat severe emphysema.
In anticipation of additional indications for a broader patient population, a company may view an HDE approval as a way to enter the market more quickly and with a baseline level of revenue. Market entry under an HDE provides a company with an opportunity to further evaluate the technology and identify next-generation design improvements. It also gives surgeons an opportunity to refine surgical techniques and protocols that may benefit future patients. In addition, an HDE could have “good will” value to the sponsor. Nevertheless, as noted above, there remain both the direct costs of supporting the HDE device and the allocation of financial and personnel resources (especially difficult for a small company) to the HDE device in lieu of another IDE device, another indication, or other research and development effort.
Some reservations about the HDE option may be moderated if a company is looking at a new indication for an already approved device. The list of HDE devices includes some devices (e.g., the deep-brain stimulation device) that are modifications of existing products approved under PMAs for other indications. The investment risk to the company for pursuing a new rare diseases indication is moderated in such cases because most of the research and development costs intrinsic to the device have already been incurred. Companies will still incur some additional incremental research and development investments to devise any modifications needed for the rare diseases application and to generate data on safety and probable benefit necessary for the HDE application.
Although the incremental costs may be relatively modest for an HDE
based on an already approved product, companies still face the limit on profits for HDE devices and uncertainties about reimbursement. They also still must consider the opportunity cost for pursuing an HDE approval rather than pursuing development of other products or pursuing approval through the regular PMA process. A company’s decision about the HDE option could also be influenced by the requirements for IRB approval and the potential for the annual market for the product to be larger than projected and exceed the annual shipment limit. In the latter situation, FDA would likely ask the company to withdraw the HDE device and seek approval through the PMA process.
Interviews conducted by Bernad (2009) suggest that device developers may sometimes decide to pursue HDE approval after the major part of product development has occurred. One company representative noted that the HDE process can save 3 to 4 years in getting a product to market, “which can be the entire product life cycle” (p. 140). An executive for a company with a device that has both PMA and HDE approvals observed that the HDE process was a means of broadening approved indications for the device that “saved 3 years, recovered $10 million from the initial research and development costs, and established good relationships with many physicians in the field” (p. 142). Others interviewed noted that the short-term benefits of the HDE process must be weighed against the negatives of the restriction on profits and the potential for insurers not to cover the device (for lack of evidence of efficacy). One official particularly cited the burdensome IRB process as involving substantial costs for meticulous record keeping, application production, and IRB fees (which could involve hundreds of sites). In addition, in an article summarizing the results of a symposium on the HDE process, Kaplan and colleagues noted that the availability of a device through the HDE process could complicate recruitment for clinical studies to evaluate the safety and efficacy of a device for a more common indication (Kaplan et al., 2005).
As noted earlier, the difference between drug and device incentives could be an issue for a combination product for a small population if the difference in incentives discouraged drug-device company cooperation on a combination product that involved a complex device and that did not intrinsically require simultaneous or coordinated clinical testing of the drug and the device. If such situations arise, Congress or FDA may need to consider a modification in the HDE policy to encourage innovation to meet unmet needs for combination devices while protecting patients from unsafe or ineffective products.
In general, however, rare conditions may be treatment targets for either a drug solution or a device solution but not both. That is, only one or the other modality holds clinical promise or clinical relevance or presents a reasonable risk-benefit ratio. In such instances, inconsistencies in incen-
tives and regulatory requirements for orphan drugs and Humanitarian Use Devices are not likely to have much practical impact on the development or use of the product. In addition, if a device is one that qualifies for clearance through the 510(k) process, then the HDE process and conditions likely will not be considered.20
It is difficult to assess the extent to which the relatively low number of HDE approvals is influenced by the profit disincentive and the limit on yearly shipment or is, rather, a function of limited opportunities for devices—compared to drugs—to meet substantial unmet health needs for small populations. As noted at the beginning of this chapter, the emphasis in discussions of rare diseases is overwhelmingly on drugs.
For devices covered by an HDE, information on the number of device units shipped is not readily available nor are the estimates submitted by companies (in support of their HDE application) of the number of affected individuals. Although indication-specific information is not available, a recent press release by Medtronic recently reported that a cumulative 75,000 patients worldwide had been treated with its implanted deep-brain stimulation technology, including for the four indications described earlier (Medtronic, 2010).
RECOMMENDATIONS
Based on information presented to the committee by representatives of companies and FDA and provided by a review of past proposals for policy change, the committee concluded that the development of reasonable and effective incentives specific to device development for small populations has proven difficult. The incentives relevant for drug development, particularly the protections from market competition, are not well matched to the realities of device development. Although recent initiatives to promote the development of pediatric medical devices modify the HDE process by allowing profits, they do not move toward either the marketing protections or the stricter approval requirements applicable to orphan drugs.
In contrast to pediatric medical devices, relatively little attention has been directed to needs for medical devices for people with rare conditions. (Even the statement to the committee from the Advanced Medical Technology Association focused as much or more on pediatric devices as on devices
for rare diseases [AdvaMed, 2009].) CDRH recently held a meeting to discuss unmet devices needs, but it did not specifically address the needs of people with rare conditions.
Without the time for a very focused examination, the committee found it difficult to assess the possible extent of unmet device needs for adults with rare conditions and the extent to which changes in FDA policies (e.g., an increase in the criterion of 4,000 patients per year for HDE approval) might promote innovation to meet these needs). A first step in understanding the potential areas for device innovation is a needs assessment for adults with rare conditions. Such an assessment, which should involve patient groups, clinicians, biomedical engineers, and device developers, can also illuminate impediments to innovations to meet those needs.
RECOMMENDATION 7-1: FDA and NIH should collaborate on an assessment of unmet device needs and priorities relevant to rare diseases. That assessment should focus on the most plausible areas of unmet need, identify impediments to meeting these needs, and examine options for overcoming impediments and stimulating high priority innovations.
The identification of needs, priorities, and impediments should help inform the consideration by government, private foundations, and others of additional incentives and supports for medical device development for small populations. Beyond simplifying some aspects of the HDE process as suggested in Recommendation 7-4, options to encourage device innovation for rare diseases include
-
the provision of additional orphan products grants and NIH awards for the development of devices to meet priority needs;
-
the authorization of tax credits for certain research and development costs, similar to those available for companies developing orphan drugs; and
-
the creation of inducement prizes for the design and initial testing of novel devices in areas of unmet need.
In addition, the changes in the HDE incentives for pediatric devices, including the relaxation of the restriction on profits, provide an opportunity to examine the case for similar changes to encourage innovative devices for adults with rare conditions. Also, as experience with the revised incentives for pediatric devices is gained, including through the GAO evaluation due in 2010, that knowledge should be applied to the encouragement of devices for adults with rare conditions. One alternative to eliminating the profit restriction altogether would be the development of a cost-plus option that
would allow companies to charge a specified amount over certain costs of development. The committee did not examine this idea in depth and recognizes that it would need careful investigation of potential unintended consequences and consideration of safeguards.
RECOMMENDATION 7-2: Congress should consider whether the rationale for its creation of additional incentives for pediatric device development also supports the use of such incentives to promote the development of devices to meet the needs of adults with rare conditions.
A modest step to encourage additional company interest in devices for small populations would involve greater flexibility in the limits on annual shipments of HDE devices. For devices covered by an HDE, information on the number of units shipped is not readily available nor are the estimates submitted by companies (in support of their HDE application) of the number of affected individuals. Such information might help in assessing how often the 4,000-per-year shipment limit is approached and thus how often the limit might restrict access within the existing framework of HDE policy. Such information would not, however, help in determining what applications might be attracted by a higher cap given that the HDE unlike the PMA does not require demonstration of efficacy. Rather than authorizing an increase in the yearly limit of device shipments that are allowed for an HDE device, Congress could provide for the cap to be raised in specific situations when CDRH determined (based on medical, demographic, and scientific information submitted to it) that an increase in the cap or annual distribution number would benefit patients with a rare disease. This policy would require the specification of boundary criteria (e.g., how large a deviation in the cap could be approved). An analysis of experience with company estimates of affected populations and annual shipments could help in evaluating this idea.
RECOMMENDATION 7-3: As a basis for possible congressional action, the Center for Devices and Radiological Health should analyze the supporting justifications offered in successful and unsuccessful Humanitarian Device Exemption applications related to the 4,000-person-per-year limit and should evaluate the subsequent experience with actual device shipments for approved applications, including any communications about projections that a company might exceed the limits. Taking the findings into account, Congress should consider authorizing FDA to permit a small, defined deviation from the yearly limit on shipments for a specific device when the agency determines that such a deviation would benefit patients with a rare disease.
In addition, FDA could take other steps to make the HDE process worth further consideration by potential sponsors. It could offer additional guidance and assistance to make it easier for sponsors and IRBs to manage the requirement for IRB review of HDEs. As discussed earlier, recent CDRH guidance is helpful but still leaves areas of confusion and uncertainty. The agency could also clarify existing guidance on the very specific details of the process for applying for designation of a Humanitarian Use Device. For example, it could provide further guidance on the evidence needed to support claims of probable benefit and the calculation of the size of the target patient population, and it could also offer consultation to help sponsors understand what data will be responsive and justifiable.
RECOMMENDATION 7-4: FDA should take steps to reduce the burdens on potential sponsors of Humanitarian Use Devices, including
-
assigning an ombudsman to help sponsors navigate the regulatory process for these applications;
-
providing more specific guidance and technical assistance on the documentation of the size of the patient population as required for humanitarian use designations; and
-
developing better guidance (including step-by-step instructions and sample documents) for sponsors and IRBs on their roles and responsibilities related to IRB review of HDEs.
In another area, CDRH could also develop new guidance on the use of surrogate endpoints in medical device trials (see discussion in Chapter 5). As noted earlier in this chapter, the planned analysis of the clinical safety and efficacy data submitted in support of PMAs and the planned guidance document on device clinical trials could contribute useful information and perspectives in this area, even though neither activity will focus on HDEs specifically.




































