
5
New Vaccine Development and the Future Needs of the Special Immunizations Program
5.1
THE PROCESS OF VACCINE DEVELOPMENT
As discussed in detail in Chapter 4, vaccine development and licensure are regulated by the Food and Drug Administration (FDA)’s Center for Biologics Evaluation and Research, according to the regulations contained in Title 21 of the Code of Federal Regulations.
Vaccine development leading to a licensed product typically involves a series of steps, starting with initial research, development, and testing for immunogenicity and protective immunity in animal models. After those preclinical studies, investigators submit data to FDA to receive an Investigational New Drug (IND) approval and move into testing in human volunteers. Clinical testing is accomplished in several phases:
-
Phase I. Testing in relatively small groups of healthy adults to obtain initial safety and human immunogenicity data.
-
Phase II. Testing in larger groups to continue refining information on safety, efficacy, and dosing.
-
Phase III. Trials usually conducted in large groups of the likely user population with a focus on obtaining information on infrequent adverse events and on demonstrating efficacy often through measurements of immune correlates of protection.
After obtaining successful results in clinical trials, the sponsor submits a Biologic License Application (BLA) to FDA to seek product licensure. Once a product is licensed, Phase IV, or post-market surveillance, continues to be conducted, and adverse events that occur are reported. Included along the continuum of the process is the need to manufacture a candidate vaccine under
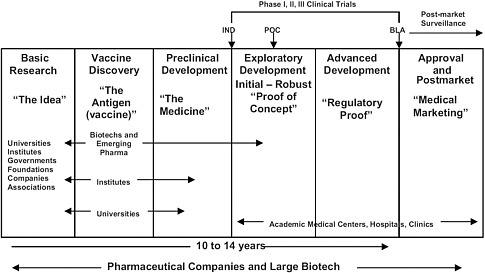
FIGURE 5.1 Vaccine research from idea to market. SOURCE: Adapted from IOM 2009. IND = Investigational New Drug application; POC = proof of concept; NDA = New Drug Application; Pharma = pharmaceutical companies.
current Good Manufacturing Practices standards on a pilot scale for preclinical testing and early clinical studies and on larger scales for testing in Phase III trials and ultimately for commercial production (Figure 5.1).
Candidates for enrollment in the Special Immunizations Program (SIP) include researchers studying fundamental biology and pathology of highly hazardous pathogens or working in the initial stages of development of new candidate vaccines and medical countermeasures (MCM). Preclinical animal challenge and testing studies also potentially expose laboratory researchers and animal technicians to pathogen infection. And personnel working on the pilot and scale-up manufacturing of a potential vaccine candidate may benefit from SIP vaccination, particularly if at risk of exposure to large quantities of live pathogen strains.
5.2
NEW VACCINE DEVELOPMENT AND THE FUTURE NEEDS OF THE SPECIAL IMMUNIZATIONS PROGRAM
Except for several licensed vaccines, the vaccines now used in the SIP are IND products and are no longer being manufactured (information on the available stocks of these vaccines is provided in Chapter 3). At the current rate of use, these stocks will be adequate for a number of years. However, a number of risks are associated with the lack of current manufacture and the current status of SIP vaccines:
-
Existing stocks could be found to be out of specification because of loss of potency, contamination with an adventitious agent, appearance of particles, or other product-quality problems.
-
There could be a need to increase use in laboratory workers in the event of an expanded research, development, or manufacturing effort associated with a specific threat agent.
-
There could be an unexpected need to use the vaccines in a larger number of people because of deployment of military or public health personnel to an area with a natural disease emergence or biological threat.
Such events could necessitate renewed manufacturing. As time passes, and the existing stocks age, there is also increasing concern that degradation might result in changes that are not detected in the stability program but could adversely affect product quality or biological performance or lead to adverse reactions.1 The committee therefore believes that it may be prudent to have fresh materials prepared for some or all products and to have bridging clinical trials performed. Such a program could be rolled out in order of priority considering aspects such as the amount of available stocks, the likely size of the researcher user population, the age of the vaccine stocks, the vaccine product profile, and concerns about potency or other product-quality issues.
In undertaking such a program, careful consideration should be given to the question of whether the legacy vaccine has a product profile that warrants the effort of new stock manufacture. For example, it is widely known that the attenuated Venezuelan equine encephalitis (VEE) TC-83 vaccine safety and immunogenicity profiles are deficient. Alternative vaccine candidates have already been developed, and clinical data on them are available (for example, the replicon vaccine developed by the U.S. Army Medical Research Institute for Infectious Diseases and AlphaVax, Inc.), so consideration should be given to replacement of VEE TC-83 vaccine in the SIP rather than making new stocks of it. Similarly, as pointed out elsewhere in the report, a licensed Q fever vaccine is manufactured by CSL, Ltd. in Australia and could replace the legacy vaccine now being used in the SIP. Although it is also used with a prevaccination skin test, this has been accomplished by intradermal administration of diluted vaccine (Gidding et al. 2009). The committee believes that the need for new manufacturing of legacy vaccines, priority-setting, improvements in vaccine
manufacture, and replacement vaccines with improved product profiles for given indications should be analyzed case by case as a separate exercise.
As research priorities in biodefense and emerging infectious diseases shift and evolve, researchers may find themselves working with highly hazardous pathogens that are not addressed in the current SIP. Thus, the committee considered the process of new vaccine development and how vaccine development issues might be related to the future of the SIP.
5.2.1
Types of Vaccines and Manufacturing Methods
Many technologies are applicable to the development and manufacture of vaccines (see Table 5.1). The risk of adverse reactions to a vaccine is highest for live, attenuated or live-vector vaccines because the full spectrum of reactogenicity cannot be known at an early stage of development and adverse reactions may occur in people who have inherited or acquired susceptibility factors. Those risks are significant, but live vaccines may permit use of very low doses (and thus allow small-scale production of many doses), usually trigger both innate and adaptive immunity, and may confer long-lasting immunity with a single dose. A substantial safety database on such products may be required before they can be included in an immunization program like the SIP. The risk of adverse reactions posed by inactivated or subunit vaccines is inherently lower. However, the inclusion of novel adjuvants with an inactivated or subunit vaccine may increase safety concerns and require additional clinical experience before they can be included in the SIP.
The equipment and facilities used for biomanufacturing are changing, and low-capital, rapid-turnaround, single-use disposable bioreactors and process systems are enabling low-cost manufacture of small volumes of vaccines and other biologics. Although fixed, hard-piped stainless-steel bioreactors and clean-in-place and sterilize-in-place facilities are still important for high-volume or dedicated product manufacture, the use of flexible, single-use equipment can reduce initial investment cost and support rapid manufacture of clinical supplies. For example, Bavarian Nordic manufactures a new investigational modified vaccinia Ankara smallpox vaccine in GE WAVE bioreactor bags. The benefits of single-use bioreactors include lower cost of facility buildout and operations, shorter changeover times, increased productivity, lower risk of bacterial or mold contamination, and less scrap. Single-use process trains can support flexibility in the operation of a multiuse facility. Several companies have developed single-use stirred-tank bioreactors,2 mixers, and downstream purification systems. The bioreactors are now large enough to meet the needs of almost all vaccines for the Department of Defense (DOD) and the Department
of Health and Human Services (HHS). No manufacturer makes a commercial product with these systems yet, but the technology will soon be applied to vaccine production; several large companies are using disposable systems for products in the last stages of development. For SIP products with requirements for small numbers of doses, traditional pilot-scale manufacturing with disposable technology—such as WAVE bioreactor bags, roller bottles, and cell factories—may suffice. However, manufacturing on a pilot scale (for example, 25–1,000 L) initially in a stirred-tank bioreactor with disposable liner bags or in a fixed tank cleaned in place ensures that scale-up can be rapid if the need arises. Roller bottles, WAVE bags, and cell factories may require replication of small units to achieve larger manufacturing capacity or may present a more challenging transition to larger bioreactors.
The requirements (or at least expectations) for product quality, purity, and freedom from animal products in new vaccines are more stringent than they were when the older SIP vaccines were manufactured. Except for vaccines prepared in diploid human cells, residual host-cell DNA concentrations must be less than 10 nanograms per human dose, and the size of residual DNA should be no more than the size of a gene. Host-cell protein concentrations are of less concern, but new vaccines generally contain less than 1 microgram per dose. Other residual contaminants from materials used in or added to the process may need to be measured. There is now a strong preference for avoiding any animal product (such as fetal bovine serum or porcine trypsin) or human blood-derived proteins in the manufacturing process. Tests for adventitious agents are more extensive and complex today than ones that were in place when the older products were made. Porcine circovirus contamination of Vero cells, African green monkey kidney cells used in the manufacture of rotavirus vaccines, is a case in point (Victoria et al. 2010).
Standards for testing for adventitious agents are very different today from when the SIP products were manufactured. Moreover, the cell culture–based SIP vaccines would have been manufactured with fetal bovine serum in the medium used to expand cells before viral infection. As a result, from 2000 to 2003, the SIP repeated complete lot release testing and tracked down the sources of fetal bovine sera to document that the vaccines meet current requirements and to exclude the possibility of bovine spongiform encephalopathy contamination.
Table 5.1 describes a variety of strategies used in the development of vaccines and compares some of their characteristics.
5.2.2
Replenishing and Expanding Existing Vaccine Stockpiles
For existing SIP vaccines, developed and made many years ago, there are numerous opportunities to improve on method of manufacture and control, product quality, stability, and potentially even efficacy (for example, by the
TABLE 5.1 Examples and Characteristics of Major Concepts for Design and Scale-up of New, Improved Vaccines
|
Platforms |
Whole Microorganisms |
Subunits |
Bulk Process Complexity |
Formulation Complexity |
BSL Required |
Transmissible to Nonvaccinees |
Immunologic Advantages |
|
Live attenuated bacterial and viral vaccines (e.g., S. typhi, shigella, varicella, measles) |
Attenuated wild-type microorganisms |
|
Minimum complexity; fermentation, cell culture, often without purification |
May require lyophilization with stabilizing agents |
Minimum BSL-2+ |
Yes |
Very low dose; innate and adaptive immunity; long duration of immunity |
|
Inactivated wild-type bacterial vaccines (e.g., DTP, animal vaccines) |
|
|
Minimum complexity; fermentation, cell culture, followed by inactivation, often without purification |
|
BSL-2/3 |
No |
Not infectious; innate and adaptive immunity |
|
Inactivated wild-type viral vaccines (e.g., animal vaccines, annual influenza |
Egg-adapted current strains; cell culture may use wild type |
“Split” vaccines processed to remove core components, leaving surface antigens |
Egg-based process-complex; cell-culture process-less complex |
|
wild-type requires BSL-3; attenuated, egg-adapted, BSL-2/3 |
No |
Not infectious; innate and adaptive immunity; “split” vaccines may have less severe injection-site reaction |
|
Replicating nonpathogenic bacterial vectors (e.g., recombinant commensal bacteria, oral typhoid, animal vaccines, experimental cancer vaccines) |
|
Recombinant heterologous antigens |
Minimum complexity; fermentation |
May require lyophilization with stabilizing agents |
|
Yes |
|
|
Replicating nonpathogenic viral vectors (e.g., cancer vaccines, HIV vaccines, animal vaccines) |
|
Recombinant heterologous antigens |
|
May require lyophilization with stabilizing agents |
|
Yes |
Recombinant-DNA vaccine platforms that stimulate both innate and adaptive immunity; may be very-low-dose systems |
|
Nonreplicating viral vectors (e.g., alphavirus replicons, RNA and DNA vaccines) |
|
Recombinant heterologous antigens |
|
|
Infectious wild-type viruses may require BSL-3 |
No |
Recombinant-DNA vaccine platforms that stimulate both innate and adaptive immunity |
|
Particulate vaccines (virus-like particles, bacterial-cell ghosts, self-assembled subunit antigens) |
|
|
Moderate to complex |
May require stabilants or cross-linking; used in conjunction with adjuvants |
|
No |
Mimic whole-virus vaccines in recruiting innate and adaptive immunity |
|
Purified proteins, peptides, polysaccharides |
|
|
Moderate to complex |
Minimal; many adsorbed to alum |
|
No |
Generally poor immunogens |
|
Conjugated proteins, peptides, polysaccharides (e.g., pneumococcal, meningococcal, H. influenzae conjugates) |
|
|
Complex; two fermentations, isolations, purifications; separate conjugation chemistry and purification steps |
Minimal; many adsorbed to alum |
|
No |
Improved immune response to purified antigens |
|
Platforms |
Whole Microorganisms |
Subunits |
Bulk Process Complexity |
Formulation Complexity |
BSL Required |
Transmissible to Nonvaccinees |
Immunologic Advantages |
|
DNA vaccines |
|
Typically recombinant genes amplified in bacterial plasmids |
Minimum complexity |
Minimum to moderate complexity |
|
No (?) |
Can be designed and scaled up in very little time; recruit innate and adaptive immune response; effective as “primer” in “prime-boost” strategies |
|
SOURCE: Drew 2007. |
|||||||
addition of an adjuvant). The priority for replacing an existing product will be driven by a case-by-case assessment of the following factors:
-
Current supply.
-
Assessment of future needs (for example, in an expanded SIP program).
-
Stability trending data that may indicate a loss of potency.
-
Potential issues of product quality (as noted, complete lot release testing was redone in 2000–2003).
-
Whether an improved product for the same indication already exists (for example, as a licensed or investigational product) or is in development with an expected timeline for availability within the lifespan of existing vaccine stocks.
-
Current product profile versus ideal product profile for a vaccine for the same indication.
-
Specific problems and issues with the existing vaccines (for example, substrate acceptability), use of animal products in culture media or manufacturing steps (for example, use of fetal bovine serum or porcine trypsin), incorporation of human-blood–derived materials in the product (for example, human serum albumin), lack of controls for adventitious agents, genetic stability, safety, and immunogenicity, to name a few concerns likely to arise given current manufacturing and regulatory standards.
-
Whether there is an existing sponsor for redevelopment and manufacturing of replenishment vaccine.
-
Existence and completeness of documentation and materials (for example, seed stocks and analytic reagents) that allow technology transfer to a contract manufacturer.
-
Extent of proposed changes in manufacturing and control methods.
-
Scope of clinical trials required for bridging past data on safety, immunogenicity, and potential efficacy to a new vaccine stock.
Changes in the manufacture of a biological product must be carefully considered because regulatory approval necessary for human use focuses on control of the process rather than on analysis of the end product (as would be the case for a well-defined chemical entity or drug). Even if a replenished supply of an older vaccine were made with methods as close as possible to the original process, there would be many changes in raw materials, cell banks, and procedures for production and downstream processing. In essence, a redevelopment program would be required to reproduce clinical-trial material with engineering or pilot batches to ensure manufacturability and quality of the product. Depending on whether FDA views changes in the production process or product as material changes, there may need to be additional preclinical
toxicology studies. Certainly, some bridging clinical trials will be necessary to ensure comparability with the original vaccine. The costs of technology transfer, manufacturing, quality control, and bridging studies for new production of an existing SIP vaccine may be substantial and should be carefully weighed case by case against an investment in innovative or improved technology for vaccine development and production.
Some substrates used to produce the original vaccine may not be acceptable for use today, such as guinea pig heart cells in the case of VEE TC-83 vaccine. However, use of a new substrate would constitute a substantial product change. For live, attenuated vaccines, such as tularemia LVS and VEE TC-83, and some other vaccines that might be considered for inclusion in an expanded SIP, such as Chikungunya 181 clone 25, genetic stability may present a challenge for production of new vaccine lots. Molecular tools that allow assessment of genetic stability were not available when these vaccines were originally developed, and genetic variability when new lots are produced would have uncertain regulatory implications. This issue will be a concern, particularly for vaccines against RNA viruses, which have high mutation rates.
5.2.3
Near-Term Requirements
Even with the complexities described above, the timeline for manufacturing, releasing, and testing a new supply of an existing product may be far shorter than the timeline for developing a new vaccine to the point where it could be integrated into a program like the SIP. Assuming that there is a need for replenished supply within a short timeline (such as 3 years), new vaccine lots of selected existing investigational vaccines that have a reasonably straightforward manufacturing path and are not likely to be eclipsed by new technology within that period could be made. That applies principally to inactivated vaccines against eastern equine encephalitis (EEE), western equine encephalitis (WEE), VEE, and Rift Valley fever (RVF), and to botulinum toxin. Assuming the availability of batch-production records, cell banks, and seed stocks for the legacy vaccines, it should be possible to bring a new lot of any of them to the point of release in about 12 months. To enable production of existing vaccines, it would be useful to prepare a product-development plan for each product that defines task-specific timelines, resource requirements, and costs.
5.2.4
Choice of Manufacturing Site, Methods, Scale, and Scalability
Vaccines in the SIP were manufactured on a small scale in multiuse facilities, such as the Salk Institute Government Services Division (GSD), which is no longer in existence. A number of laboratories are engaged in early-stage research on and development of new vaccines of interest to the SIP, but they are generally small biotechnology companies or academic institutions, and only
a few have the facilities and capability to develop vaccines to the point where they could be included in the SIP or to produce or maintain vaccine supplies over a longer period.
Since closure of the Salk GSD, the DOD has had few options for obtaining vaccine development and production services. Other contractors, for example, declined to compete for the vaccine work that had been done by the Salk Institute, and discussions in 1991 with five commercial vaccine producers confirmed the lack of contractor interest (GAO 1991). In the absence of a commercial market for biodefense vaccines in the United States, commercial vaccine producers have not invested in the construction of the containment facilities needed to produce them. Commercial manufacturers explained that such facilities are expensive to construct and shockingly expensive to operate and maintain and that they would therefore seek a guarantee from the DOD before any construction effort that they would recoup their investments. One official estimated that it would cost about $25 million to construct a facility that could produce 1–2 million doses each of seven or eight vaccines a year (GAO 1991).
In many respects, the Salk GSD, which operated as a nonprofit foundation funded by government contracts, may be one model for future facilities for making vaccines against special pathogens that lack commercial markets. Other groups have called for establishment of government–industry partnerships and the creation of additional incentives to encourage greater industry participation in advanced development and manufacturing of medical countermeasures (IOM 2004; HHS 2010b; NBSB 2010a,b). Recent requests for information and requests for proposals from both HHS and DOD address components of this vaccine manufacturing gap.
In 2009, HHS issued a request for proposals for establishment of new multiuse facilities based on single-use flexible manufacturing formats for developing and manufacturing new vaccines (HHS 2009a). The request focused on a facility to handle the array of preclinical to clinical development of recombinant influenza vaccine and might potentially provide some surge capacity for MCM manufacturing more broadly. The projected costs would be split between government and industry and two contracts have recently been announced for approximately $100 million each. However, the emphasis on production of civilian pandemic influenza vaccine limits the application of such a potential development and manufacturing facility for the SIP. Recently, HHS also issued a request for proposals more explicitly focused on the U.S. MCM program (HHS 2011). The request seeks public–private partnerships in “Centers for Innovation in Advanced Development and Manufacturing” to expand capabilities for producing MCM against potential known or unknown threats, as well as additional pandemic influenza surge production capacity.
DOD has also issued a request for information on the development and manufacture of MCM as an effort complementary to the HHS undertaking
(U.S. Department of the Army, 2010). The request seeks to gather information to enhance the ability to develop countermeasures against threat agents for military needs and may also have some ability to enhance the development of countermeasures for civilian use. The request emphasizes government–industry partnership, a flexible production scale of up to 100 million doses per month, and a focus on innovative technologies and platform development. If the planning for the DOD and HHS facilities moves forward, the committee believes that new vaccines are likely to enter into development for a wider array of pathogens and toxins, including ones against hazardous pathogens of relevance to the SIP. As discussed in Chapter 2, the Army at one time considered expanding the biocontainment facilities at the Pilot Bioproduction Facility of the Walter Reed Army Institute of Research. This facility currently remains at BSL-2, but could potentially also be modified and expanded to increase options for advanced development and manufacturing of small amounts of MCM.
5.2.5
New Technologies and Longer-Term Strategies
A number of exciting innovative technologies bear directly on the future objectives of the SIP.
Platform technologies are applicable to the development of multiple products for different indications using a single, foundational technical approach. The best example in vaccinology is the use of a single vector virus, bacterium, or yeast to deliver foreign genes against the pathogen to which immunity is desired. The same vector backbone, with or without some modifications, can be reused to deliver other genes. Such chimeric constructs have been successfully developed with a variety of vector backbones. A common theme is that the vector itself provides important biological functions that are critical for efficacy of the chimeras: the ability to carry large foreign genes or multiple genes, the ability to infect or transduce cells to generate high yields of translated foreign protein, the ability to target the foreign gene to relevant cells (such as dendritic cells) for immunity induction, in some cases the ability to elicit cells that traffic to mucosal sites and induce mucosal immunity, and the ability to enhance the response by activating toll-like receptors. Examples of successful viral vector platforms include adenoviruses, poxvirus, flaviviruses (yellow fever 17D and dengue), alphaviruses (VEE and Sindbis), vesiculoviruses (VSV-Ind and VSV-NJ), measles virus, and lentiviruses. The principal problem in the use of most vectors has been anti-vector immunity, but this has been overcome in many systems, particularly in the alphavirus system, which has been shown to be insensitive to vector immunity. Most of these systems also include technology for rendering the vaccine candidates safe (by gene deletions that prevent replication in vivo or the use of replicons that generate protein or subviral particles.
Several vector platforms are essentially ready for use in generating new candidates that could be used in the SIP against existing and new target indi-
cations, including improved VEE vaccine, and vaccines against Ebola, Marburg, and others. Manufacturing methods have been developed and include well-established complementing cell lines (for example, PerC6 in the case of adenoviruses) or methods for large-scale electroporation of recombinant and helper RNA (e.g., alphavirus replicons). Cell lines used for production are widely available, and multiple products produced with the methods used for manufacturing chimeric viruses have approved INDs and are in clinical trials.
Other important platform technologies for vaccine development include the use of plasmid DNA with or without enhanced delivery systems, including cationic liposomes or electroporation devices. For some indications, it is possible to generate virus-like particles by expressing a single viral protein or by co-expressing the target viral proteins with other molecules that form particulate structures.
Many examples indicate that optimal immune responses may require priming and boosting strategies by using two different modalities. Priming with DNA followed by boosting with a live vector, priming with one vector and boosting with a different vector, or priming with a live vector followed by a protein or subunit boost may elicit the optimal response.
5.2.6
H5N1 Avian Influenza Vaccine
H5N1 influenza virus can cause life-threatening human disease and may pose a risk to laboratory workers and the public in the event of an escape of the virus from laboratory containment (via an infected laboratory worker).3 According to Biosafety in Microbiological and Medical Laboratories (BMBL), although “LAI [laboratory-acquired infections] have not been routinely documented in the literature … informal accounts and published reports indicate that such infections are known to have occurred, particularly when new strains showing antigenic shift or drift are introduced into a laboratory for diagnostic/research purposes” (CDC/NIH 2009). As noted in Section 3.5.7, a licensed, nonadjuvanted H5N1 vaccine is available in the United States, but stocks are controlled by HHS. The vaccine needs to be adjuvanted (for example, with MF59 or AS03 emulsions) for maximal immune responses and several investigational adjuvanted vaccines exist. Personnel working on H5N1 vaccine development and manufacture may be able to gain access to investigational vaccine candidate strains by other mechanisms. H5N1 vaccines are relevant to broader discussions on the use of investigational vaccines against pathogens and
toxins for the potential protection of laboratory workers, and vaccines against H5N1 could be considered for inclusion in the SIP as part of multistakeholder strategic discussions on SIP priorities.
5.3
THE INTERNATIONAL CONTEXT OF THE SPECIAL IMMUNIZATIONS PROGRAM
Infectious disease research and the development of vaccines are not limited to the United States. Many of the pathogens that are studied in the context of biodefense are exotic pathogens, and some pose public health threats in other countries and have resulted in the production of vaccines for protecting the general population from natural threat of infection. In addition, just as in the United States, a few vaccines have been developed only to an early developmental stage and might be considered for risk amelioration for potential protection of laboratory workers or for emergency response. Examples of these are presented to show the availability of alternatives that might be useful in the context of the SIP.
5.3.1
Vaccines Developed in Other Countries
A number of vaccines, approved for use for public health purposes in their countries of origin, have been used in the United States for special immunization purposes in laboratory settings under IND status. They have included vaccines against Japanese encephalitis (JE), tickborne encephalitis (TBE), and Argentine hemorrhagic fever (AHF) vaccines. Vaccines against JE have since been licensed in the United States for use in military and civilian travelers. The TBE vaccine was used briefly under the umbrella of DOD’s operations in deployment of forces to Bosnia and is now the subject of an IND held by the National Institutes of Health for use in laboratory workers. TBE and AHF vaccines are no longer available in the SIP program. As a result, some U.S. researchers travel to the countries where those vaccines are available to be vaccinated (for example, TBE vaccine is available in Canada and in many European countries).
As can be seen in the table and the discussions above, numerous vaccine candidates of potential value to the SIP either are under development in the United States or abroad or are already licensed for use in other countries. A partial list of such vaccines, focused on arboviruses, is provided in Table 5.3.
5.3.2
Vaccines Developed in the United States and Later Used in Other Countries
Some of the vaccines that were developed at the Salk Institute have proved useful in countries that have endemic diseases and disease emergencies. A vac-
TABLE 5.2 Vaccines That Are Approved or Have Been Clinically Tested in Other Countries but Not Currently Licensed for Use in the United States
|
Pathogen |
Disease |
Country |
Type of Vaccine |
|
Yersinia pestis |
Plague |
Russia |
Live, attenuated |
|
Francisella tularensis |
Tularemia |
Russia, UK |
Live attenuated |
|
Coxiella burnetii |
Q fever |
Australia (Ackland et al. 1994) |
Inactivated |
|
Tickborne encephalitis virus |
Tickborne encephalitis |
Germany (Novartis), Austria Baxter) (Fischer et al. 2009) |
Inactivated |
|
Crimean-Congo hemorrhagic fever virus |
Crimean-Congo hemorrhagic fever |
Bulgaria (Christova 2009) |
Inactivated suckling mouse brain |
|
Japanese encephalitis virus |
Japanese encephalitis |
China, India, South Korea, Nepal, Sri Lanka (PATH 2007) |
SA14, tissue culture, modified live |
|
Kyasanur Forest disease virus |
Kyasanur Forest disease |
India |
Inactivated suckling mouse |
|
Hantaan virus |
Hemorrhagic fever with renal syndrome |
South Korea, China |
Rat brain, tissue culture, inactivated |
|
Junin virus |
Argentine hemorrhagic fever |
Argentina |
Live, attenuated |
|
Ebola Zaire virus |
Ebola hemorrhagic fever |
Canada |
Vesicular stomatitis virus replicating vector |
|
Severe acute respiratory syndrome (SARS) virus |
SARS |
China |
Inactivated cell culture |
cine against Argentine hemorrhagic fever, Junin candidate 1 (Candid 1) vaccine underwent Phase III clinical testing in Argentina (sponsored by DOD), and the vaccine seed stock and manufacturing technology were transferred to an Argentine manufacturer (Instituto Nacional de Enfermedades Virales Humanas, J. I. Maiztegui, Pergamino, Argentina) in the 1990s. In that case, co-development with a foreign government and initial manufacture in the United States resulted in the successful deployment of an investigational vaccine in its endemic area (Maiztegui et al. 1998). The vaccine is now used to immunize the human population in a large area of Argentina in which the disease is endemic and has reduced the number of cases from hundreds to a few cases each year in the endemic area, has effectively controlled the disease, and has saved many lives. Even though the first immunizations in human volunteers occurred in the
TABLE 5.3 Examples of New Vaccines Under Development That Could Be Considered for Incorporation into the Special Immunizations Program
|
Indication |
BSL Level |
Lab Infections Reported |
Vaccine |
|
West Nile |
3 |
Many |
ChimeriVax-WN (YF/WN chimera, live attenuated) |
|
|
|
|
Recombinant subunit-alum |
|
|
|
|
Live, attenuated, chimeric DEN4/WN |
|
|
|
|
Live, attenuated chimeric DEN2/WN |
|
Tick-borne encephalitis |
3 |
Many (deaths) |
Inactivated whole virus-alum |
|
|
|
|
Live, attenuated DEN4/Langat |
|
Chikungunya |
3 |
Many |
Live, attenuated |
|
|
|
|
Live, attenuated, chimeric |
|
St. Louis encephalitis |
3 |
Yes |
ChimeriVax-SLE (YF/SLE chimera, live attenuated) |
|
Murray Valley encephalitis |
3 |
Yes |
Imojev® (ChimeriVax-JE, YF/JE chimera, live attenuated) |
|
Kyasanur Forest disease |
4 |
Many (deaths) |
Inactivated whole virus (cell culture) |
|
Ross River |
2 |
Yes |
Inactivated whole virus (cell culture) |
|
Dengue |
2 (3 in EU) |
Yes |
ChimeriVax-DEN |
|
|
|
|
Recombinant subunit-alum |
|
|
|
|
Live attenuated |
|
|
|
|
Live attenuated, chimeric |
|
Japanese encephalitis |
3 |
|
Imojev® (ChimeriVax-JE, YF/JE chimera, live attenuated) |
|
EEE |
3 |
|
Live, attenuated chimeric Sindbis/EEE |
|
WEE |
3 |
|
Live, attenuated chimeric Sindbis/EEE |
|
VEE |
3 |
|
Live, attenuated chimeric Sindbis/EEE |
|
Ebola/Marburg |
4 |
Yes (deaths) |
Ad5 recombinant live vector |
|
|
|
|
VSV recombinant live vector |
|
|
|
|
VEE replicon |
|
Rift Valley fevera |
4 |
Yes (deaths) |
Live attenuated MP12 |
|
aAs noted in Chapter 3, the inclusion of Rift Valley fever MP-12 in the SIP has been discussed. |
|||
|
Company |
IND |
Development Stage |
Comment |
|
sanofi pasteur (Acambis) |
Yes |
Phase II |
|
|
Hawaii Biotech |
Yes |
Phase I |
|
|
NIH |
No |
Preclinical |
|
|
Inviragen |
No |
Preclinical |
|
|
Novartis |
No |
Licensed (Europe) |
|
|
Baxter |
No |
Licensed (Europe, Canada) |
Previously under IND (Yugoslav conflict) |
|
NIH |
Yes |
Phase I |
|
|
(USAMRMC) |
Inactive |
Phase II |
Redevelopment, sanofi pasteur |
|
(UTMB) |
No |
Preclinical |
|
|
sanofi pasteur (Acambis) |
No |
Preclinical |
|
|
sanofi pasteur (Acambis) |
Yes |
In licensure registration |
Cross protects against MVE |
|
Haffkine Institute (Mumbai) |
|
Licensed (India) |
|
|
Baxter |
No |
Preclinical or Phase I |
|
|
sanofi pasteur (Acambis) |
Yes |
Phase III |
|
|
Hawaii Biotech |
Yes |
Phase I |
monovalent vaccine in clinic) |
|
NIH |
Yes |
Phase I |
|
|
Inviragen |
Yes |
Phase I |
|
|
sanofi pasteur (Acambis) |
Yes |
In licensure registration |
|
|
(UTMB) |
No |
Preclinical |
|
|
(UTMB) |
No |
Preclinical |
|
|
(UTMB) |
No |
Preclinical |
|
|
NIH, GenPhar |
No |
Preclinical |
|
|
Profectus |
No |
Preclinical |
|
|
Alphavax |
No |
Preclinical |
|
|
(UTMB) |
No |
Preclinical |
|
United States (Barrera Oro et al. 1988), the product is now being manufactured and used in Argentina and is not available in the United States through the SIP. As a result, U.S. investigators working with the virus travel to Argentina to avail themselves of the vaccine.
In 2005, Chikungunya virus caused large epidemics in countries around the Indian Ocean and ultimately spread to Europe. The Army’s Chikungunya vaccine seed stock was transferred to the French National Institute of Health and Medical Research (INSERM) and was under investigation as a potential means of controlling this disease in future outbreaks (Powers and Logue 2007). It was transferred to additional countries and development efforts are being pursued in India (Ellen Boudreau and Judy Pace Templeton, USAMRIID, personal communication).
Another investigational vaccine developed by the U.S. Army, VEE strain TC-83, was instrumental in controlling the extensive epizootic of VEE that occurred in northern South America (Colombia, Ecuador, Peru, and Venezuela), Central America, Mexico, and Texas in 1969–1971. Vaccine stocks produced by Merrell-National Laboratories were used to immunize equids, and the vaccine was transferred to a number of veterinary-vaccine manufacturers in the United States (Jochim et al. 1973). The vaccine was licensed for the vaccination of equids at risk of infection but remained available as an investigational product for human use through the SIP. The investigational formalin-inactivated RVF vaccine developed in the United States was used to help protect laboratory workers in Egypt in the outbreak of 1977–1978 (J.M. Meegan, personal communication) and deployed peacekeepers in the Sinai conflict (Niklasson et al. 1979). RVF vaccine was again requested by and provided to the Kingdom of Saudi Arabia to help protect laboratory workers and other high-risk people in the 2000 outbreak there (CDC, unpublished material). Similarly, Kenyan laboratory and field workers were afforded the use of the vaccine in the 2006–2007 outbreak in East Africa.
Willingness of the United States to provide limited uses of some investigational products has resulted in a great deal of goodwill and has not resulted in any recognized adverse events in the relatively small number of instances of use of the vaccines. The attenuated Junin virus vaccine has yielded the most benefit: the number of cases of this serious disease has been reduced from hundreds to a few each year in the endemic area. The import or export of vaccines requires that FDA be involved.4 In the examples noted above of the use of some vaccines in epidemic settings, FDA approval of export was necessary, and the materials used in clinical trials required certificates for movement.
5.4
COOPERATION WITH THE VETERINARY COMMUNITY
Most of the diseases included in the SIP are zoonoses, and many are important diseases of livestock—VEE, EEE, WEE, JE, RVF, anthrax, and Q fever. The animal health industry currently manufactures veterinary vaccines against some of those diseases. The use of a human vaccine (VEE TC-83) in equids in the face of a public health emergency in 1969–1971 has been noted above and the Salk Institute GSD-produced EEE vaccine was tested for potential protection in whooping cranes (Olsen et al. 1997). Moreover, new vaccines against some diseases of possible future interest for inclusion in the SIP (such as new VEE and RVF vaccines, and vaccines against brucellosis and glanders) are of great interest to or are in development by the veterinary community. The committee recognizes the value of closer interactions between the human-health and animal-health scientific communities in the development, production, and testing of effective vaccines against those indications. The Animal Rule for regulatory approval, which relies on animal models of human disease, is an obvious subject for such collaboration. The importance of collaboration between human-health and animal-health research communities is the basis of the One Health Initiative,5 a worldwide effort to integrate the fields of human medicine and veterinary medicine in ways that improve public health, including industry efforts to develop new medicines and vaccines.
5.5
FINDINGS AND CONCLUSIONS RELATED TO FUTURE VACCINE NEEDS IN THE SPECIAL IMMUNIZATIONS PROGRAM
On the basis of the above discussions, the committee offers the following findings related to vaccine development and use in the SIP.
-
Finding 10: In the absence of such a facility as the previous Salk Government Services Division or establishment of a new government-owned, contractor-operated vaccine facility, vaccines of interest to the SIP are unlikely to be manufactured or replaced, particularly if the pharmaceutical industry is reluctant to commit its resources to them.
-
Finding 11: Numerous vaccine candidates of potential value to the SIP either are under development in the United States or abroad or are already licensed for use in other countries (for example, Q-Vax, a Q fever vaccine developed in Australia).
-
Finding 12: Investigational vaccines developed in the United States and initially tested on human volunteers have proved valuable in other countries where the diseases may be endemic. This use of IND vaccines serves a public health goal, is a source of international goodwill,
-
and is thus an additional benefit of MCM research. The data on vaccine safety and immunogenicity obtained by immunizing SIP participants with these IND vaccines help to provide the clinical basis for such uses.
The committee concluded that a systematic assessment of the need for new manufacturing of vaccines that are now in the SIP and a systematic assessment of potential candidates that could be entered into the SIP should be undertaken as a separate exercise. Vaccines and vaccine candidates developed in other countries should be considered as part of this assessment process for possible inclusion in the SIP.
Efforts to increase vaccine manufacturing capacity under HHS and DOD contracts appear to be under way. The committee encourages such efforts to include the SIP’s current needs and capacities in making decisions on expanding the manufacturing capacity of a comprehensive U.S. MCM effort. The committee also recognizes that manufacturing new stocks of existing SIP vaccines or incorporating some or all of the additional vaccines into the SIP must occur in the context of U.S. regulatory policy, as discussed in Chapter 4.




















