
6
Pathogenesis
The pathogenesis of a disease describes the mechanisms by which it develops, progresses, and either persists or is resolved. Understanding pathogenesis of an infectious disease at the cellular and molecular levels is critical for discovering, developing, and implementing methods to prevent infection, and to improve patient outcomes after treatment.
By determining which microbial molecules establish infection by binding to and entering human cells or tissues, for example, scientists can develop vaccines against tick-borne diseases (TBDs)—as they already have for influenza. The pathogenesis of tick-borne diseases can also reveal why some individuals are more prone to severe disease, or fail to resolve infection.
Scientists rely on several methods to study the pathogenesis of TBDs. These include in vitro studies, based on cultured cells; animal studies, based on tracking animals with a disease; and patient studies, based on clinical trials and specimens from biopsies and autopsies. While no one approach can represent the full spectrum and complexity of human disease, the ability to “reduce” or “control” the number of variables by using in vitro and animal models allows more rapid and less equivocal determination of key variables in disease progression—knowledge required to improve prevention, diagnosis, and treatment of TBD in patients.
Animal models have been especially useful in shedding light on the key features of tick-borne infectious diseases. These models include naturally occurring infectious disease, such as neuroborreliosis in horses and Rocky Mountain spotted fever in dogs, and infections introduced into animals such as mice. Mice are particularly helpful in revealing the pathogenesis of infectious disease because scientists can study mice that differ only in a
single gene, and because they can use imaging to track the progression of infection and cellular trafficking in real time.
In this chapter, six scientists presented the state of the science regarding the pathogenesis of tick-borne infections—specifically those caused by pathogens in the Anaplasma, Borrelia, Ehrlichia, and Rickettsia genera.
PATHOGENESIS OF BORRELIA BURGDORFERI INFECTION AND DISEASE
Janis J. Weis, Ph.D., Department of Pathology, University of Utah School of Medicine
In humans, the bite of the infected tick is required for introduction of the pathogen through healthy skin. This extracellular pathogen starts in the dermal tissue where it begins to adapt to life in the mammalian host by changing the expression of its surface glycoproteins. At the same time, the bacterium stimulates responses of inflammatory cells and their secreted mediators that cause acute-phase lesions such as the classical erythema migrans (EM) lesion. The bacterium also activates proteases and other induced host cell molecules to allow for dissemination through the blood and into other tissues, including secondary skin lesions, joints, the heart, and nervous tissue (Coleman et al., 1997; Gebbia et al., 2004; Rosa et al., 2005).
Differences in the severity and spectrum of disease among patients infected with Borrelia burgdorferi is one of the hallmarks of Lyme disease (Steere and Glickstein, 2004). The reasons for this variation include both genetic differences among strains of the bacterium and differences in the host responses. On the bacterial side, genetically distinct strains, identified by ribosomal spacer types and outer surface protein C (OspC) heterogeneity, have been associated with invasive versus localized cutaneous disease (Wang et al., 2002; Wormser et al., 2008a). Furthermore, B. burgdorferi is characterized by a large and complex plasmid content, some of which are essential for infection and others which can vary among strains (Rosa et al., 2005). Similarly, the host response has significant differences in the host response. Among human patients, approximately 60 percent of infected patients who do not receive early treatment develop clinical arthritis (Steere et al., 1987). This difference between those patients who do and those who do not develop arthritis reflects, at least in part, genetic differences in host response. These effects can be studied using inbred strains of mice with defined genetic differences and with clearly reproducible difference in the severity of carditis and arthritis following B. burgdorferi infection (Barthold et al., 1990).
The use of gene knockout mice has begun to unravel the genetic contribution to the spectrum of the disease. Severely combined immunodeficient C3H/HeJ mice (SCID)—which lack B and T lymphocytes—develop severe
arthritis and carditis independent of the number of B. burgdorferi used to experimentally infect the mice. In contrast, C57BL/6 mice with the same SCID mutation develop only mild arthritis and carditis, again independent of the infectious dose (Barthold et al., 1992). These results suggest that although B cells and T cells are important in clearing B. burgdorferi, the adaptive immune response (mediated by B and T lymphocytes) itself does not drive severe disease. Furthermore, C57BL/6 mice with mild disease and severely affected C3H mice have equal numbers of bacteria in their ankle tissues, evidence that arthritis severity does not correlate with the bacterial load (Ma et al., 1998; Morrison et al., 1999).
Understanding how B. burgdorferi traffic to and colonize various tissues is important in shedding light on the reasons for differences in the organ-specific manifestations and severity of disease. B. burgdorferi expresses outer surface proteins that selectively interact with endothelial cells, platelets, chondrocytes, and extracellular matrix via specific interactions with integrins, glycosaminoglycans, fibronectin, and collagen (Coleman et al., 1997; Gebbia et al., 2004; Coburn et al., 2005). These interactions are important in homing to and colonization of tissues, including the skin, joint, and heart. Bacterial ligands, such as DBPA/B, p66, BBk32, and OspC, promoting heart and joint invasion have been identified by genetic and immunological techniques (Coburn et al., 2005). These receptor-ligand interactions also contribute to inflammatory responses in resident cells (Behera et al., 2005, 2006a).
The host response to B. burgdorferi plays a key role in disease pathogenesis. B. burgdorferi does not produce toxins or proteases that are directly responsible for tissue damage upon colonization. In contrast, the bacterium produces multiple molecules that activate host responses and can lead to localized and generalized inflammatory pathogenic responses. Most of these host responses normally function to contain or clear infections and are components of the innate defense and/or inflammatory response (Liu et al., 2004; Benhnia et al., 2005; Behera et al., 2006a; Oosting et al., 2010). Although their purpose is to clear infection, if continually activated, they lead to lesion development and disease.
Numerous signaling pathways have been identified that are responsible for Lyme disease arthritis (see Table 6-1). The Pam3Cys-lipid-modified proteins of B. burgdorferi, which are abundantly expressed by the bacteria, are the best characterized. These lipoproteins interact with the host, specifically through toll-like receptor (TLR)-2 and TLR-1 heterodimers, and activate signaling through the adaptor molecule MyD88. This signaling pathway results in activation of numerous proinflammatory cytokines, chemokines, and matrix metalloproteinases (Hirschfeld et al., 1999; Alexopoulou et al., 2002). In addition, the bacterial flagellin and peptidoglycan also activate host TLRs, again connecting to the MyD88 pathway (Bolz et al., 2004;
TABLE 6-1 Numerous Signaling Pathways Have Been Implicated in Pathogenesis
|
B. burgdorferi Ligand |
Host Receptor |
Signaling Pathway |
Type of Response |
|
Pam3Cys-outer surface lipoproteins (Osps) |
TLR2/TLR1 |
MyD88 & NF-kB dependent, MAP Kinases |
Pro-inflammatory cytokines, MMPs chemokines, anti-inflammatory (1, 2, 18) |
|
Flagellin |
TLR5 |
MyD88 |
Cytokines, etc. (33) |
|
Peptidoglycan |
TLR2, NOD2 |
MyD88 |
Cytokines, etc. (29) |
|
RNA |
TLR7 & 2nd unidentified PRR |
MyD88 dependent and IRF 3 dependent |
Type I IFN (α/β)(26, 30) |
|
Secreted molecules |
Unknown |
IRF3 |
Type I IFN (α/β)(26) |
|
BBB07 |
α3β1 integrin |
Endosome |
Cytokines, MMP(5) |
|
Diacyglycolipid-BbGL-IIc |
CD1d |
iNKT-TCR |
IL-2, IFNγ(19, 20) |
Liu et al., 2004; Behera et al., 2006a; Shin et al., 2008). The result of this MyD88 stimulation is production of pro-inflammatory products such as cytokines, chemokines, and matrix metalloproteinases.
Using knockout mice that lack individual components of the pathways, the contribution of specific pathways to control of infection and resolution of disease can be studied. The TLR/MyD88 pathway is important for host defense and for controlling the bacteria numbers in tissues. However, knockout mice that lack either TLR2 or MyD88 still develop arthritis (Wooten et al., 2002; Bolz et al., 2004; Liu et al., 2004; Behera et al., 2006a). These results suggest that although this pathway is important for host defense and control of the bacterial numbers, it is not essential for arthritis development. Consequently, global gene expression profile analysis was used to find pathways specific to arthritis development (rather than host defense) in C3H and C57BL/6 mice. These experiments focused on early time points in pathogenesis, 1 week after infection prior to the arrival of inflammatory cells in joint tissue. As noted previously, C3H mice had severe ankle swelling beginning at week 1, while C57BL/6 mice had minimal swelling. In C3H mice, there is an early and transient induction of genes associated with an interferon signature profile at one week that decreases by week 2. Furthermore, immunodeficient (lacking interleukin-10 or IL-10) C57BL/6 mice showed a delayed increase in the same interferon panel that remained elevated through the infection (Crandall et al., 2006).
One hypothesis was that type I interferons (IFNs), which are normally associated with host response to viral infections, are important for the development of arthritis following B. burgdorferi infection. To test that hypothesis,
the C3H mice were treated with an antibody that blocked the receptor for type I IFNs. Arthritis was reduced by 50 percent in these C3H mice given a single injection of interferon-blocking antibody before infection (Miller et al., 2008). A second study with mutant CH3 mice deficient in interferon receptors confirmed the involvement of type I IFNs. These studies provide functional evidence for the involvement of type I IFNs in the development of arthritis. This result was unexpected as most bacteria known to induce host type I interferon are intracellular, whereas B. burgdorferi are extracellular. Notably, this type I interferon pathway can be induced by at least three distinct ligands from B. burgdorferi, some of which function independently from the MyD88 adaptor molecule pathway (Petzke et al., 2009; Miller et al., 2010).
Importantly, the host responses and inflammatory pathways have organspecific differences. In contrast to arthritis, which is characterized by infiltration of neutrophils, carditis is characterized by influx of macrophages and T lymphocytes at the base of the heart where B. burgdorferi infiltrates connective tissue (Barthold et al., 1990; Ruderman et al., 1995; Bockenstedt et al., 2001). Also unlike arthritis, the numbers of infectious bacteria in the heart are correlated with the severity of inflammation (Morrison et al., 1999). Furthermore, T lymphocytes help resolve Lyme disease carditis by production of interferon gamma and other cytokines, which in turn activate the macrophages to clear B. burgdorferi from the heart, and therefore suppress the carditis (McKisic et al., 2000; Olson et al., 2009). Disruption of this interferon gamma pathway in C57BL/6 mice results in more severe carditis.
Knowledge Gaps and Research Opportunities
Weis noted three key questions for future study:
-
What is responsible for the variability in individuals’ response to infection by Borrelia burgdorferi?
-
Why do some symptoms persist in some patients?
-
What is responsible for the pathogenesis of neuroborreliosis in patients?
DURATION OF SPIROCHETE INFECTION FOLLOWING ANTIBIOTIC TREATMENT IN ANIMALS
Linda K. Bockenstedt, M.D., Yale University School of Medicine
Bacterial infections have a number of outcome determinants, including pathogen factors, host genes, host co-morbid conditions, host immunity, and the effects of antibiotics. A current debate is how effective antibiotics are in vivo against B. burgdorferi. Antibiotic treatment failures occur
occasionally in all animal models of Lyme borreliosis and in humans. Evidence for treatment failure comes from persistence of B. burgdorferi DNA after treatment for Lyme disease arthritis and, in animal models, the ability to culture spirochetes from tissues. Several studies have found that B. burgdorferi DNA can be detected in tissues for extended periods of time after antibiotic treatment of laboratory animals even though cultures of tissues may be negative (Straubinger et al., 1997; Bockenstedt et al., 2002; Hodzic et al., 2008). This raises the question of how to interpret the significance of B. burgdorferi DNA in tissues.
In our published study (Bockenstedt et al., 2002), we used xenodiagnosis with ticks to determine whether bacterial DNA detected in mouse tissues after antibiotic treatment indicated the presence of spirochetes that could replicate and cause infection. In this approach, uninfected laboratory-reared ticks were allowed to feed on mice that had previously been infected with B. burgdorferi and treated with antibiotics (doxycycline or ceftriaxone). Immunofluorescent staining was then used to determine whether the ticks had acquired B. burgdorferi spirochetes. The results were equivocal in that spirochete forms were detected microscopically in the tick midguts, but, on further study, these spirochetes appeared to be attenuated because genes on specific plasmids required for B. burgdorferi infectivity could not be detected by polymerase chain reaction (PCR) (Bockenstedt et al., 2002).
Similarly, spirochetes could not be cultured from tissues of mice that had been treated with antibiotics. No other method was used to assess viability and infectivity of the spirochetes visualized in ticks. However, larval ticks that had fed on antibiotic-treated mice could not transmit B. burgdorferi infection to uninfected mice. In a subsequent study (Hodzic et al., 2008), ticks used for xenodiagnosis of ceftriaxone-treated mice were able to transmit B. burgdorferi DNA to uninfected immunodeficient SCID mice, which lack T and B cells, and are highly susceptible to B. burgdorferi infection. In addition, B. burgdorferi DNA could be detected in some SCID mice that had received tissue transplants of skin from antibiotic-treated mice. Although viable spirochetes could not be cultured from the ticks, the antibiotic-treated donor mice, or the recipient mice, rare spirochete forms were visualized microscopically in specific connective tissues of some donor mice (Hodzic et al., 2008). These findings raised two questions. First, does the persistent DNA indicate continued infection, or is it simply residual debris? Second, are the rare residual spirochete forms viable and infectious?
To begin to address these questions, we used two-photon (multiphoton) confocal microscopy to directly visualize the location and motility of spirochetes in living, anesthetized mice in real time. C57BL/6 MyD88-deficient mice were infected with B. burgdorferi strain 297, a strain previously isolated from a Lyme disease patient but subsequently genetically modified to express a green fluorescent protein. The C57BL/6 mice are relatively disease resistant, but deficiency in MyD88 results in higher bacterial load so that
infected mice have 100- to 1,000-fold more spirochetes in the skin and other organs, thus enhancing the ability to image the spirochetes. After 21 days of infection to allow dissemination of spirochetes throughout the tissues, mice were treated with either ceftriaxone or doxycycline. Multiphoton microscopy was then used to image spirochetes in ear skin and tendons, two easily accessible sites where spirochetes often reside. After imaging, mouse tissues were tested by culture to detect viable organisms, PCR to detect residual spirochete DNA, and direct fluorescent antibody (DFA) staining to detect residual spirochete antigen.
In the experiments using ceftriaxone, mice were treated twice daily with ceftriaxone for 5 days or sham treated. We began analyzing mice during the antibiotic treatment period and up to 9 days after completion. Spirochetes could not be cultured from tissues of mice treated with antibiotics at any time point. Multiphoton microscopy revealed a large number of spirochetes moving in the ear skin of sham-treated mice, but after just two doses of ceftriaxone (1 day of treatment), only a few spirochetes remained in antibiotic-treated mice. Fewer spirochetes were seen in the tendons of sham-treated mice, and only two stationary spirochetes were found in the tendon of one mouse treated with two doses of ceftriaxone (Bockenstedt et al., 2011). The spirochetes in the tendons of sham-treated mice were less motile than those in the skin. With the exception of mice analyzed after one day of antibiotic treatment, no spirochetes could be visualized in ear skin or tendons of antibiotic-treated mice at any time. At the end of the experiment, however, DFA revealed green fluorescent material adjacent to the ear cartilage in all of the antibiotic-treated mice.
In the doxycycline experiments, mice were given a one-month course of antibiotics supplied in drinking water to maintain serum levels above the minimal concentration necessary to inhibit B. burgdorferi growth in vitro. This method of antibiotic administration sustains therapeutic serum drug levels analogous to levels achieved in humans treated with oral doxycycline. Mice were analyzed between 2 and 10 weeks after the last dose of antibiotics. Similar to the ceftriaxone-treated mice, spirochetes could not be cultured from mice treated with doxycycline. Ticks used for xenodiagnosis also tested negative by culture after feeding on antibiotic-treated mice. In sham-treated (control) mice, multiphoton microscopy revealed motile spirochetes in the skin, as well as large, amorphous collections of fluorescent debris near ear cartilage, a finding not seen in uninfected mice. Similar non-motile fluorescent material adjacent to the ear cartilage was also visualized in the treated mice (Bockenstedt et al., 2011), and these mice tested positive for B. burgdorferi DNA by PCR in both skin and joints.
To determine whether the amorphous fluorescent material contained viable and infectious spirochetes, ear tissue was transplanted into MyD88-deficient mice, which may provide a “permissive” environment for attenuated organisms. When analyzed up to 5 months after the transplant,
sera from mice transplanted with tissues from antibiotic-treated mice only showed reactivity to single bands on immunoblots. Sera from mice transplanted with tissue from sham-treated mice, in contrast, showed a banding pattern typically found in mice and humans infected with B. burgdorferi. Neither viable spirochetes nor spirochete DNA were observed in mice that had received tissue transplants from treated mice. In contrast, mice that received ear transplants from sham-treated mice did have spirochete DNA and were culture positive (Bockenstedt et al., 2011). In a separate experiment, infected immunocompetent C57BL/6 mice given a month-long course of oral doxycycline were evaluated similarly for persistent infection by skin transplantation into MyD88-deficient mice. Only one of the five mice that received ear transplants from an antibiotic-treated donor mouse developed a serologic response to B. burgdorferi as indicated by a single immunoblot band. Mice that received transplants from sham-treated donor mice, in contrast, developed evidence of infection, as revealed by tissue culture, PCR, and serologic conversion (Bockenstedt, unpublished observations).
Bockenstedt noted that a number of conclusions can be drawn from this work:
-
Antibiotics are effective in eliminating B. burgdorferi infection in immunocompetent C57BL/6 mice and even immunodeficient C57BL/6 MyD88-deficient mice. Because C57BL/6 mice are relatively disease resistant, similar studies are in progress in immunocompetent and MyD88-deficient C3H mice, a mouse strain background that is more susceptible to B. burgdorferi-induced infection and disease.
-
Spirochete debris may persist for some time after B. burgdorferi-infected MyD88-deficient mice are treated with antibiotics. More extensive analyses need to be performed to determine whether such debris occurs in different tissues throughout the host and whether this debris could serve as a nidus for stimulating inflammation.
-
Tissue transplants containing the debris may elicit an antibody response in the new host, but if the donor mice were treated with antibiotics, the tissue transplants induce responses to only one or two B. burgdorferi proteins. Only mice that receive transplants from sham-treated mice become infected, as shown by tissue culture, PCR, and seroconversion. Transplants from mice treated with antibiotics do not introduce infection into recipient mice.
DISCUSSION
The discussion session focused on the pathogenesis of Lyme disease and how these studies inform us about human disease. A participant questioned if the spirochetes were able to change their morphology and “hibernate”
under periods of stress. Bockenstedt noted that there are changes in the morphology of spirochetes when they are exposed to different stressful conditions in culture. For example, if the spirochetes are nutrient deprived, they can actually stop making peptidoglycan and fold up on themselves. However, the same phenomenon has not been observed in vivo.
Schutze questioned whether the observed residual pieces of organisms could stimulate an inflammatory cascade. Bockenstedt noted that this material does contain DNA, which might stimulate an inflammatory cascade through TLRs. The fluorescent material in the tissue specimens has not been isolated for testing, and there may be other components in the sample that would trigger inflammation. However, the current research has not been able to answer these questions.
Another participant inquired about the genetic differences between the C57BL/6 and the C3Hmice and what these differences meant for TBDs. Dr. Weis noted that some genes transcend the genetic differences among the mice strains, for example, MyD88, TLR-2, and interleukin 10. Mutations in those genes on either the C57BL/6 or the C3H background would result in a compromise either in host defense, resulting in elevated levels of bacteria in tissues, or in the case of IL-10, an increase in inflammation (Brown et al., 1999, 2008; Wooten et al., 2002). The MyD88-deficient knockout mice are particularly interesting because the bacterial number in tissues is significantly elevated compared to bacteria loads in wild-type mice (Bolz et al., 2004). She further noted that there is no indication of a change in the bacteria, but rather that they persist longer because the host cannot clear the bacteria.
Research is currently being done to understand the difference in arthritis severity between C57BL/6 and C3H mice through the use of genetic intercross populations. Weis noted that she has identified at least 12 different loci that are different between these two mice strains (Ma et al., 2009). One area of research will be to look for genes that regulate type I interferon because there is a difference in the induction of type I interferon in infected joints between the two mouse lines.
ANTIGENIC VARIATION AS A MECHANISM FOR PERSISTENT BORRELIA INFECTION
Steven J. Norris, Ph.D., Department of Pathology & Laboratory Medicine, University of Texas Medical School at Houston
Pathogens can vary in their ability to be invasive and toxic to an organism (see Figure 6-1). For example, Clostridium botulinum is very toxigenic and produces a powerful neurotoxin, but is not invasive. In contrast, C. perfringens, which causes gas gangrene, is both highly invasive and
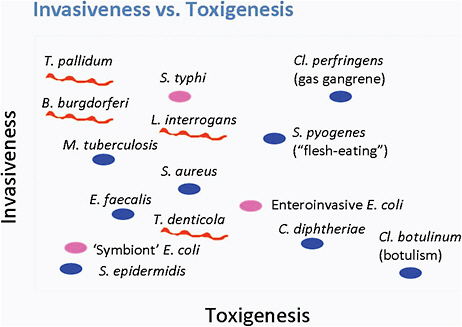
FIGURE 6-1 The relationship between invasiveness and toxigenesis.
SOURCE: Norris et al., 2010.
highly toxigenic. A group of pathogens, including Treponema pallidum and Mycobacterium tuberculosis that produce no known toxins, are highly invasive organisms that can persist for the lifetime of the host.
Lyme disease Borrelia is a highly motile organism. In animal models, B. burgdorferi disseminates early during infection into numerous tissues, including skin, joints, heart, bladder, and spleen, and persists in the tissues for up to 2 years. The persistence of infection in humans is not well understood, but likely can last months to years; however, the Borrelia produces no known toxins or enzymes that cause tissue damage. Thus, Lyme disease Borrelia falls into the group of highly invasive, non-toxigenic pathogens.
To cause persistent infection, B. burgdorferi must have multiple ways of evading a host’s immune response (see Norris et al., 2010). One common mechanism is protective niches through sequestration of the pathogen in dense tissue, such as tendons. A second cellular process is through down-regulation of antigen expression. During infection of the mammalian host, B. burgdorferi down-regulates the expression of the surface protein antigen OspA. This protein is important during the tick part of the B. burgdorferi life cycle, but the organism usually does not express OspA at high levels during mammalian infection. A third mechanism is the inhibition of a
host’s innate immune response. For example, B. burgdorferi inhibits the complement cascade by complement regulator-acquiring surface proteins. A fourth mechanism is antigenic variation, which causes a change in a surface structure that usually occurs at a higher rate than expected from mutation. Antigenic variation and, specifically, the vls gene system will be the focus of this presentation.
A 28-kilobase linear plasmid of B. burgdorferi B31 called lp28-1 contains a variable membrane protein-like sequence locus, which resembles a similar system in the relapsing fever spirochete, a prototypical antigenically variant pathogen. The plasmid contains both an expression site called vlsE and a set of silent cassettes upstream from vlsE. Alignment between the expression site and the silent cassettes reveals regions of sequence identity or relative invariant sequence and other regions of variation. Approximately 92 percent of the genetic sequences of the silent cassettes are identical to those of the central part of the expression site. In contrast, the areas of variation are important in determining the structure of the antigen expressed by the vls system.
The initial hypothesis was that each of the silent cassettes could exchange into the expression site, and therefore result in approximately 15 variants of the antigen. Subsequently, however, segmental recombination via a gene conversion mechanism was discovered in which the silent cassettes donate genetic sequences of different lengths and locations into the expression site. This recombination event appears to occur only within the mammalian host and has not been detected in standard liquid culture or in ticks. The recombination process continues as long as mammals are infected and can theoretically produce as many as 1032 different sequences of amino acids. Most variations in sequences consist of only one or two amino acids. In fact, so much variation occurs that it is rare to find the same vlsE sequence twice in a given tissue 28 days after infection (Zhang et al., 1997; Zhang and Norris, 1998; Coutte et al., 2009). The VlsE protein is anchored to the outer membrane of the organism, with the variable regions being accessible on the surface of the protein. Thus, the sequence differences in those regions provide a mechanism so that the organism can effectively evade the immune response through continuously changing the amino acid sequence of the exposed region.
The invariant regions also elicit a host immune response during infection, one of which is now used to diagnose Lyme disease (see Chapter 8). The IR6 region of the protein, also called C6, induces a particularly high antibody response in humans and other animals. This region is not the only reactive invariant region, but the one that is best characterized. Overall, it is not understood how this protein can permit evasion of the immune system while also inducing a high antibody response.
In a landmark study, the portion of the B. burgdorferi plasmid lp28-1
that carries the vls locus was deleted (Bankhead and Chaconas, 2007). These modified organisms could not infect immunocompetent mice, which quickly eliminated them within 3 weeks. These results indicate that this locus is required in enabling B. burgdorferi to evade a mammal’s adaptive immune response.
Little is known about the mechanism of vlsE recombination. Although it involves gene conversion, it does not require RecA—a protein commonly involved in recombination. The vlsE gene conversion is greatly reduced in Borrelia strains lacking the Holliday junction resolvase, which is encoded by proteins RuvA and RuvB (Dresser et al., 2009; Lin et al., 2009). As noted above, vlsE recombination occurs during animal infection but has not been detected during standard test-tube cultures of B. burgdorferi. Recent work has made progress in understanding what occurs during vlsE recombination using tissue explants from skin, heart, liver, spleen, and bladder. Explants were cultured on Gelfoam and then inoculated with B. burgdorferi to mimic what happens in mammalian tissue. After 16 days, the tissue retained normal appearance, although with loss of some lymphocytes. There was multiplication of B. burgdorferi, which was dependent on the type of tissue and the culture medium. Because vlsE recombination is a rare event, occurring in about 1 of 106 cells per generation, a PCR technique was used that distinguished between the parental and recombinant sequences in the region of the central cassette sequence where most of the replacements would change the sequence. In three of the four explant samples, the specific PCR amplicon indicative of recombination was detected. The first explant showed a recombination with cassette 7, while the second and third explants showed a recombination in cassette 2 and cassette 1, respectively. While still preliminary, this method may provide a model to study antigenic variation in vitro.
The vls antigenic variation system appears to be present in all Lyme disease Borrelia but exhibits a high degree of sequence diversity. In comparing 13 strains of B. burgdorferi, these vls sequences vary more than any other homologs in Lyme disease Borrelia (unpublished data). The identity and similarity in vlsE sequences is as low as 54 and 69 percent, respectively. That contrasts with the 69 and 79 percent values for OspC—touted as one of the most heterogeneous proteins. These results indicate that the vls system is under a high degree of evolutionary pressure and selection. Differences between the vls loci in different strains are therefore substantial and may be responsible for differences in the infectivity and virulence of the strains. There are 22 silent cassettes in B. burgdorferi strain 64B as opposed to the 15 in the initially characterized strain, B31. These 22 silent cassettes have three frameshifts. Strain 29805 has 17 silent cassettes, with a single long open reading frame. Finally, Far04 of Borrelia garinii contains 18 silent cassettes. However, most of the frameshifts in this strain occur between
silent cassettes rather than interrupting them. This meta-analysis called into question many theories about how the vls system works because invariant regions are not as conserved between different species and strains as expected. One possible next step will be to replace the vls system in one strain with the vls system of other strains to observe the effect on pathogenesis.
This work provides several conclusions. First, the vls system of antigenic variation is an important mechanism allowing Lyme disease Borrelia to evade a host’s immune system—and allowing infection to persist. Second, while the vlsE gene conversion process is not well understood, it is known to involve the RuvAB Holliday junction resolvase. Third, the tissue explant model may allow investigators to identify the factors required to regulate and carry out vlsE recombination. Finally, the vls system shows greater sequence diversity among different Borrelia strains than any other genetic component, including OspC.
Knowledge Gaps and Research Opportunities
Norris noted the following key questions for future study:
-
What are the cis- and trans-acting factors—that is, DNA segments and proteins—that regulate and carry out vlsE recombination?
-
Can investigators use tissue explant models to study the mechanisms of immune evasion and other aspects of the pathogenesis of Lyme disease?
-
Do differences in the vls antigenic variation systems in Borrelia correlate with distinct manifestations of the disease, such as arthritis and neurologic effects?
-
How can a Borrelia protein that induces strong antibody responses—now used to diagnose Lyme disease—help the organism evade a host’s immune system?
-
What other mechanisms of immune evasion are involved in persistent infection by Lyme disease Borrelia?
COLLAGEN SEQUESTRATION AS A MECHANISM FOR PERSISTENCE OF BORRELIA BURGDORFERI
Stephen W. Barthold, D.V.M., Ph.D., University of California, Davis
The persistence of B. burgdorferi in mammals is integral to the bacteria’s natural cycle of infection. Unlike relapsing fever Borrelia, which requires antigenic variation to maintain high levels of spirochetes in the blood (spirochetemia) for transmission by rapidly feeding soft ticks, B. burgdorferi needs to survive for long periods of time in its reservoir mammalian
hosts for transmission by slower feeding hard ticks. This is an inefficient mechanism of transmission, which requires that the Borrelia spirochetes disseminate widely in its reservoir host in order to maximally populate and persist in the skin, which is the host–vector interface. Persisting spirochetes become intercalated within collagen fibers of the skin and other tissues, which appears to be a unique mechanism of host immune evasion. After persisting in collagen-rich tissues, especially skin, B. burgdorferi can emerge and replicate as a host’s antibodies to the bacteria’s key antigens wane. In humans and animals, such periodic emergence of persisting spirochetes allows inflammation to recur periodically. Despite the fact that persistence is a key characteristic of B. burgdorferi, the mechanisms underlying that attribute are not well understood.
Humans are not competent reservoirs hosts because their infections are localized and multifocal, rather than disseminated and uniform, as occurs in small rodents. Erythema migrans occurs relatively early in infection after an infected tick has fed, transmitted the bacterium, and dropped off. The rash results from a person’s adaptive immune response: infiltration of lymphocytes, plasma cells, and other types of cells. The EM is a transitional point in the infection. With the evolution of the host immune response, the erythema migrans will spontaneously resolve, but spirochetes persist in the same tissue, without eliciting inflammation. Thus, one of the earliest clinical signs of Lyme disease, erythema migrans, signals the onset of the immune phase of persistent infection, when spirochetes are sequestered in collagen with minimal or no inflammation.
Various laboratories have found adhesions on B. burgdorferi that are specific to ligands in the host’s extracellular matrix (Cabello, 2007). For example, an undefined Borrelia protein was found to adhere in vitro to type I collagen lattices stripped of glycosaminoglycans (Zambrano et al., 2004). The adherence allowed the bacterium to invade the collagen lattices, where it underwent a burst of replication. Studies in the mouse model suggest similar activity in vivo (Barthold et al., 2006). For example, SCID mice are being used to study the infection process by accentuating events in the absence of adaptive immunity and then interrupting these events with the transfer of immune components back into the mouse through passive immunization or adoptive transfer.
Based on mouse model studies, there are two phases of infection (Barthold et al., 2010). The first is the pre-immune phase, when B. burgdorferi spirochetes disseminate in blood (spirochetemia) and the extracellular matrix. In this phase, spirochetes populate extracellular matrix of loose connective tissue throughout a host’s body, particularly skin, vessel walls, nerves, muscle, and myocardium (the heart’s muscular wall). Arthritis and carditis occur during this phase. In the second phase (adaptive immune phase), spirochetemia ceases and there is a generalized reduction of spirochete populations in the
host’s tissues. Disease, including the inflammation characteristic of arthritis and carditis, also resolves with the evolution of the immune response. This occurs in immunocompetent mice, mice deficient in T cells, and SCID mice reconstituted by adoptive transfer of naïve lymphocytes. Inflammation can also be resolved with passive transfer of immune serum from persistently infected immunocompetent C3H mice into SCID mice (Barthold et al., 2006). Using the antibodies from immune sera to screen an expression library, a number of antigens have been identified that may be important in disease resolution, including decorin-binding protein (Dbp) A, and arthritis-related protein (Arp), found on linear plasmid 28-1, upstream from the vlsE locus of B. burgdorferi strain B31, and elsewhere in other strains. After hyperimmunizing mice with non-lipidated recombinant DbpA and Arp proteins, and transferring the resulting hyperimmune serum into SCID mice, there is resolution of both carditis and arthritis (Barthold et al., 2006). Transfer of immune serum from infected C3H mice into SCID mice mimics what happens in immunocompetent animals: a global 10- to 100-fold reduction in spirochetes occurs. Transferring immune serum into immunocompromised MyD88 mice can produce an even greater reduction in spirochetes. However, as occurs during the evolution of the host immune response in immunocompetent mice, complete elimination of spirochetes is ineffective and spirochetes persist within collagen fibers of skin, tendons, and vascular adventitia.
In the initial pre-immune phase of infection, spirochetes are ubiquitous and present in loose connective tissue all over the body, around the vessels and nerves, in the dermis, in the myocardium, and in the muscle. As the adaptive immune response begins, most of these spirochetes will be cleared; however, as the immune response evolves, translocation of spirochetes occurs. Spirochetes move from sites of inflammation, where they are eliminated by host immune factors, into a “safe zone” of the more collagenous type I milieu of adjacent tendons, ligaments, and vessel walls. This translocation coincides with formation of multifocal colonies of spirochetes, similar to that seen in vitro within collagen lattices. This phenomenon of spirochete proliferation is not specific to mice, but appears to occur in other mammals, such as during neuroborreliosis in horses. In these examples, spirochetes appear to be responding to the adaptive inflammatory environment that they encounter during the first stage of infection.
To understand the role of DbpA/B and Arp proteins in the translocation process, SCID mice were infected with B31 spirochetes from which either the Dbp A and B locus or the Arp locus had been knocked out. Similar to the normal infection, spirochetes were observed in the adventitia, the myocardium, and the media of the vessels. However, when the infected SCID mice were treated with immune serum from normal persistently infected mice, the antibodies cleared the DbpA/B-spirochetes from all three sites and the animals became PCR- and culture-negative. In contrast, in SCID mice infected
with Arp-spirochetes and treated with immune serum from normal infected mice, large numbers of Arp-spirochetes remained in all three sites, each mouse was still spirochetemic, and carditis worsened. These findings reveal very specific interactions between spirochetes and different types of tissue.
In the immune phase of persistent infection, when spirochetes are sequestered within collagen and inflammation is absent, spirochetes seem to be no longer motile and are not replicating. Initial analysis of their RNA transcription profiles also reveals transcriptional changes—such as down-regulation of flagellin, for example. This apparently dormant persistent state within dermal collagen allows spirochetes to wait for a tick vector to initiate feeding. This dormancy is maintained by the host immune response. For example, passively transferred immune serum administered to SCID mice will induce this state of persistence without inflammation, but if the antibody is allowed to decay, spirochetes become active again, disseminate, and reinduce inflammation. In persistently infected immunocompetent mice, biologically active antibodies (those that induce disease regression and spirochete reductions in tissues) peak at 60 days and then progressively decline. Thus, the host immune response wanes, despite the presence of persisting spirochetes. This suggests that persisting spirochetes, by virtue of their collagen-sequestered dormant state, are not apparent to immune surveillance. Despite these findings, scientists are far from a complete understanding of the process by which B. burgdorferi spirochetes persist and evade immune clearance.
Knowledge Gaps and Research Opportunities
Barthold noted that a number of key questions need future study:
-
Does collagen have a specific B. burgdorferi ligand?
-
What is the role of innate immunity in the sequestration and persistence of B. burgdorferi?
-
What is the role of adaptive immunity in the sequestration and persistence of B. burgdorferi?
-
What replicative and metabolic activity do sequestered B. burgdorferi undergo?
-
What is the antibiotic tolerance of sequestered B. burgdorferi?
DISCUSSION
Following the presentation, a number of participants questioned the panelists about the studies of persistence in animal models. The range of the type of questions asked is summarized for the reader.
One participant noted that it is stated frequently that Borrelia burgdorferi is an extracellular bacterium, but wanted the speakers to discuss
the studies that show that Borrelia is found intracellularly under certain conditions. Barthold said he consistently observes extracellular forms of spirochetes, fully elongated and in association with collagen, but any intracellular organism are observed in a macrophage, where they are believed to be killed. Norris agreed, but noted that in rare instances Treponema pallidum, also a spirochete, can become an intracellular bacterium.
Another participant questioned how neurologic symptoms occur if the bacterium is just in collagen, even if it is associated with neural tissue. Barthold noted that mice do not get central nervous system disease, possibly because they don’t have much connective tissue in their brain. However, central nervous system disease is seen in larger mammalian species that have more collagen in their meninges and perivascular spaces. Under those circumstances, Barthold has observed spirochetes in the collagenous areas and along the perivascular spaces into the brain. Because there is a dearth of good analysis of human neuroborreliosis cases, it is not known if the spirochetes are located in other areas. He noted that a tissue bank or biorepository would be very valuable to allow for these types of analysis.
Another participant noted that the studies by Bockenstedt had affirmed that 28 days of doxycycline was effective in treating the newly infected immunocompetent mice, but that the transplant debris only tested positive in one out of five mice, or 20 percent. The individual questioned if studies were planned that looked at animals where treatment was delayed. Bockenstedt noted that she is planning to do such studies in which infection is first established and allowed to progress to a persistent stage before administering antibiotics. These experiments will be done in C3H immunocompetent and C3H MyD88-deficient mice. She further commented on where spirochetes can be seen by multiphoton imaging in wild-type mice and MyD88-deficient mice that have been infected for 6 months. She noted that while spirochetes may be more difficult to find in immunocompetent mice at 6 months of infection in comparison to earlier periods, they are always found in extracellular collagen-rich matrixes, not inside cells.
Gerber noted a need to understand the phenotypic expressions of disease in humans and to identify the physiological, metabolic, and genetic determinants that affect disease expression. She questioned if the research involving C3H and C57BL/6 mice had incorporated both males and females and what metabolic differences (e.g., fat metabolism, obesity, propensity to develop tumors) had been identified. Weis noted that both male and female mice are used and there is not a gender difference in the arthritis severity. For many experiments, female mice are used for simplicity, but the results are consistent with male mice. To begin to understand the metabolic differences, Weis suggested the use of Genome Wide Association Studies in humans infected with Borrelia burgdorferi and displaying different symptoms.
These studies have provided clues to genes involved in hypertension, diabetes, and other conditions regulated by a complexity of traits.
PATHOGENESIS OF EHRLICHIA AND ANAPLASMA INFECTION AND DISEASE
Nahed Ismail, Ph.D., M.Sc., Department of Pathology, Meharry Medical College
Ehrlichia and Anaplasma are small obligate, intracellular gram-negative bacteria with a characteristic dimorphic appearance and cell wall ultrastructure. They reside in cytoplasmic endosomes generally within hemopoietic cells that have evolved in close association with ticks and reservoir hosts. There are several species of Ehrlichia and Anaplasma, with E. chaffeensis being the causative agent of human monocytic ehrlichiosis (HME) and A. phagocytophilum being the causative agent for human granulocytic anaplasmosis (HGA). In the mammalian hosts, including infected humans, the primary target cells of E. chaffeensis and A. phagocytophilum are, respectively, monocytes and neutrophils.
E. chaffeensis is a small bacterium with a 1.2–1.5 mb genome. Unlike classical gram-negative bacteria, this pathogen lacks both peptidoglycan and lipopolysaccharide (LPS), but it uses cholesterol acquired from the host to maintain membrane integrity. A similar mechanism is used by Anaplasma. The E. chaffeensis P28 outer membrane protein family stimulates specific antibody responses in humans. P28 is also immunoprotective: Antibodies against it protect mice from fatal infection. However, the presence of a large family of P28 proteins may also enable the bacteria to evade the host’s immune system and adapt to different hosts such as ticks and mammals. Several secreted Ehrlichia proteins have tandem repeats associated with interaction between the pathogen and the host. Furthermore, several proteins with eukaryote-like ankyrin domains, which influence transcription and translation of genes in the host, have been described. The mechanism for delivering secreted proteins into the host cell cytosol is not completely understood but in part uses the type IV secretion system (TFSS).
Ehrlichia and Anaplasma have developed mechanisms for evading a host’s immune response. For example, Ehrlichia down-regulates cytokines essential for stimulating a protective Th1 phenotype of acquired immune response and subsequent elimination of the bacteria. These cytokines include IL-12, IL-15, IL-18, and MHC class II. Ehrlichia and Anaplasma also down-regulate the TLR2 and TLR4 receptors that the innate immune system uses to recognize and respond to Ehrlichia. Furthermore, Ehrlichia and Anaplasma also down-regulate several bactericidal mechanisms of monocytes and neutrophils, including degradation of p22phox, inhibition
of superoxide generation, and inhibition of phagolysosomal fusion. To survive and replicate inside cells, the bacteria induce apoptotic inhibitors or decrease expression of apoptotic inducers.
As discussed in the previous chapter, HME can manifest as either a mild, self-limited disease or a severe fulminate disease with a toxic shock–like syndrome. Patients with severe HME usually have multiorgan dysfunction that progresses to multiorgan failure. Although their symptoms may be nonspecific, patients with severe HME also present with marked leukopenia, lymphopenia, marked thrombocytopenia, and elevated liver enzymes. There is a disconnection between the number of bacteria in the blood of HME patients and the severity of the disease. This suggests that the pathogenesis of the disease and the outcome of infection have a significant immune-mediated component. The first task is therefore to characterize the molecular and cellular immune mechanisms that contribute to Ehrlichia-induced toxic shock. The long-term goal is to develop both a vaccine and an immune-based therapy.
A well-established fact is that protective immunity against several intracellular bacteria is mediated by Th1 cells that promote cell mediated immune responses (O’Garra and Murphy, 2009). Stimulation of T cells occurs when bacteria is phagocytosed by the host antigen-presenting cells (APCs), processed into small peptides and presented to naïve CD4+T cells and CD8+ T cells in the context of MHC class II and I, respectively. Following activation, T cells differentiate into either Type-1 or Type-2 cells depending on costimulatory signals cytokine environment. Intracellular bacteria stimulate IL-12 production by APCs to induce Th1-type cells that produce large amounts of IFN-γ. IFN-γ produced by Th1 cells activate macrophages to kill the bacteria, activate the bactericidal mechanisms of neutrophils, and enhance or stimulate an antibody response, mainly IgG2a antibodies. The latter allows opsonization (i.e., engulfing and digesting) of extracellular bacteria, and the killing of intracellular bacteria.
Immunocompetent mice have been used to understand how host defenses interact with the bacteria and contribute to resolution or progression of disease. Although E. chaffeensis does not accurately recapitulate human infection and disease, the related Ixodes ovatus Ehrlichia (IOE), which is highly virulent, and Ehrlichia muris, which is mildly virulent, do recapitulate key features and have been used in a series of elucidating studies. The disease is dose dependent. For example, mice receiving a large dose of IOE intradermally died on day 10 post-infection, while mice receiving a low dose of IOE survived (Stevenson et al., 2006). Specifically, the mice that died developed focal necrosis, apoptosis, and toxic shock–like syndrome. However, as in humans, despite severe tissue injury, these mice did not have evidence of overwhelming infection.
The immune mechanisms responsible for fatal disease were examined.
CD4 T cell proliferation and the frequency of CD4 Th1 cells were decreased, which, as noted, are very important in clearing Ehrlichia and intracellular bacteria from the host. The mice that died also had a concomitant increase in proinflammatory and anti-inflammatory cytokines TNF-alpha and IL-10—both implicated in tissue injury (Ismail et al., 2004, 2006). Furthermore, there was a marked expansion of CD8 T cells producing TNF-alpha in the mice that died. Mice that lacked CD8+T cells survived a lethal low-dose infection with IOE compared to similarly infected wild-type mice (Ismail et al., 2007). Survival of IOE-infected CD8+T cell deficient mice was associated with enhanced bacterial elimination, increased numbers of CD4+Th1 cells, decreased TNF-alpha production, and decreased tissue injury. These data suggest that CD8 T cells play a pathogenic role during severe and fatal monocytic ehrlichiosis by mediating apoptosis of CD4 T cells, decreasing Th1-type responses, and immunopathology.
Although fatal ehrlichiosis is associated with an increase in pathogenic CD8+T cells, which possibly mediate leukopenia and low CD4+T cell count, the mechanism by which Ehrlichia induced this pathogenic response is not yet known. It is well known that early interactions occur between the host’s antigen-presenting cells and innate lymphocytes, such as when natural killer (NK) and natural killer T (NKT) cells influence the subsequent acquired immune response against intracellular pathogens. Unlike conventional CD4+ and CD8+T cells, NKT cells recognize endogenous host self-ligands as well as foreign microbial ligands (e.g., glycolipids, lipoprotein, or even cholesterol) presented by antigen-presenting cells through a receptor called the CD1d molecule, which is a non-polymorphic MHC class I-like molecule. For gram-negative bacteria that have LPS such as Salmonella, NKT cells are stimulated via signals mediated by endogenous self-ligands presented in the context of CD1d and signals generated by toll-like receptors (TLRs are pattern recognition receptors). In contrast, alpha protobacteriae, including Ehrlichia that lack LPS, appear to have a specific bacterial ligand that directly stimulates NKT cells (Mattner et al., 2005). NKT cells are essential for eliminating the bacteria, thus NKT-deficient mice succumb to an overwhelming bacterial infection (Stevenson et al., 2006). Moreover, NKT cells prevent chronic joint inflammation after infection with Borrelia. A recent study has shown that Lyme disease patients seem to have a low number of NKT cells and low migration of those cells to joints, which was postulated to be an etiologic factor that contributes to arthritis in Lyme disease patients. One recommendation from this study was to propose enhancing the stimulation of NKT cells, and their migration to peripheral tissues, so they would suppress joint inflammation (Tupin et al., 2008).
One question that remained was whether NK cells are functionally
similar to NKT cells, or whether they have different roles during infection. In fatal ehrlichiosis, NK cells expand in the liver by day 7 post-infection and produce most of the cytokines produced during fatal ehrlichiosis, including TNF-α, IFN-γ, and IL-10 (Stevenson et al., 2010). Furthermore, these NK cells are also highly cytotoxic. The next step was to show a causal association between NK cells and development of immunopathology and fatal disease. After depleting NK cells from mice, there was a significant decrease of the systemic cytokine production, mainly IL-10 and TNF-alpha, and a decreased number of apoptotic cells and necrotic foci, suggesting that NK cells directly or indirectly mediate tissue injury during fatal ehrlichiosis. Even more surprising, the absence of NK cells enhanced the elimination of bacteria (Stevenson et al., 2010). That suggested, conversely, that NK cells inhibit effective elimination of bacteria. Together, those findings suggest that interaction of virulent Ehrlichia with antigenpresenting cells following high-dose lethal infections results in strong stimulation of cytotoxic NK cells. These data suggest that NK cells are possibly the main inducers of the harmful/pathogenic immune responses seen in ehrlichiosis, including generation of pathogenic CD8+T cells and development of CD4+Th1 hyporesponsiveness. The mechanism by which NK cells promote pathogenic responses following ehrlichial infection is not completely known, however, it is possible that this occurs via stimulation of IL-10 production, as well as via pro-inflammatory cytokines. Human patients with fatal HME had increased Th2 immunosuppressive cytokines, mainly IL-10, as compared to those with mild disease (Ismail et al., unpub. data). Patients with fatal outcomes also had a higher level of NK and monocyte chemokines, IP-10 and MCP-1, and decreased T cell chemokines, including RANTES. In addition, these patients had increased pro-inflammatory cytokines, including IL-1 alpha, IL-6, and TNF-alpha, and increased neutrophil chemokines, including IL-8 and granulocytic colony-stimulating factor.
In conclusion, cytokine dysregulation and expansion of pathogenic NK and CD8 T cells are the main immunopathological mechanisms in ehrlichiosis that mediate tissue injury and organ dysfunction. On the other hand, hyporesponsiveness of CD4+ T cells, decreased number of CD4+T cells, and a late-stage apoptosis (programmed cellular death) of CD4+T cells also contribute to severity of disease, possibly by failure to control continuous microbial stimulation of NK and CD8+T cells. These findings are consistent with Dumler’s findings in a murine model of HGA, in which pathogenic innate responses consisting of uncontrolled macrophage activation, NK, and NKT play a role in the immunopathology caused by Anaplasma phagocytophilum infection in mice.
Knowledge Gaps and Research Opportunities
Ismail noted the following areas are critical ones for future study:
Understanding the Bacteria
-
The regulatory mechanisms that control the developmental cycle of E. chaffeensis;
-
Proteomic analysis of the biphasic forms of E. chaffeensis, to identify the determinants of invasiveness and virulence;
-
The mechanistic details of how the T4SS and other secretion mechanisms secrete Ehrlichia and Anaplasma effectors, and their subcellular sites of action; and
-
Identification of effector candidates—including ankyrin-motif bearing proteins and cognate partners secreted via T4SS or other secretion apparatus. This will provide a molecular basis for understanding pathogen subversion of host defense, and disease.
Understanding the Host
-
Immune defense mechanisms and regulation at the peripheral sites of tick-borne Ehrlichia infection, such as the skin, liver, and lung;
-
The relative contribution of specialized Langerhans cells, hepatocytes, Kupffer cells, and endothelial cells to immune surveillance, immunity, and pathology;
-
Local factors influencing dendritic cell, NK, and T cell recruitment and differentiation;
-
The mechanisms controlling the cross-presentation of endosome/phagosome-derived Ehrlichia antigens to CD8+T cells; and
-
The role of regulatory T cells in controlling immune responses to Ehrlichia.
Potential Therapeutics
-
Molecular and cellular profiles of mild and fatal infections in patients with HME;
-
Collection of human samples, such as blood, cerebral spinal fluid, and tissues;
-
Development of screening tests, including biomarkers, to identify individuals at early stages of infection, and those at risk for progressive disease;
-
Studies of the efficacy of highly promising interventions in animal models of disease; and
-
Characterization of host defenses and immune responses in models of tick-transmitted Ehrlichia and Anaplasma infections that mimic mild and severe HME and HGA.
PATHOGENESIS OF RICKETTSIAL INFECTIONS
Gustavo Valbuena, M.D., University of Texas
Rickettsia are small, obligate intracellular bacteria in the Class α-Proteobacteria. Ticks serve as both vectors and primary reservoirs of spotted fever group Rickettsia as they can transmit the Rickettsia between stages and transovarially to the next tick generation. In general, small mammals act as amplifying hosts and human infections are accidental. The main target tissue in mammalian hosts, including humans, is the endothelium, which lines the interior of the vascular system.
Taxonomically, Rickettsia can be subdivided into four groups: typhus, spotted fever, transitional, and ancestral. In North America, the spotted fever group and the typhus group are of most concern. Rickettsia rickettsii cause the most severe rickettsiosis in North America, Rocky Mountain spotted fever. However, the newly discovered R. parkerii also produces an important disease syndrome, although apparently less severe than that produced by R. rickettsia.
Ticks can survive for long periods while harboring Rickettsia, although the organisms may decrease the fitness of the tick. When a tick attaches to a host, the Rickettsia are “reactivated”—a poorly understood process that requires 12 to 18 hours and results in the Rickettsia acquiring an infectious phenotype. Because hard ticks take several days to feed on a vertebrate host, they produce substances that inhibit the host’s immune and coagulation systems, possibly allowing infection to become established.
Rickettsia enter endothelial cells rapidly through a process of receptor-induced endocytosis: enzymes produced by the bacteria rapidly lyse the endocytic vacuole and move into the cytoplasm, where they replicate (Weiss, 1973). Several mechanisms are likely involved in damaging the endothelium. The first is cell death, necrosis (Silverman, 1984), in which the replicating Rickettsia lyse the cell. Second, there is evidence of increased oxidative stress as cells respond to the intracellular infection(Rydkina et al., 2004). Third, although cells could also die through apoptosis, there is evidence that rickettsia can inhibit apoptosis to favor its own survival (Bechelli et al., 2009). In addition, during the infection, Rickettsia induce increased production of nitric oxide and several lipid mediators derived from the cyclooxygenase system, particularly COX-2 (Rydkina et al., 2010).
Once infection is established, endothelial cells acquire an activated
phenotype that can trigger coagulation and activate the host’s inflammatory response. The endothelial cells can express cytokines and other immunomodulatory substances, and adhesion molecules, which recruit leukocytes to the infection sites (Valbuena and Walker, 2009). The inflammatory response is mediated, in part, through NF-kappa B, an important transcription factor that regulates many immune response genes (Sahni et al., 1998). Other mechanisms in the host’s inflammatory response include synthesis of inflammatory cytokines, including IL-1 and IL-8. When endothelial cells interact with immune cells, especially if the latter are producing interferon gamma and TNF-alpha, as in the case of NK cells or CD8 T cells, the endothelial cells become activated and kill Rickettsia. A goal is the harness the mechanisms that allow the endothelium to kill the pathogen for use in treating disease.
There are a number of reasons why Rocky Mountain spotted fever often becomes severe and results in a high case fatality rate. Endothelial cells normally form a barrier in the vasculature and balance the movement of fluid between the intravascular and extravascular spaces. The disruption of this barrier due to rickettsial infection of the endothelium affects these functions and results in leakage of fluid. When this occurs in organs such as the brain or lungs, the disease can rapidly progress. Rocky Mountain spotted fever may also be severe because it is a systemic infection involving cells that regulate the coagulation and immune systems. Furthermore, clinicians often confuse Rocky Mountain spotted fever with viral illnesses—for example, influenza in North America and dengue fever in Latin America. This confusion can have severe consequences because early suspicion of spotted fever can result in effective treatment with the antibiotic doxycycline. However, once patients develop the full spectrum of disease, physicians may refer them to higher level hospitals, which may treat the patients with newer broad-spectrum antibiotics—to which R. rickettsia are frequently constitutively resistant. Another barrier to combating Rocky Mountain spotted fever is that current diagnostic tests rely on antibodies, which are produced after the infection has already disseminated.
Knowledge Gaps and Research Opportunities
Valbuena noted that the number of key areas for future study include:
-
Determination of the mechanism by which Rickettsia are reactivated in the tick to an infectious state. (The fact that the bacteria must be reactivated allows for public health intervention. For example, because ticks must remain attached to a host for at least 6 to 8 hours to transmit R. rickettsii, people at risk for exposure could prevent infection by checking their bodies daily for ticks.)
-
Further definition of the cells that are initially infected and the underlying early pathology. Rickettsia could be transmitted directly into vessels and cause rapid systemic infection or, alternatively, the bacteria could move into lymphatic vessels, and from there into local lymph nodes, triggering an early response of the immune system.
-
Understanding of the preference of Rickettsia rickettsii to infect endothelial cells in vivo, given that they can infect numerous cell types in vitro.
-
Better understanding of the roles of autophagy and of the activated innate intracellular mechanisms is needed.
-
Identification of the metabolic pathways used by Rickettsia during growth and replication in the cytosol.
-
Identification of genes and proteins differentially expressed and required for growth in mammalian hosts versus tick vectors.
-
Identification of the R. rickettsii antigens that stimulate a protective immune response. This will be essential for development of a vaccine against Rocky Mountain spotted fever.
-
Development of better animal models including those that better recapitulate the natural mode of transmission via tick bite and include human tissue and immune systems.
-
Study of Rickettsia from a systemic approach that considers the response of the vector to the host, the response of the host to the vector, the response of the vector and host to the bacteria, and the response of the bacteria to the vector and host.
Three overall priorities for addressing rickettsial diseases were highlighted:
-
Diagnostics. People now die of these diseases because clinicians have no effective way to diagnose them at the early stages of disease when antibiotic treatment is most effective.
-
More studies of the disease pathogenesis. These would allow scientists to develop treatments for severe disease, such as when complications arise because of delayed treatment.
-
Vaccine development. This would allow prevention of infection and disease in endemic areas and for individuals at high risk of exposure.
DISCUSSION
One participant asked how much is known about the intracellular tick-borne pathogens in the tick and the triggers for reactivation. Valbuena noted that in the Rickettsia, research has observed that all tissues in the ticks are infected. Thus when the ticks bite, their salivary glands are already infected.
Those rickettsiae in the salivary glands undergo a mechanism of reactivation by a process that is still poorly understood. He further noted that in nature Rickettsia can be transmitted through stages of the tick life cycle, as well as to the next generation by transovarial passage. In general, ticks do not require an amplifying host, unlike Ehrlichia, to maintain the infection in nature. Ismail noted that studies in animal models of HME and patients infected with E. chaffeensis have identified infection-induced production of chemokines, which are attractants for monocytes and other host target cells such as neutrophils. The influx of these cells into the skin would provide a niche for further replication of ehrlichiae within the mammalian host. However, it is not yet clear whether a similar process occurs in infected ticks.
Another participant asked if there may be a potential therapeutic approach by targeting the mechanism where the infected host has to get the pathogen back to the tick. Ismail noted that in the knowledge gaps there is a need to study different parameters of the immune responses at multiple time points in humans, reservoir host (e.g., white-tailed deer), and vector host. This would include examining the different stages of infection to see how the bacteria progress from initial infection until the tick again acquires the bacterium.
Gerber asked if Valbuena had done dose responses and then looked at the CD4 or CD8 T cell response in mice. Valbuena noted that these experiments were done in Walker’s laboratory. In the mouse model, when R. conorii, which is similar to R. rickettsii, is injected at a relatively low dose into the mouse, the animals become ill but do survive and establish a solid immunity. If these mice are subsequently challenged with extremely high doses, they will not succumb to the disease. In contrast, a very high dose will result in death of a naïve animal. These results may imply that a dysregulation in the immune response occurs when the dose is too high but also indicate that protective immunity, as a basis for a vaccine, is feasible. Ismail stated that for Ehrlichia infection, they noted that the dose can control the magnitude of immune responses and determine the outcome of infection. For example, a strong correlation between the dose of Ehrlichia and decreased CD4+T cell count and apoptotic death of activated CD4 T cells and NKT cells had been established in murine models of fatal monocytotropic ehrlichiosis. In fact, when mice were treated with doxycycline, the number of NKT cells could be restored.
CONCLUDING THOUGHTS ON PATHOGENESIS
Guy Palmer, D.V.M., Ph.D., College of Veterinary Medicine, Washington State University
The overriding lesson from these scientists is the yin and yang of the immune response to tick-borne pathogens in the Anaplasma, Borrelia,
Ehrlichia, and Rickettsia genera. On the one hand, the immune response controls the number of pathogens and helps people and other animals avoid massive systemic infection. On the other hand, the immune effectors themselves—especially those of the innate immune system—cause inflammation and tissue damage. These lessons apply to both early and persistent phases of infection, corresponding to acute and chronic disease.
Scientists have clearly identified the innate immune system, and specific immune effectors, as mediating inflammation in Lyme disease. In fact, the mechanisms underlying inflammation and damage can be organ specific. That is, the mechanism producing arthritis differs from that leading to carditis.
Studies based on genetically defined lines of mice have clear relevance to pathogenesis in humans, as individual patients may suffer severe symptoms in some organs and not in others. Comments from patient advocates throughout the discussion—and indeed throughout the workshop—underscored this variation in symptoms. Presenting scientists and discussants alike emphasized the need for better markers of the progression in severity and chronicity of tissue damage. This is a notable translational gap between basic research on the science of Lyme disease and help for patients.
The persistence of B. burgdorferi infection is complex and involves both antigenic variation and sequestration. That is, the bacteria’s ability to generate novel variants that display new antigens on their surface allows them to escape the host’s adaptive immune system. How these variants may alter the response of cells and tissues and inflammatory immune responses remain unanswered questions. Other knowledge gaps include how the bacteria’s repertoire of surface proteins varies among strains, and how those variants affect disease.
The evidence that infectious spirochetes sequester in sites protected from antibodies also raises important questions. Are these spirochetes truly quiescent in replicating and in stimulating inflammation? How similar are these spirochetes to “persister” cells described in bacterial infections? Do these sequestered bacteria reemerge during actual infection—as suggested by passive serum transfer experiments? Experimental approaches can likely help close these knowledge gaps, but applying the findings to human infection will again prove challenging.
Unlike passive techniques such as PCR, scientists can use imaging to detect viable organisms. Imaging has therefore provided new answers to vexing questions regarding whether or not infection persists even after antibiotic therapy. These questions concern the migration of bacteria to and from transmission sites, and the responses of cells to viable as well as nonviable bacteria. Indeed, the detection of “remnant” material at infection sites raises questions about whether an antigenic stimulus persists even after viable, replicating bacteria are killed. The use of imaging technology may
also allow scientists to examine host pathogen interactions concerning the progression of Lyme disease in deeper tissues such as the joints and heart.
Infection with rickettsial pathogens, including those in the Anaplasma, Ehrlichia, Orientia, and Rickettsia genera, can progress so rapidly that patients require immediate hospitalization and intensive care—along with antimicrobial therapy—to prevent death. The acuteness and severity of these infections highlight the need for better educating medical professionals in regions where the organisms are endemic. Investigators also need to better define these endemic regions and determine the risk that infectious bacteria and their animal hosts as they shift their range and distribution, and the likelihood that new pathogens will emerge.
Finally, we need more accurate tools for clinical and laboratory diagnosis of these diseases. The reasons underlying differences in the severity and rapidity of progression in patients is a major scientific gap—both on the pathogen side (diversity of species and strains) and the host side (genetic background and immune status).
With only a few exceptions, the pathogenesis of the broad group of rickettsial diseases is understudied—typical of many neglected diseases of significant but underappreciated significance for public health. Workshop presenters and discussants emphasized all these challenges.
However, two experimental models reveal the progress that scientists can achieve. Experiments using the Ixodes ovatus Ehrlichia are some of the best so far and underscore the dominant theme of the session: that the immune system is responsible for controlling infection but also producing the severe toxic shock–like syndrome when that control gets out of hand. A better understanding of immune mechanisms and effectors is critical to improving therapy once infection has progressed to severe acute disease.
Research on Rickettsia in the spotted fever group has similarly begun to elucidate the pathogenesis of severe disease. Progress in developing animal models illustrates the possibilities. Still, the knowledge gaps regarding the pathogenesis of the rickettsial pathogens are numerous and wide, and the need for experiments that lay the groundwork for translating that knowledge to human disease is strong.
In fact, such translational studies are essential for the full spectrum of tick-borne pathogens. To avoid a “translational canyon” between experimental studies and human treatment and prevention, scientists should consider studying B. burgdorferi in naturally occurring models, such as neuroborreliosis in horses and Rickettsia rickettsii and Ehrlichia canis in dogs. The use of “humanized” organs such as human skin in mouse models—as noted by Valbuena—can accelerate scientists’ understanding of pathogenesis, and speed the application of that understanding to treating patients and preventing infection.




























