
External Factors That Affect the Medical-Device Regulatory System
Chapters 3, 4, and 5 review and analyze how the Food and Drug Administration (FDA) regulates medical devices. However, that regulatory process does not exist in isolation. It is a part of a broad landscape consisting of such additional factors as the increasing complexity of medical devices, the process of innovation, the business environment in which medical devices are developed, and the international medical-device regulatory arena. This chapter explores those factors and how they have the potential to affect the regulation of medical devices in the United States.
THE GROWING NUMBER AND COMPLEXITY OF MEDICAL DEVICES
The number of 510(k) submissions to the FDA varies from year to year (see Figure 6-1). In 1976, fewer than 1,000 510(k) submissions were received by the FDA’s Center for Devices and Radiological Health (CDRH). The number of submissions reached about 7,000 in 1989 (in part because of a change in the status of examination gloves from 510(k)-exempt to non-exempt). In 2009, about 4,000 submissions were received (FDA, 2010a). The number of 510(k) submissions declined most dramatically from the early 1990s to the middle 2000s. At least three changes occurred during this time, which might have impacted 510(k) submission numbers.
The first change was publication of the Temple report in 1993, a review of the quality of clinical science submitted to CDRH in support of 510(k) submissions and PMA applications (FDA, 1993). The report was critical, observing (albeit on the basis of a small sample of PMA applications and
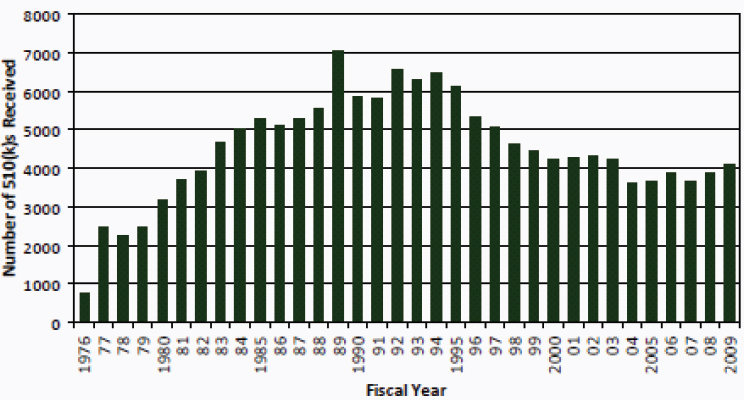
FIGURE 6-1 Original 510(k) submissions to CDRH, FY 1976–2009.
SOURCE: FDA, 2010a.
510(k) submissions containing clinical data) that studies submitted in support often failed to meet fundamental scientific standards. In response, the FDA initiated recruitment of additional scientists, including physicians and scientists, to perform premarket review of devices. While the review standard for 510(k) submissions was unchanged (that is, substantial equivalence), the review determinations began to shift from a descriptive to a data-driven base. The second change was issuance of FDA’s final guidance, in January 1997, advising industry as to when modifications to a cleared device could be made without submitting a new 510(k) notification (FDA, 1997).1 This guidance gave manufacturers autonomy in making decisions about devices with changes that were sufficiently minor as to preclude the need for premarket review. Finally, passage of the FDA Modernization Act in 1997 resulted in exemption of most Class I and many Class II devices from premarket review.2 This Act resulted in decreased numbers of low-risk and, in some cases, moderate-risk devices subject to premarket review.
Those changes in administration, regulation, and statue might have had an effect on the volume of 510(k) submissions. The committee was unable to draw conclusions, however, on the basis of the available data as to whether innovation was influenced in any way by these changes.
___________________
1Updated guidance was issued by the FDA on July 27, 2011, after the committee had completed its work.
2Food and Drug Administration Modernization Act of 1997, Pub. L. No. 105-115, 111 Stat. 2296 (1997).
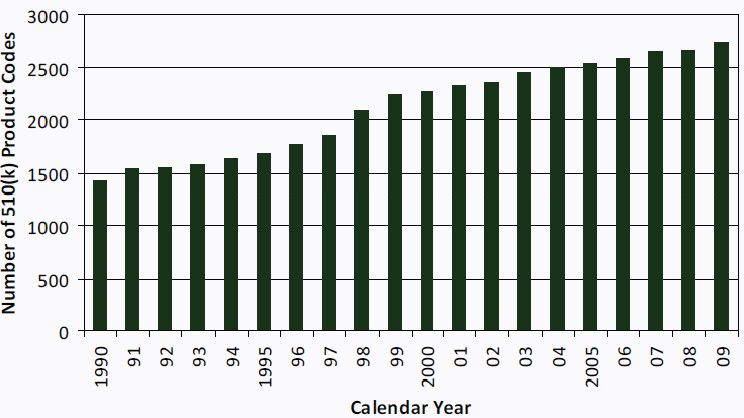
FIGURE 6-2 Number of 510(k) product codes, 1990–2009.
SOURCE: FDA, 2010a.
The number of types of medical devices also has grown. The FDA uses product codes to identify generic categories of devices. The product codes are organized by 16 medical specialties (for example, cardiovascular, general and plastic surgery, and orthopedic), and the medical specialties are listed in the Code of Federal Regulations.3 From 1990 to 2009, more than 1,000 product codes were added (see Figure 6-2).
In addition to the increase in 510(k) submissions to CDRH and the greater variety of types of products that CDRH must review, submissions have become longer and more detailed. As shown in Figure 6-3, the average number of pages per 510(k) submission in 2008 was more than seven times the number in 1983. In 2008, CDRH staff reviewed nearly 1.4 million pages of 510(k) submissions. Reasons for the increase in the length of 510(k) submissions are not readily apparent. The committee was not able to determine how often some types of data (for example, clinical data, bench-testing data, software-validation data, and labeling-comprehension studies) are included in 510(k) submissions, nor was it able to review a representative sample of complete (that is, not redacted) 510(k) submissions.
The technologic complexity of medical devices has increased substantially over the past 35 years as well. A 2010 FDA report states that “devices are unique among medical products in that they are defined by innovation, either through incremental evolution or disruptive revolution”
___________________
321 CFR §§ 862–892.
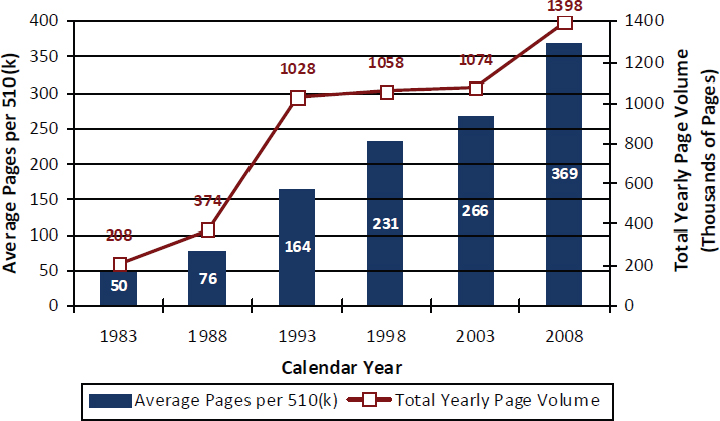
FIGURE 6-3 Pages per 510(k) submission and total page volume received, 1983–2008.
SOURCE: FDA, 2010a.
(FDA, 2010b). Examples of new technologies in medical devices are software (incorporated in medical devices and as stand-alone medical devices), nanotechnology, and medical robotics. The evolution (and revolution) of science and technology creates many challenges related to the regulation of medical devices.
As medical devices become more complex, so do 510(k) submissions. As a result of the complexity and the increasing number and size of 510(k) submissions received each year, the burden on CDRH review staff is increasing (see Chapter 3). The growing complexity of medical devices also affects how the medical-device industry approaches the 510(k) clearance process. For example, industry representatives have indicated that they view the increased length of 510(k) submissions as a response to the lack of predictability in decision-making by CDRH. To avoid delays in clearance of their products, industry includes more information in the submissions than it might have in the past.4
Multiple Predicates and Split Predicates
As detailed in Chapters 3 and 4, predicates are used in the 510(k) clearance process as the basis for demonstrating substantial equivalence. The increasing complexity of medical devices is reflected in how predicates
___________________
4FDA Docket Number FDA–2010–N–0054.
are selected and used. Applicants may submit more than one predicate for several reasons, including these:
• The applicant is not sure which predicate is the most appropriate.
• The submission is part of a bundled submission.
• The new device combines functions of more than one predicate device, so the applicant submits more than one predicate (termed multiple predicates) to demonstrate substantial equivalence of the new device.
• The new device has the same intended use as one predicate and the same technologic characteristics as another predicate (termed split predicates).5
Multiple predicates and, in some cases, split predicates are cited in more than half the 510(k) submissions (FDA, 2010a). In general, multiple and split predicates are used in 510(k) submissions for new devices that are more complex than the predicates. Using more predicates leads to longer CDRH review times (FDA, 2010a). It is not clear whether the increase in review time of devices that have multiple predicates is related to the number of predicates or to the increased complexity of the devices. It may be appropriate for multiple and split predicates to play a role in premarket review of Class II devices if the CDRH review team has the necessary expertise to ensure a high-quality review and if appropriate postmarket activities (for example, postmarketing surveillance) are used.
Combination Products
Combination products are therapeutic and diagnostic products that combine drugs, devices, and biologic products (FDA, 2008). As new technologies emerge and older technologies evolve, combination products are increasingly complex. Over the past decade, it has been increasingly common to enhance the performance of medical products by using multiple products together. For example, a genetic test that is a 510(k)-cleared or a premarket-approved (PMA) device may be used with a drug or biologic with the aim of identifying patients whose genetic characteristics place them at heightened risk for drug-related injuries, or a stent that is a medical device may be coated with a drug with the aim of reducing complications associated with the stent or altering the underlying pathologic condition for which the stent was placed. Combined uses of medical products present complex regulatory issues because the resulting treatment or diagnostic test
___________________
5In January 2011, CDRH announced that it no longer intends to use the term split predicate. It plans to issue guidance to clarify the circumstances under which it is appropriate to use multiple predicates to demonstrate substantial equivalence (FDA, 2011a).
incorporates multiple components that cut across the traditional categories of FDA regulation—drug, device, and biologic.
In response to a growing trend toward combined uses of medical products, Congress called for the FDA to establish an Office of Combination Products (OCP) in the Medical Device User Fee and Modernization Act of 2002. Combination product is a term of art that has a specific meaning.6 To qualify as a combination product, the two (or more) constituent products need to be integrally combined or mixed with one another, packaged together in a single package as a unit, or, if packaged separately and in the particular circumstance of being used in combination, cross-labeled in a way that makes it clear that one is specifically intended for use with another product that is “individually specified” in the labeling. By that definition, the mere fact that products happen to be used together in clinical practice does not necessarily make them combination products for purposes of the FDA’s regulations. If products are simply used together in practice without constituting a combination product, each product retains its separate identify and is regulated separately. For example, a drug would be regulated as a drug and a device as a 510(k)-cleared or PMA-approved device. However, even in that circumstance, OCP may play a role in the clearance or approval process for the device, as is discussed in more detail later in this section.
When a pair or set of products does meet the definition of a combination product, special regulatory provisions apply. The aim is to reconcile conflicts that would otherwise make it difficult to comply with drug, device, and biologic regulations. On the basis of the combination’s primary mode of activity—that is, whether the combination achieves its medical purpose primarily through the action of the drug, the biologic, or the device—primary responsibility for premarket review is assigned to one of the FDA’s centers—the Center for Drug Evaluation and Research, the Center for Biologics Evaluation and Research, or CDRH. The responsible center, while having primary jurisdiction over the combination, will work closely with the other centers to ensure appropriate oversight of issues related to the combinations of other components. Because postmarket regulatory requirements also differ for drugs, biologics, and devices, OCP in 2008 prepared a proposed rule on postmarketing safety reporting for combination products (FDA, 2008).7 A final rule has not been published as of May 2011.
Information for FY 2008 indicate that a large percentage of combination products involve 510(k)-cleared devices. There were 330 original applications related to combination products. Of those, 120 were original 510(k) submissions, 2 were original PMA applications, 14 were original new drug applications, and 4 were original biologic license applications. The remainder
___________________
621 CFR § 3.2(e).
774 Fed. Reg., 50,744 (October 1, 2009).
consisted of 158 applications for investigational new drug status and 32 for investigational device exemptions (FDA, 2008). Thus, many combination products enter the market on the basis of having the primary mode of activity of the incorporated 510(k)-cleared device.
OCP also has a role in situations in which such products as medical devices and drugs are used in concert but not as true combination products. OCP may act as a convener and facilitator in developing guidance documents. Guidance documents can influence the regulatory review of medical devices. For example, imaging tests use devices, such as magnetic resonance imaging (MRI) and computed axial tomography machines; they may use contrast agents, such as gadolinium and iodinated contrast agents that are regulated as drugs; and they may use software, which is regulated as a device, for the reconstruction and processing of the resulting data. Guidance coordinated through OCP has influenced the review of software devices on the basis of perceived limitations imposed by the specific language that describes the indications for use of the contrast agents (FDA, 2009a). For example, the use of MRI contrast agents that are approved for use in imaging of the abdomen, but not specifically imaging of the liver or kidneys, might limit the ability of CDRH to review a software tool that analyzes data produced by approved MRI machines after administration of the MRI contrast agents if the data analyzed pertain to the liver or kidneys. Guidance documents may stymie innovation in software tools that would be used merely to analyze data that are being acquired daily in a clinical setting. Because the contrast agent is readily available for use in clinical practice, there is no practical mechanism whereby the software company can influence the supplier of the contrast agent to apply for expanded or specific new label indications.
Finally, the acceleration of device evolution is leading to increasingly novel devices that could not have been conceived of when the 1976 Medical Device Amendments and later amendments were written and enacted. For example, nanodevices include particles that are activated by biologic and pathologic processes, and some of them depend on metabolic pathways for the mechanism of action. Atomic and molecular computational platforms are under development. Complete “laboratories on a chip” are being delivered and used in vivo. Even previously contemplated combination products, such as drug-eluting stents, have evolved in ways that stretch the capabilities of the current system.
Some novel device–drug–biologic products—not truly combination products—are not well managed on the basis of the current combination-product concept. For example, nanoparticle drug-delivery systems (combination products of a drug or biologic encapsulated in a device) could be activated by external ultrasonographic energy (delivered by a device) and monitored by software analysis of resulting real-time images (a device).
Other than arbitrary assignment, there is no obvious lead agency for the review and approval of such a multipart product; in fact, the review might best be accomplished by multiple centers and branches.
Finding 6-1 Medical-device technologies have evolved rapidly and devices have become increasingly complex since the 1976 Medical Device Amendments.
Software
Software is used in medical devices, as medical devices (for example, medical-device data systems), and as a tool in producing medical devices. Manufacturers are increasingly using software in their medical devices. An analysis by Fu showed that a milestone was reached in 2006: since then, more than half the medical devices on the market have relied on software in some way (IOM, 2011). Software offers many benefits over hardware in some situations, for example, flexibility, ease of change, and usability in other applications. To reduce costs, many software vendors produce “software product lines,” that is, software designed to allow ease of variation to accommodate similar, and perhaps tailored, product lines. In some cases, general-purpose, off-the-shelf (OTS) software is incorporated directly into medical devices. The FDA’s software validation guidance addresses that situation, noting that “the use of off-the-shelf software in automated medical devices and in automated manufacturing and quality system operations is increasing” (FDA, 2002).
Software Is Responsible for an Increasing Number of Recalls
As software becomes more common in medical devices, it also is increasingly responsible for device failures and recalls. For example, CDRH (FDA, 2002) notes that
the FDA’s analysis of 3140 medical device recalls conducted between 1992 and 1998 reveals that 242 of them (7.7%) are attributable to software failures. Of those software related recalls, 192 (or 79%) were caused by software defects that were introduced when changes were made to the software after its initial production and distribution.
Fu noted that from 1999 to 2005, 11.3% of recalls were attributed to software, and 49% of recalled devices relied on software in some way (IOM, 2011). In the period 2002–2010, at least 537 recalls of software-based devices affected at least 1,527,311 devices. However, because of the limitations of recall data discussed in Chapter 5, there is insufficient information to determine the underlying issues in the increased rates. Because there is no
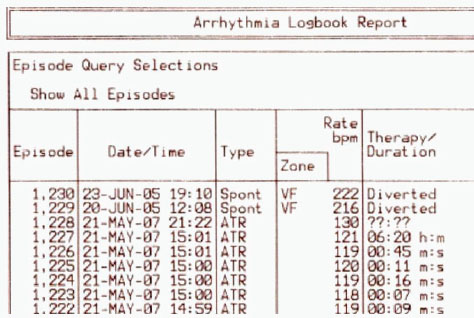
FIGURE 6-4 Example of a medical-device software malfunction.
SOURCE: Reprinted with permission from Halperin et al., 2008. © 2008 IEEE.
way to know the number of devices on the market or how many of them use software, it is not possible to know whether the change is related to the increasing proportion of software in medical devices or whether it is a signal of new or different types of problems and vulnerabilities in medical-device software.
A drawback of software is that it can be difficult to recognize problems, find their source, and fix them without adverse consequences. When software is used in medical devices, at least three outcomes are to be avoided:
• Unreliable and unavailable devices.8
• Insecure devices.
• Unpredictable behavior.9
Figure 6-4 is an example of how a software failure can be manifested. Here, the arrhythmia log is read from the bottom up. The episodes are in
___________________
8Reliability means that a system works as expected almost all the time; it rarely fails. Availability means that the system works whenever it is needed to work. There is no delay, and it is usable on demand.
9A device has unpredictable behavior if, for the same inputs, it gives different answers at different times.
BOX 6-1
Example Recall Announcement
URGENT: Medtronic Announces Nationwide, Voluntary Recall of Model 8870 Software Application Card
MINNEAPOLIS, Sept. 22, 2004 - Medtronic, Inc. (NYSE: MDT) today announced a voluntary recall that involves all Version AAA 02 Model 8870 software application cards in the U.S. that are used in conjunction with all Model 8840 N’Vision™ Clinician Programmers. This action has been classified by the Food and Drug Administration (FDA) as a Class I Recall…. a reasonable probability that the use of or exposure to the product will cause serious adverse health consequences or death.
Medtronic became aware in August 2003 that some users had mistakenly entered a periodic bolus interval into the minutes field, rather than the hours field, potentially resulting in drug overdoses. Data entry errors have been related to seven serious injuries and two deaths. The previous model 8870 software application card did not provide a label for the hours/ minutes/seconds fields; the new software has this labeling.
SOURCE: FDA, 2009b.
chronologic order, and they inexplicably and suddenly move from 2007 back to 2005. Such an error can occur only when software is faulty.
Similar software failures are noted frequently in recall announcements. For example, Medtronic issued a press release related to the Class I medical-device recall of the Medtronic 8870 Software Application Card Version AAA 02 (see Box 6-1).
The Guidance for Premarket Submissions for Software Contained in Medical Devices for industry and FDA staff issued on May 11, 2005, poses questions about problems with OTS medical-device software (FDA, 2005a):
What is it about “network-connected medical devices” that causes so much concern? … Vulnerabilities in cybersecurity may represent a risk to the safe and effective operation of networked medical devices using OTS software. Failure to properly address these vulnerabilities could result in an adverse effect on public health. A major concern with OTS software is the need for timely software patches to correct newly discovered vulnerabilities in the software.
The “vulnerabilities” are problems with the software that can be exploited by a malicious agent or can simply malfunction if the software is subjected to particular conditions. For instance, poorly designed software
may malfunction if it receives a type of input that it does not expect or if it conflicts with other devices that are communicating along the same channels. A patch is the application of a software correction; the resulting modified software should work as desired. However, the FDA’s request for “timely patches” suggests that patching is straightforward. In the committee’s opinion, it is not.
Beattie et al. (2002) analyzed the timing of patch availability and application. The researchers concluded, “Patch too soon, and you may suffer from instability induced by bugs in the patches. Patch too late, and you get hacked by attackers exploiting the vulnerability.” And patches are not always available as soon as a problem is discovered. The second Tuesday of each month is known to Microsoft Corporation’s customers as “patch Tuesday”: the day on which patches are made available for application to existing systems. Only when a problem is considered an emergency is a patch offered off-cycle to customers.
Microsoft is not alone. For example, Oracle (2010) notes that “Oracle Sun releases over 4,500 patches every year, for Solaris 8, 9, and 10, SPARC and x86, Solaris Cluster, Middleware, Developer, Storage, and other products. Just 17 have been withdrawn after release in the last year due to serious issues.”
Software Is Different from Hardware
Pfleeger et al. (2002) have written extensively on how software is different from hardware. The authors identify three key characteristics of software that make it different from hardware:
• Software developers are overoptimistic. Careful empirical studies have shown that software developers, particularly testers, often assume that they have found the last problem in the software under scrutiny. That is, they commonly stop looking once they find a problem, not recognizing that other problems remain. Their optimism results in overconfidence in both testing techniques and the degree to which the tests exercise all the functions implemented by the software. Such optimism is shared by hardware testers but seems to be extreme in software testers. Indeed, Beck (2004) noted that “optimism is an occupational hazard of programming, feedback is the treatment”; and Jorgensen (2010) has shown that, counterintuitively, identification of more risks can lead to developers’ overconfidence and overoptimism.
• Software is discrete, not continuous. Unlike hardware, software is extremely sensitive to small errors. Off-by-one errors, negligible in hardware, can result in huge changes in software. Just as an off-by-
one telephone number can send a call around the world, an off-by-one software error can lead to potentially dangerous outcomes. And because software is discrete, interpolation is difficult and sometimes impossible. Whereas hardware that passes a test at two different values can be assumed to pass for all values in between (as with bearing a load, for instance), software must be tested for every possible class of values that it can handle. Finally, there are no safety margins in software akin to those in hardware; software developers cannot double the strength of a material. Instead, every possible adverse situation must be anticipated and designed for, and resilience takes the form of multiple systems that perform the same critical functions.
• Software is immature and subject to rapid change. The National Research Council Committee on Information Systems Trustworthiness notes that “because a typical Networked Information System is large and complex, few people are likely to have analyzed one, much less had an opportunity to study several. The result is a remarkably poor understanding today of design and engineering practices that foster NIS trustworthiness” (NRC, 1999).
For those reasons, software is substantially different from hardware and should be treated and tested by different means. In particular, there is more uncertainty in software test results than there would be in testing an equivalent hardware implementation. An implication of the difference is that hardware should not be considered a predicate for software.
Software Validation Requires Interpretation Within the System Context
The FDA provides guidance on how software can be validated to ensure that it is performing its functions correctly (FDA, 2002):
Software validation is a requirement of the Quality System regulation, which was published in the Federal Register on October 7, 1996 and took effect on June 1, 1997.10 Validation requirements apply to software used as components in medical devices, to software that is itself a medical device, and to software used in production of the device or in implementation of the device manufacturer’s quality system…. FDA considers software validation to be “confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled.”
___________________
1061 Fed. Reg., 52,602; 21 CFR pt. 820.
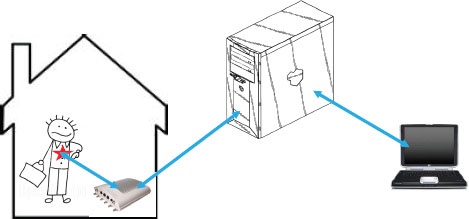
FIGURE 6-5 Transmission of medical data.
SOURCE: Adapted from Figure A, Halperin et al., 2008.
In other words, the FDA requires objective evidence that demonstrates predictability and consistency of function. To provide that evidence, a medical device must be viewed as part of a larger system in which it functions. Figure 6-5 illustrates what that larger system might contain. In the figure, the patient has a medical device that transmits data to a receiver in his home. In turn, the data are sent to a server that eventually places the data on a physician’s laptop. The communication may work in two directions; after the physician evaluates the patient data, she may transmit signals to change the settings in the medical device, for instance, changing dose amount or frequency.
It is essential that the “system boundary” be defined to include all devices and situations that may interact with each other. To see why, consider the findings of Lee et al. (2008). The researchers examined an MP3 player to see whether there were interactions with pacemakers and implantable defibrillators. Although no serious electromagnetic interference by the MP3 player was found, there were other irregularities:
• Device interference by headphones (not the player itself) was documented in 14 of 60 (23%) patients.
• Inappropriate (asynchronous) pacing was observed in 4 of 27 (15%) pacemaker patients.
• Inhibition of implantable cardioverter defibrillator detection occurred in 10 of 33 (30%) patients who had those devices.
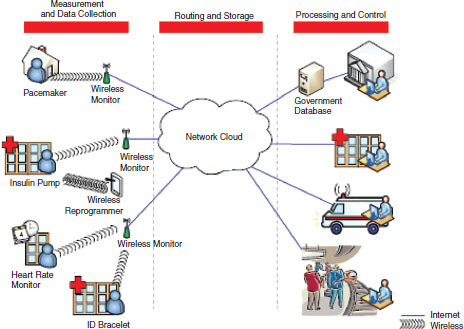
FIGURE 6-6 Vision of future data transmission.
SOURCE: Adapted from Figure 1, Defend et al., 2008. Reprinted with permission.
The researchers concluded that clinically significant electromagnetic interference could occur when headphones were placed close to implanted pacemakers and implantable defibrillators.
Figure 6-6 illustrates a vision of medical-device interaction in the not-so-distant future. Devices will be collecting, analyzing, storing, and transmitting data in many ways to large numbers of people, institutions, and other devices. It is essential that software validation take all those system elements into account in an effort to anticipate interactions among the elements that may at least add to uncertainty and at worst cause patient harm.
Software Validation Evidence Should Be Organized as an Assurance Case
The differences between hardware and software mean, among other things, that software can never be tested completely. As a consequence, ensuring the safety of software in a medical device requires presenting a convincing argument that the actions taken, when viewed collectively, support the claim that the device is likely to be safe. Similar arguments are made in other disciplines, such as nuclear power–plant safety, in which a “safety
case” is presented by the developers and reviewed by a body of experts who determine the argument’s soundness.
The case usually has three parts:
• One or more claims that some set of properties is satisfied.
• A body of evidence (from a variety of sources) supporting the claims.
• A set of arguments that link the claims to the evidence.
However, software developers and device manufacturers often focus too much on producing evidence and too little on building the associated arguments, analyzing their soundness, and accounting for uncertainty (Pfleeger, 2005). Schum (1994) identified four distinct categories of evidence, some with more uncertainty than others: tangible, testimonial, accepted facts, and missing evidence. Each has an associated degree of credibility, depending not only on its type but also on its source and its sensitivity to error.
To address soundness, several key questions can be asked:
• What kinds of evidence (and how much evidence) are needed to demonstrate that a device works? Some technologies show their effects even before the software is fielded, but some (such as those affecting reliability) can be evaluated only after a system is in use. Evidence must demonstrate the effects, both before and after. The evidence must also enable comparison of outcomes when the technology is used and when it is not used.
• Who provides the evidence, and who vets it? Many vendors provide evidence of their products’ effectiveness; many are eager to demonstrate that their new tools can make a favorable difference to developers or users. But that enthusiasm can bias the results even when vendors take great pains to eliminate it. Independent assessment is almost always preferable.
• If a technology works in one domain, what does that say about how it works in other domains? Evidence gathered in one context or environment might or might not apply to others.
Rather than be dismayed by the nature and variability of the uncertainty of evidence, one can use the uncertainty to advantage. Evidential force is the degree to which each piece of evidence contributes to or detracts from an argument that uses it. Ideally, each piece of evidence adds to an argument’s overall force.
Bloomfield and Littlewood note that arguments involving diversity of evidence are stronger than arguments involving multiple replications of the same kind of evidence. Their work is motivated by the need for strong evidence in making decisions about safety-critical systems. The “multilegged
argument,” in which each leg handles a different type of evidence, can be easier to analyze than one comprehensive argument. Moreover, the legs need not be independent. The extra legs usually provide more confidence than one leg alone, but the extra cost of the extra confidence must be justified (Bloomfield and Littlewood, 2003).
Finding 6-2 Manufacturers are using increasing amounts of software in devices and as devices; the increase is expected to continue. Software offers many benefits over hardware, including flexibility, ease of change, and the possibility of use in other devices.
Finding 6-3 Software is responsible for an increasing number of recalls. There are insufficient data, however, to determine whether the increase reflects the increasing proportion of software in medical devices or a new and different set of problems and vulnerabilities.
Finding 6-4 Software is different from hardware and therefore requires a different kind of evaluation.
INNOVATION AND THE 510(K) CLEARANCE PROCESS
In considering the role or impact of the 510(k) clearance process on innovation, it is first necessary to define innovation. It has been defined as “the introduction of something new,” “a new idea, method, or device,” or “a new method, idea, product, etc.” (Merriam Webster, 2011; Oxford Dictionaries, 2011). Doris Estelle Long offers this regarding innovation: “It has been defined as everything from ‘introducing something new’ to ‘a scientific approach for finding newer better ideas and solutions to problems, which make life easier and simpler to live’” (Long, 2008). Long went on to quote Joseph Schumpeter’s definition of innovation as “the introduction of a new good … a new method of production … the opening of a new market … the conquest of a new source of supply … [and] the carrying out of the new organisation of any industry.” Everett Rogers, author of Diffusion of Innovations, defines innovation as “an idea, practice, or object that is perceived as new by an individual or other unit of adoption” (Rogers, 2003). Susan Bartlett Foote in her book Managing the Medical Arms Race: Public Policy and Medical Device Innovation cites several variations, including “certain technical knowledge about how to do things better than the existing state of the art” (Teece, 1986) as cited in (Foote, 2005). Ahn et al. (2009) discussed the criteria for the adoption of new technology by posing the following question: “Can this technology improve clinical care for my patients?”
Scott Berkun, author of The Myths of Innovation, cited one definition of innovation in his book as “significant positive change” but went on to sug-
gest how such a relatively simple definition “burdens creators to understand the recipients’ perspective of whatever they make” (Berkun, 2010). That is, what may have been intended as a favorable change by an innovator may not be similarly perceived as favorable by the recipient of such an innovation, and thus simply associating innovation with a perception of positivity may not be sufficient. Berkun suggests resisting overuse of the word as a stand-in for a more specific description: rather than state something as innovative, describe its benefits in more precise detail. For considering the utility, practicality, or overall goodness of innovation, Berkun offers the following criteria: an innovation can be good for you, good for others, good for industry or economy, good for a society, good for the world, and good for all time.
Although those general definitions may be technically accurate, they lack the nuance of the term as it is related to technology and the medical-device industry. Innovation is broadly viewed as describing new favorable changes, but more precise definitions of innovation may indeed require the consideration of the context in which it is being discussed. In the context of medical-device regulation, it is reasonable to use patient care and utility as a test for innovation. Foote discusses innovation in the context of medical devices and says that it “embraces the frontiers of science and engineering, adapting computer technology, nanotechnology, and biotechnology to medical applications. Many of these technologies are delivered by physicians in in-patient and out-patient settings and are embedded in the service delivery system. Physicians’ needs and experiences in clinical settings often trigger innovation and incremental improvements; physicians also require ongoing training to effectively use innovative therapies” (Foote, 2005).
Although a specific definition of innovation from the FDA was not evident, the FDA does discuss innovation in various ways. For instance, the FDA defines its charge on its Web site under the banner “What We Do.” Here, the FDA defines its role in terms of innovation as follows: “FDA is also responsible for advancing the public health by helping to speed innovations that make medicines more effective, safer, and more affordable and by helping the public get the accurate, science-based information they need to use medicines and foods to maintain and improve their health” (FDA, 2010d). It is assumed that the use of the word “medicines” in this context does not exclude devices. In the context of devices, a whitepaper from CDRH in February 2011 titled CDRH Innovation Initiative states that “new scientific discoveries or novel ideas are often at the root of innovative medical device development—whether the product is a transformative technology, a modified version of an already marketed model, or a novel application of existing tools or scientific approaches” (FDA, 2011b). Those statements suggest that the FDA considers an attention to innovation to be at the heart of its overall mission and, more specifically, that CDRH consid-
ers innovation to encompass new ideas that lead to both incremental and sweeping changes in technology.
Process of Innovation
An assessment of innovation produces not only a number of definitions but also various strategies, processes, and principles. Innovation is found to exist not in isolation but as part of a larger structure of both thinking and doing. Berkun discusses the process of innovation and how it is often incorrectly paired with the term epiphany and how most innovative advances were not the product of any singular epiphany but “a combination of things that existed before…. For most, there is no singular magic moment; instead, there are many smaller insights accumulated over time…. It’s simply the final piece of a complex puzzle falling into place” (Berkun, 2010, xvii and 6-9). Foote also offers that innovation is “process leading to technical change” (Foote, 2005). The author goes on to describe innovation as consisting of two stages; stage 1, discovery, which encompasses science, invention, and development; and stage 2, distribution, which encompasses adoption and distribution. Foote goes on to suggest that the progression of innovation cannot exist without each element in place; nor is it a purely linear process, but rather an iterative one. This last point was supported by Privitera et al. (2009) by examining the interconnectivity of basic-science research and product development in designing medical devices when the appropriate design of a new device allows and facilitates the translation of a seemingly complex product into “easy-to-use, innovative products.”
In April 2009, The Lancet published a three-part series summarizing a study of surgical innovations that provided an overview of how the innovations initially occur and are eventually adopted (Barkun et al., 2009; Ergina et al., 2009; McCulloch et al., 2009). This model, termed the IDEAL model, divided the process into five stages: innovation (or idea), development, exploration, assessment, and long-term study. Stage 1, innovation, was defined as “the first use of a new procedure in a patient, prompted by the need for a new solution to a clinical problem” (McCulloch et al., 2009). Linehan and Chaney (2010) similarly concluded that “clinical needs provide opportunities for medical device innovation” through identifying an unmet or undermet clinical need, harnessing new insight into the physiology of a disease or a new diagnostic approach, new developments in technology, or some combination of such factors.
In its 2004 report, updated in 2010, Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products, the FDA outlined a critical path for medical-product development from basic research to FDA approval (FDA, 2010c). In describing this critical path from scientific innovation to commercial product, the FDA identified three
distinct scientific and technical dimensions: (1) assessing safety for each stage of development, (2) demonstrating medical utility or, more simply, that the product benefits people, and (3) having an industrialization component that considers whether a product can go from concept or prototype to a manufacturable product (endorsing the concept that innovation is a process, not one defined event).
Given the above discussion, the committee made a few determinations about the concept of innovation in a general sense:
• There is no single accepted definition of innovation.
• Innovation most likely is a result of several factors that coalesce toward a single objective.
• Innovation often occurs in small increments rather than quantum leaps.
• Innovation is generally considered to be a change toward the greater good rather than the opposite.
The Committee’s Working Definition of Innovation
It is with those ideas in mind that the committee agreed on a definition of innovation through which it would attempt to assess whether the current 510(k) clearance process optimally protects patients and promotes innovation in support of public health. The committee believes that given the broad interpretation of the term it should define innovation not simply as a change but as a favorable change in the context of public health. The committee also believes that given the complexity of the process of innovation and the iterative and combinatorial nature of the process, a broad definition would be more accurate than one that sought to create a list of specific attributes. In the context of this report, the committee defined innovation as improving the quality of, efficiency of, or access to health care. To revisit the charge given by the FDA to the committee, the question that needs to be answered is, Does the current 510(k) clearance process optimally protect patients and promote innovation in support of public health? Answering that question requires a general understanding of whether and how CDRH promotes innovation.
Does the 510(k) Clearance Process Facilitate or Inhibit Innovation?
In testimony to the US House Subcommittee on Health of the Committee on Energy and Commerce in February 2011, Jeffrey Shuren, director of CDRH, indicated that it was the role of the FDA to “ensure the safety and effectiveness of the medical products that Americans rely on every day, and also facilitate the scientific innovations that make these products safer,
more effective, and more affordable” (Shuren, 2011). In February 2011, CDRH launched the CDRH Medical Device Innovation Initiative, a multipronged program that proposes actions that CDRH could take to “help accelerate and reduce the cost of development and regulatory evaluation of innovative medical devices safely and based on sound science” (FDA, 2011b). Those actions include facilitating the development and regulatory evaluation of innovative devices through new priority review programs and a streamlining of the pre-existing de novo pathway, strengthening the US research infrastructure, and improving its preparedness for transformative innovative technologies and scientific breakthroughs. The FDA’s initiative explicitly outlines a strategy to promote innovation in CDRH, but what is less clear is whether the 510(k) process promotes innovation in support of public health. A cursory look at the original legislation and later changes, additions, and modifications may provide some insight.
The structure of the Medical Device Amendments of 1976 rejected premarket approval for all devices, limiting PMA to Class III devices. That approach balanced the adverse effects of preclearance on innovation with the public-health benefits of premarket review of products. For new products with a lower degree of risk, it would be sufficient to demonstrate that the products were already classified in a lower class and, for Class II devices, complied with performance standards. During the transition period and thereafter in showing that the device was already classified, market entry was based on substantial equivalence. The substantial-equivalence standard was vague and arguably required equivalence but precluded, or at least did not mandate, superiority. The Office of Technology Assessment commented on the concept of substantial equivalence as a tool for device clearance, stating that as devices “diverge more and more from their preamendment antecedents, it will be harder for manufacturers to use the substantial equivalence method of market entry” (OTA, 1984). It could be reasonably argued that allowing market entry on the basis of the equivalence of one device to a pre-existing device is not in support of innovation. Congress showed some concern during a House Subcommittee on Health hearing in 1987 that equivalence would freeze the world as it was, indicating that technologic improvements had continued to occur in the 10 years since the initial legislation but that under the concept of substantial equivalence “new devices need not incorporate these improvements: they need only be as safe and effective as similar devices on the market before 1976.”11
In the late 1980s, the FDA redefined the term substantially equivalent and Congress endorsed this interpretation with the Safe Medical Devices
___________________
11Memorandum Re: Medical Device Hearing, May 4, 1987, Medical Devices and Drug Issues: Hearing Before the Subcomm. on Health and the Env’t of the H. Comm. on Energy and Commerce, 100th Cong. 336-47 (1987) (referred to in the statement of Rep. Henry A. Waxman, Chairman, H. Subcomm. on Health and the Env’t).
Act of 1990 (SMDA) that allowed for devices to be substantially equivalent if they had different technologic characteristics from the predicate device but were “as safe and effective as a legally marketed device.” In so doing, it explicitly sought to promote evolution of the marketplace (new devices) toward improvements. The law, however, did not force innovation if it is defined as bringing about change for the greater good, in that the new definition of substantial equivalence still allowed manufacturers to use potentially inferior predicates as long as they had the same technologic characteristics as a predicate device.
As discussed in Chapter 2 and Appendix A, the Food and Drug Administration Modernization Act of 1997 had various implications for innovation in the FDA. With respect to Class II devices and the 510(k) process, the effect of the 1997 amendments on innovation were indirect, lessening regulatory burdens and limiting the scope of FDA preclearance requirements. Specific provisions that lessened regulatory burdens included the de novo process, which permitted devices determined not to be substantially equivalent to another device to be classified in Class I or Class II rather than going through the PMA process; the least burdensome rule, which specified that the FDA could request information only if it were necessary to make a determination of substantial equivalence (allowing the FDA to down-classify devices in light of postmarket controls); reaffirming the 90-day period for 510(k) reviews; and introducing priority reviews for important new devices. Finally, the various user-fee laws, beginning in 2002, were intended to ensure that the FDA had adequate resources to complete reviews within the statutory time limits, increasing predictability of review.
In reducing regulatory burdens and providing additional resources to allow for timely review, the changes in the original legislation over the past 35 years neither forced nor rewarded innovation. Although it may be argued that such changes may improve the likelihood of innovation, it remains unclear—and the committee argues that it is indeterminable, given current data—whether those changes promoted innovation in balance with public health rather than at its expense.
The medical-device ecosystem includes the environment that affects medical-device development, commercialization, and availability to consumers. The regulatory aspect of the ecosystem, including the 510(k) clearance process, is covered in detail in earlier chapters and is not repeated here.
Medical devices are developed by commercial interests that range from small startup companies to established conglomerates. Publicly traded US medical-device companies generated revenues of $188.8 billion in 2008
(Ernst and Young, 2009). Most of the revenue from medical devices is earned by 30 companies (Kalorama Information, 2010).
Hundreds or thousands of companies operate in the medical-device market, and most of them are small: more than 80% have fewer than 50 employees (MDMA and NVCA, 2009). Startup companies, generally supported by investments made by venture capitalists and other investors, are viewed as the drivers of innovation in the medical-device industry (Platzer, 2006).
510(k) clearance or PMA often is cited as an event that triggers transactions that result in change in ownership of intellectual property and return of investment to venture capitalists and other investors. Larger companies often are willing to wait to acquire smaller companies or their intellectual property after 510(k) clearance or PMA approval even if they have to pay a premium.
Many startup medical-device companies generate little or no sales revenue during the development phase before they receive permission to commercialize a device, and the funds invested in such companies by venture capitalists and other investors must sustain the companies until regulatory approval or clearance. Another potential barrier to the economic viability of both small and large companies is supportive insurance for coverage, payment, and reimbursement. In general, for nearly all device-based therapies and many diagnostic tests, a device must be cleared or approved and have valid current procedural terminology codes for a medical service before an insurer or payer will consider covering the service.
Delays during any of those phases extends the “burn rate” of capital (that is, the amount of funds spent in a given period) and can create financial difficulties for companies, particularly startup companies, which often have limited financial resources. A positive return on invested capital is important for maintaining a robust environment for innovation of medical devices (Platzer, 2006). Investment return is affected by several factors, including the state of the economy, changes in patent law, and challenges in getting medical devices through regulatory and reimbursement pathways (MDMA and NVCA, 2009). According to company representatives, the medical-device development environment is “fragile and extremely sensitive” to changes in the cost of innovation (IOM, 2010). Because achieving access in the market is critical in defining investment return, any delay in the ability to commercialize can have serious consequences, not only to specific devices and device companies but to the entire medical-device ecosystem.
The vast majority of medical devices subject to FDA premarket review—more than 90%—are brought to market via the 510(k) clearance process. The FDA receives about 4,000 510(k) submissions each year (FDA, 2010a). Because the 510(k) pathway is less expensive and less time-consuming than the PMA pathway, medical-device companies view it as a useful mechanism
for bringing moderate-risk devices to market. Representatives of medical-device companies have indicated that in recent years it has been increasingly difficult to move devices through the 510(k) pathway (Ernst and Young, 2009; IOM, 2010, 2011; Makower et al., 2010; MDMA and NVCA, 2009). Medical-device industry representatives believe that overall the 510(k) clearance process works well but that there is increasingly a lack of predictability and transparency in CDRH (IOM, 2010; Makower et al., 2010). They have voiced concern that those factors have led to increased time to 510(k) clearance of products, which has created an unfavorable environment for development of medical devices. Additional time to 510(k) clearance has the potential to affect a large number of medical-device companies and, by extension, medical-device innovation. Other industry concerns related to the 510(k) process include inadequate communication with CDRH staff, lack of publicly available information about predicate devices (needed to demonstrate substantial equivalence), issues surrounding the de novo process, and issues surrounding the use of standards and other types of evidence to show safety and effectiveness in the absence of a predicate device.
Reports from the venture-capital community have indicated that funding availability for device development has been adversely affected, in part by the regulatory environment and economic circumstances (IOM, 2010; Makower et al., 2010). However, the fiscal environment is more favorable once a device has achieved regulatory permission for commercialization and favorable coverage, payment, and reimbursement policies than before that benchmark in the device life cycle is achieved. As previously stated, a company that benefits from that more favorable economic environment is often a different company from the one that must contend with the development of the device, including premarket regulatory clearance or approval.
One of the critical economic pressures in the device ecosystem is obtaining favorable coverage, payment, and reimbursement policy decisions from payers, such as the Centers for Medicare and Medicaid Services (which manages the Medicare and Medicaid programs) and private health insurers. With rare exception, however, payers do not contribute to the collection of clinical data (for example, by funding clinical care during clinical trials) that could be used in making reimbursement decisions. One of the notable exceptions is the Medicare Coverage with Evidence Development program, which includes several examples of data collection for diagnostic and therapeutic devices as used in the course of clinical care.
As Feigal and others have opined, the medical-device life cycle includes not only premarket evaluation but continued postmarket evaluation, which might lead to the development of new devices or improvements in marketed devices (IOM, 2010). (Postmarketing surveillance is discussed in more depth in Chapter 5.) High-quality postmarketing surveillance data are useful for several parties. In addition to industry use of the data to support future
device development and improvement, regulatory agencies could use them to track how devices are used in clinical settings (for example, compared with labeled intended use)12 and how devices function (for example, identifying adverse events and effectiveness); healthcare providers increasingly need them for the Maintenance of Certification process as mandated by the American Board of Medical Specialties; institutional providers need them for quality-review assessments; patients and patient-advocacy organizations could use them to assist in educational efforts; and professional organizations could use them to assess their members’ needs (for example, educational and strategic planning needs).
Industry has several potential partners in the ecosystem for collection of postmarket information. And the clinical environment in which devices are used is quite varied, ranging from increasingly high-technology inpatient to underdeveloped outpatient sites of service. However, new and evolving tools could be used in varied clinical environments to conduct postmarketing surveillance. Professional societies could establish registries by using information extracted from electronic medical registries in the high-technology inpatient setting, and mobile computing devices, such as “smart phones,” facilitated by the availability of Unique Device Identification codes, might have utility in collecting and transmitting data in many outpatient settings. Developing and understanding a holistic view of the device ecosystem could contribute substantially to improving various aspects of it, including the regulatory mechanisms.
Finding 6-5 Industry-funded assessments of the effects of the 510(k) clearance process report that implementation of the process leads to a lack of predictability and transparency, which in turn has an adverse effect on venture-capital investment.
Finding 6-6 The committee did not find assessments of how much and in which way innovation is influenced by the 510(k) clearance process.
Finding 6-7 There is little collaboration in collection of postmarketing surveillance data among the FDA, healthcare facilities, healthcare providers, the medical-device industry, professional societies, payers, and patient-advocacy groups.
___________________
12For example, incorporation of an off-the-shelf product, such as a smart phone or commercial software application, might stretch proposed use beyond intended use and require evaluation of the technology by the FDA.
GLOBALIZATION OF THE MEDICAL-DEVICE INDUSTRY
The United States is the largest consumer and producer of medical devices, with about half the world’s market. The European Union (EU), Japan, Canada, and Australia also have large, stable medical-device markets, and the developing world is rapidly increasing both its consumption and its production of medical devices. In addition, medical-device manufacturers are increasingly multinational—some companies based in one country produce the majority of their devices in another (ITA, 2010). Of the top 10 medical-device manufacturers, 7 are American and 3 are European. The market share in the Indian subcontinent and China is increasing rapidly. The global market for medical devices was $290 billion in 2009 and is expected to be $312 billion in 2011 (Kalorama Information, 2010). Studies sponsored by industry have suggested that premarket review times for medical devices, particularly more complex devices, are longer in the United States than in the EU and that some companies have shifted toward obtaining approval for their devices in the EU first (Davis et al., 2011; Makower et al., 2010). A study of comparable recall data from the United States and the EU found similar recall rates in the two. The study authors concluded that the EU’s approval process and shorter time to market for medical devices did not affect patient safety adversely (Davis et al., 2011).
Foreign establishments exporting medical devices to the United States must register with the FDA (FDA, 2009c). Medium-risk and high-risk imported devices must be cleared or approved for marketing by the same standards and by the same processes as equivalent US-made devices. Low-risk imported devices do not require premarket review, only registration and listing and, as appropriate, conformity with other regulations, such as quality-system regulations and labeling requirements.
Medical-device regulations in the United States were developed largely at a time when the medical-device industry was primarily domestic, but now medical devices are global products in a global market. Medical-device companies are increasingly multinational. In this environment, regulators and consumers can reasonably assume that any given device may have been manufactured in a jurisdiction different from the one in which it is being used. Device components may also be made in several countries. To bring a medical device to market in multiple jurisdictions, manufacturers must navigate the regulatory environments of each jurisdiction. Additional time to market and costs occur if the jurisdictions’ regulatory frameworks differ substantially. Given the multinational nature of the market, the committee found it instructive to examine the international environment of medical-device regulations when considering its charge.
The FDA and medical-device manufacturers operate in the wider context of international markets and regulatory structures. All developed nations have some regulatory frameworks in place to ensure medical-device
safety and effectiveness. Recognizing the multinational environment of medical devices, governments and industry joined to form the Global Harmonization Task Force (GHTF) to attempt to harmonize regulation across borders. The GHTF (discussed below) has issued many guidance documents and promoted a pilot program for harmonized regulatory approval. Several countries, including Japan and India, are moving toward using the GHTF principles, although there has yet to be a large-scale movement toward standardization among regulators or industry (IOM, 2010).
International Approaches to Medical-Device Regulation
In examining other systems, the committee focused on the EU as the nearest counterpart of the United States in the structure and scope of the market. However, it is important to note key differences between the two. First, although the EU acts as a single entity with respect to trade, it is made up of 27 member states, 16 of which use a common currency. Member states are obliged by treaty to conform to communitywide laws and regulations but retain sovereignty over their own territories. Second, the US system has been in place much longer than the EU system. Although some member states of the EU, most notably the United Kingdom, had long regulated medical devices, most had not. Thus, the FDA has greater experience than most countries in regulating medical devices. Beginning with a clean slate, however, gave the EU the opportunity to put into place a logic model that used the latest thinking in risk analysis. In the United States, the standard for clearance is substantial equivalence to a previously cleared device; but the EU and other countries that tightly regulate medical devices do not rely on this standard. Finally, the primary role of the EU is to promote trade among its members and between Europe and the rest of the world.
Unlike the United States, the EU has different governmental structures for the regulation of medical devices and of pharmaceuticals. Pharmaceuticals are overseen by a centralized authority, the European Medicines Agency. Medical devices are regulated by the Directorate General for Health and Consumers, which is responsible for a wide variety of consumer products. The legislation governing medical devices is written as a directive. Member states are bound by treaty to enact directives by transposing them into national law, but governments are given some latitude as to the details of the law as long as the intended effect is as laid out in a directive. The directives are in contrast with regulations, which have the effect of becoming law throughout the European Community as written immediately on adoption by the European Commission. Regulations have been used to cover pharmaceuticals but have not been used for medical devices. In the case of directives, it is the national law that is binding, not the directives themselves. That
approach leads to some variation among member states, particularly in enforcement procedures and the imposition of sanctions (Grubb et al., 2011).
The Directives
Three European Commission directives govern the regulation of medical devices:
• Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD).
• Council Directive 93/42/EEC on Medical Devices (MDD).
• Council Directive 98/79/EC on in Vitro Diagnostic Medical Devices (IVDD).
The directives lay out the goals, structure, and standards for both safety and marketing. They are closely tied to one another in content and structure, but they differ in their requirements according to the category of device. As noted earlier, before the passage of the directives (in 1990, 1993, and 1998, respectively), medical devices in the EU were regulated at the national level if at all (Grubb et al., 2011). The impetus for the change was the so-called new approach of 1985 that aimed to standardize the technical requirements for a wide array of products. Its goal was to remove barriers in the internal market caused by differing regulatory standards among the member states. The AIMDD, issued in 1990, was the first directive concerning the regulation of medical devices in the EU. Among its objectives were that “active implantable medical devices must give patients, users and other persons a high level of protection and achieve the intended level of performance when implanted in human beings” and that “national provisions ensuring that safety level should be harmonized in order to guarantee the free movement of active implantable medical devices without lowering existing and justified levels of safety in the Member States” (European Commission, 2001). Patient safety and confidence were seen as necessary components of the free movement of goods within the internal market and thus important to the promotion of free trade. In 1993, the AIMDD was amended by the MDD, which covered a broader spectrum of products. The MDD also laid out the risk-classification structure that would be used in the approval process. Finally, in 1998, the council issued the IVDD, a directive covering in vitro devices and based largely on the two earlier directives.
The directives have been amended several times since their adoption to reflect changes in technology and in response to issues that have arisen over the course of implementation. In 2008, the European Commission started the process of a major revision of the entire medical-devices framework, known as the Recast of the Medical Devices Directives. The recast
began with a public consultation of stakeholders in 2008 and has continued through consultation with the European Commission’s Medical Devices Experts Group and a further public consultation concerning in vitro devices in 2010 (European Commission, 2010b). Among the changes under consideration are consolidating the three main medical-device directives and five supporting directives into one piece of legislation, tightening oversight and accreditation of notified bodies (described below), and creating an EU-level entity in line with that which exists for pharmaceuticals to oversee the regulation of medical devices. It is unclear which changes will be adopted by the European Commission. According to the Roadmap 2011 posted on the European Commission’s Consumer Affairs Web site, adoption of the initiative is expected in the first quarter of 2012 (European Commission, 2010d). In the directives, a medical device is defined as
any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, together with any accessories, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
— diagnosis, prevention, monitoring, treatment or alleviation of disease,
— diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
— investigation, replacement or modification of the anatomy or of a physiological process,
— control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means.
In 2007, Directive 2007/47/EC made extensive amendments to the MDD, including the addition of software to the definition of a medical device and making software validation part of the essential requirements that had to be met to establish conformity with safety standards.
A device must perform its “intended purpose” according to its labeling. That purpose is only what the device does, however, not what it is for. Thus, European requirements are related to safety and performance rather than to safety and effectiveness as in the US system (Kaplan et al., 2004). Many EU countries have incorporated an effectiveness standard (cost-effectiveness or cost-benefit) into purchasing decisions by healthcare insurance systems.
Essential Requirements
The three directives also lay out the essential requirements that apply to every medical device whatever its class. The essential requirements are a set of safety and quality standards for how a device must perform and be produced. Any device that meets the essential requirements laid out in the directives can receive the CE Marking13 and be distributed and sold in any member state. The essential requirements are intended to protect the health and safety of patients and providers while ensuring that there are no internal obstacles to trade within the European Community. They begin with general provisions that lay out the overall standards of safety and move into more specific requirements regarding manufacturing and labeling. Although the general provisions of essential requirements differ slightly among the categories of devices, they cover the same main points:
• A device must not cause harm to the patient, operator, or others.
• A device must do what the manufacturer says it does.
• A device must be packaged in such a way that transport does not compromise safety or effectiveness.
• A device must be able to withstand ordinary rigors of use throughout its lifetime without compromising safety or effectiveness.
• Any undesirable side effect must constitute an acceptable risk when weighed against the intended performance.
The more specific essential requirements cover design and construction of a device. These detailed requirements cover every aspect of production and the device, including chemical, physical, and biologic properties; intended and unintended radiation; and labeling.
The manufacturer must ensure that its device complies with all the essential requirements that apply to it. To do so, it may choose to provide evidence that the device meets the requirements of one or more technical standards (the use of standards in this manner is voluntary). If the manufacturer chooses to comply with a standard that is recognized by the European Commission as offering “presumption of conformity” with one or more essential requirements (known as harmonized standards), the regulatory authority (RA) or conformity assessment body (CAB) is required only to determine that evidence of compliance with the standard exists. If the manufacturer uses a different standard, one that is not “harmonized,” the RA/CAB must determine that the manufacturer had evidence that the device complies
___________________
13The CE Marking is a standardized logo that appears on a wide variety of consumer products sold within the European Community. It signifies that a product is one that is subject to communitywide regulation and that it has met all the standards laid out in the applicable legislation.
with the essential requirement itself. Harmonized standards are developed and issued by the European Committee for Standardization (CEN) and in some cases the European Committee for Electrotechnical Standardization. Those two committees are responsible for writing the standards that deal with both entire classes of products and specific products. In some cases, the standards are adopted wholesale from the International Organization for Standardization (ISO). Manufacturers are free to use whatever standards they like, but if their devices meet the CEN or, where applicable, ISO standards, they are judged to have met the essential requirements—often the easiest path to approval.
Risk Classification14
A device is assigned to one of four groups, or classes, by using a set of rules that take into account the potential of the device to cause harm to a patient or user. This system is referred to as a “risk-based classification scheme” although it does not take into account the probability that harm will occur by modifying evidence requirements at the conformity-assessment stage. Risk is determined on the basis of the risk associated with the use of the device, whether the device is invasive, and the length of time it is in contact with the body (Table 6-1) (Chai, 2000). It is incumbent on the manufacturer to determine the level of risk associated with a device on the basis of the specifications laid out in Annex IX of the MDD (Grubb et al., 2011). As in the FDA classification system, there are three risk classes, and subdivisions of classes to delineate the magnitude of risk further.
Class I devices are noninvasive, with some exceptions, and are judged to be of low risk. There is no explicit subdivision of products within Class I, but devices that are sterile or have a measuring function require greater oversight. Manufacturers are able to market Class I products that are non-sterile and do not have a measuring function without oversight from a notified body (NB, defined below) but must keep documentation and technical specifications available for audit on request. Class I devices that are sterile or have a measuring function must be certified by an NB (Chai, 2000).
Class II devices are divided into Class IIa and Class IIb. Class IIa devices require the involvement of an NB at the production stage but not the design phase. Class IIb devices are judged to have the potential to be of high risk and require NB approval at the design and manufacturing phases.
Class III devices are deemed to be of high risk and require “explicit prior authorization” from an NB for all phases of development and production (European Commission, 1998). Manufacturers can also turn to guidance
___________________
14The essential requirements, classification rules, and conformity-assessment procedures are substantially different for in vitro diagnostic medical devices, which are regulated under their own directive. Some of the information in the following sections does not apply to these devices.
TABLE 6-1 European Commission Risk Classification Scheme
|
Class I (low risk) |
Noninvasive devices except devices that are intended to store or channel blood, body liquids, or tissues to be introduced to the body at a later time |
|
Class IIa (medium risk) |
Invasive devices intended for transient (<60 min) or short-term (<30 days) use except devices used to examine ear, nose, mouth, and throat, which are in Class I; all surgically invasive devices intended for transient use unless they emit radiation or have a biologic effect, in which case they are in Class IIb |
|
Class IIb (medium to high risk) |
Surgically invasive devices for short-term or transient use if they have a biologic effect, emit radiation, or administer medicines except devices that have direct contact with the heart or central circulatory system or central nervous system, in which case they are in Class III; all implantable or surgically invasive long-term (>30 days) devices unless they are placed in the teeth (in which case they are in Class IIa) or have contact with the heart, central circulatory system, or central nervous system (in which case they are in Class III); all devices intended to prevent conception or sexually transmitted disease unless they are implantable or long-term invasive devices (in which case they are in Class III) |
|
Class III (high risk) |
All invasive surgical devices, whether for short-term or long-term use, that come into contact with the heart, central circulatory system, or central nervous system; all implantable or long-term invasive devices that have a biologic effect, are absorbed, or undergo chemical change in the body; all implantable or long-term invasive devices intended to prevent conception or sexually transmitted disease; all devices that use animal tissues or derivatives except where they come into contact only with skin |
documents released by the overseeing body in the European Commission, the Directorate-General Health and Consumer that provide detailed instructions on classifying devices, including diagrams and examples. The guidance is not legally binding but is provided for clarification purposes (European Commission, 2010a).
Notified Bodies
The directives contain provisions for the establishment of NBs, the backbone of the European regulatory structure for medical devices. An NB is a third-party private organization that is responsible for ensuring that a device meets the essential requirements laid out in the legislation. Each
member state designates a competent authority (CA), an official government regulator, that is required to oversee the accreditation and designation of third-party organizations that will perform conformity assessments and various other tasks. The CA then notifies the European Commission and the other member states as to which private organizations have been judged to be competent according to the standards laid out in the directives and for which tasks. The commission maintains an updated list of the NBs and makes this information available to the public through the New Approach Notified and Designated Organisations Web site (NANDO, 2011). NBs are accredited under individual directives, and their number varies by directive. For example, although more than 70 NBs are accredited to examine products under the MDD, only 19 are accredited for devices covered by the more specialized AIMDD (NANDO, 2011). The NBs compete with one another in that any manufacturer can go to any accredited NB in any member state and receive a CE marking.
NBs must adhere to standards regarding conflict of interest and financial incentives. NBs must be independent of manufacturers. Directors and assessment and verification staff must not be involved in the development of the device in question, nor can they represent any party involved in its development. Their compensation structure must not depend on the results of their evaluations or on the number of verifications that they provide. They must have the necessary staff and facilities to assess properly the devices for which they are designated. Any subcontractors retained by an NB must also meet all these requirements.
Although an NB is responsible for ensuring a device’s conformity with the essential requirements, it also has a contractual relationship with the manufacturer, which pays for the work. That affords some protection to the manufacturer because, in theory, an NB that is inefficient or difficult to deal with will soon find itself without any customers. The accreditation procedures and standards of the European Commission ensure that the standards of safety are met, and the private status of the NB ensures efficiency and consistency for the manufacturer.
As noted above, the level of involvement of an NB is determined by the classification of the product (Table 6-2).
The NB has the responsibility to inform the CA if it withdraws a design examination certificate from the manufacturer. The CA then ensures that the device is withdrawn from the market. However, the manufacturer has the right to respond to a decision to restrict or withdraw a device before it goes into effect unless there is an urgent need to take such a step.
TABLE 6-2 Conformity Assessment Procedure by Class
|
Class I |
Manufacturer certifies that product meets essential requirements without involvement of an NB unless device has measuring function or is sterile; documentation of product specification must be kept for auditing purposes; Class I devices must be registered with a member state |
|
Class IIa |
Manufacturer prepares technical documentation to support a “declaration of conformity” and makes it available to the NB for inspection and adopts a quality-management system that applies to manufacturing and is subject to audit by the NB, or manufacturer adopts a full quality-management system that applies to both manufacturing and design control and is subject to audit by the NB |
|
Class IIb |
Manufacturer submits to type examination by an NB in which representative sample of product along with relevant technical documentation is examined to ensure that it fulfills essential requirements and adopts a quality-management system that applies to manufacturing and is subject to audit by the NB, or manufacturer adopts a full quality-management system that applies to both manufacturing and design control and is subject to audit by the NB |
|
Class III and Active |
Manufacturer submits to type examination by an NB |
|
Implantable Devices |
in which representative sample of product with relevant technical documentation is examined to ensure that it fulfills essential requirements and adopts a quality-management system that applies to manufacturing and is subject to audit by the NB, or manufacturer adopts a full quality-management system that applies to both manufacturing and design control and is subject to audit by the NB; NB also examines design dossier and issues a certificate to confirm that the device complies with all relevant essential requirements |
Postmarket Vigilance
In addition to the postmarketing surveillance functions of the NBs, the MDD mandated the establishment of a European Databank on Medical Devices, a centralized database containing information on manufacturers, devices, conformity assessments and certificates, and clinical investigations. Its use is currently voluntary but will be required of member states in 2011 (European Commission, 2010c). The committee is not aware of any comprehensive evaluations of the European system based on postmarketing surveillance or adverse-event reports.
The Global Harmonization Task Force
The multinational nature of medical-device manufacturing and distribution has led governments, consumers, and industry to recognize the need for international cooperation regarding medical devices. In 1992, regulatory bodies and industry of the leading medical-device producer and consumer nations—Canada, the European Union/European Free Trade Association, Japan, Australia, and the United States—formed the GHTF. A collaborative body whose purpose is to improve public health and safety, promote international trade, and provide guidance to countries with developing medical-device regulatory systems, the GHTF works to form consensus among the partners on regulatory and technical standards and postmarketing surveillance efforts. Many international agreements are in place regarding medical devices and their regulatory requirements for importation. The GHTF is an attempt to bring nations that tightly regulate medical devices into line with one another to ease the flow of devices between countries, manufacturers, and consumers while ensuring safety. In addition to participants from the founding members, the task force includes participants from non–founding members and liaison bodies, which include public-health organizations and international standard-setting organizations.15
The GHTF is structured around five study groups that are responsible for examining different issues and different paths and barriers to harmonization within them. The study groups, made up of geographically diverse members from government and industry, are (GHTF, 2008)
• Study Group 1—medical-device regulatory systems, paths to harmonization, and standards for premarket submissions and labeling.
• Study Group 2—adverse-incident reporting, postmarketing surveillance, and harmonizing data-collection and reporting systems.
• Study Group 3—existing quality-system requirements.
• Study Group 4—quality-system auditing practices.
• Study Group 5—clinical safety and performance.
To date, the study groups have released 31 final documents providing technical guidance and standardized definitions and outlining generally agreed-on principles for use by regulators and manufacturers worldwide (GHTF, 2009). The GHTF has met with mixed success in its efforts. For example, the Summary Technical Document (STED) guidance is intended to
___________________
15In March 2011, the Steering Committee Chair of the GHTF announced that the organization would be developing and transitioning to a “regulator-led harmonisation and collaboration group.” According to the statement, the new entity would focus primarily on regulators of medical devices, including seeking new members in order to more accurately reflect the current medical-device market, while continuing to value input from industry and other stakeholders as appropriate (GHTF, 2011).
standardize the format whereby a manufacturer submits technical information to a regulatory authority or conformity-assessment body (such as an NB). STED would allow manufacturers to save time and effort in preparing a product for market and allow better comparison and information-sharing among regulatory jurisdictions. Although the STED guidance has been adopted or implemented as a pilot program in all five GHTF founding-member jurisdictions, it has not been widely used (IOM, 2010). Nations that have highly developed regulatory frameworks agree in principle that harmonization is desirable, but differences in legal systems and regulatory cultures make implementation challenging.
The Summary Technical Document Program
In 2002, GHTF Study Group 1 proposed a pilot program for device premarket review known as the Summary Technical Document (STED), program (GHTF, 2008). The FDA then issued draft guidance to assist the medical-device industry and FDA staff in implementing a voluntary pilot program (FDA, 2005b). Not all devices are eligible to participate in the pilot. Eligible devices are limited to those which are seen to be the subjects of common and substantial interest by the member countries of the GHTF.16
The program, begun in June 2003, is intended to evaluate STED as an alternative premarket submissions process. Its objective is to decrease the regulatory burden and increase the efficiency of the approval process for manufacturers that operate within the international market (FDA, 2005b; GHTF, 2008). Industry groups have stated their support for the STED program, but the FDA has identified only 27 STED submissions17 (IOM, 2010). Of the submissions, 24 were 510(k) submissions, and 3 were PMA applications. The PMA applications were all approved. Of the 510(k) submissions, 20 resulted in a determination of substantial equivalence, 2 were not substantially equivalent, and 2 were withdrawn. Given the lack of data needed to evaluate the program and the small number of devices eligible, it is not possible to determine what factors contribute to the lack of industry participation in the program.
Finding 6-8 Other countries that tightly regulate medical devices do not rely solely on substantial equivalence to a predicate for premar-
___________________
16A list of eligible devices and information on the alternative format (developed by the Global Harmonization Task Force) can be found in Appendix C of the Guidance on Traditional and Abbreviated 510(k)s.
17The FDA’s Document Mail Center identifies all submissions on receipt. STED submissions are flagged in the system. However, the FDA has stated that there are limitations in the system, and that the data on STED submissions may not be accurate (e-mail from Philip R. Desjardins, FDA, January 7, 2011).
ket review of medium-risk devices. The Global Harmonization Task Force also does not offer as part of its guidance a predicate-based system for premarket review of medical devices.
The Growing Number and Complexity of Medical Devices
• Finding 6-1 Medical-device technologies have evolved rapidly, and devices have become increasingly complex since the 1976 Medical Device Amendments.
• Finding 6-2 Manufacturers are using increasing amounts of software in devices and as devices; the increase is expected to continue. Software offers many benefits over hardware, including flexibility, ease of change, and the possibility of use in other devices.
• Finding 6-3 Software is responsible for an increasing number of recalls. There are insufficient data, however, to determine whether the increase reflects the increasing proportion of software in medical devices or a new and different set of problems and vulnerabilities.
• Finding 6-4 Software is different from hardware and therefore requires a different kind of evaluation.
The Medical-Device Ecosystem
• Finding 6-5 Industry-funded assessments of the effects of the 510(k) clearance process report that implementation of the process leads to a lack of predictability and transparency, which in turn has an adverse effect on venture-capital investment.
• Finding 6-6 The committee did not find assessments of how much and in which way innovation is influenced by the 510(k) clearance process.
• Finding 6-7 There is little collaboration in collection of postmarketing surveillance data among the FDA, healthcare facilities, healthcare providers, the medical-device industry, professional societies, payers, and patient-advocacy groups.
Globalization of the Medical-Device Industry
• Finding 6-8 Other countries that tightly regulate medical devices do not rely solely on substantial equivalence to a predicate for premarket review of medium-risk devices. The Global Harmonization Task Force also does not offer as part of its guidance a predicate-based system for premarket review of medical devices.
Ahn, H., M. Bhandari, and E. H. Schemitsch. 2009. An evidence-based approach to the adoption of new technology. Journal of Bone and Joint Surgery American Volume 91(Suppl 3):95-98.
Barkun, J. S., J. K. Aronson, L. S. Feldman, G. J. Maddern, S. M. Strasberg, D. G. Altman, J. M. Blazeby, I. C. Boutron, W. B. Campbell, P. A. Clavien, J. A. Cook, P. L. Ergina, D. R. Flum, P. Glasziou, J. C. Marshall, P. McCulloch, J. Nicholl, B. C. Reeves, C. M. Seiler, J. L. Meakins, D. Ashby, N. Black, J. Bunker, M. Burton, M. Campbell, K. Chalkidou, I. Chalmers, M. de Leval, J. Deeks, A. Grant, M. Gray, R. Greenhalgh, M. Jenicek, S. Kehoe, R. Lilford, P. Littlejohns, Y. Loke, R. Madhock, K. McPherson, P. Rothwell, B. Summerskill, D. Taggart, P. Tekkis, M. Thompson, T. Treasure, U. Trohler, and J. Vandenbroucke. 2009. Evaluation and stages of surgical innovations. Lancet 374(9695):1089-1096.
Beattie, S., S. Arnold, C. Cowan, P. Wagle, and C. Wright. 2002. Timing the application of security patches for optimal. Presented at LISA 02: Sixteenth Systems Administration Conference, Berkeley, CA.
Beck, K. 2004. Extreme programming explained: Embrace change. Boston, MA: Addison-Wesley.
Berkun, S. 2010. The myths of innovation. Sebastopol, CA: O’Reilly Media, Inc.
Bloomfield, R., and B. Littlewood. 2003. Multi-legged arguments: The impact of diversity upon confidence in dependability arguments. Presented at 2003 International Conference on Dependable Systems and Networks (DSN 03).
Chai, J. Y. 2000. Medical device regulation in the United States and the European Union: A comparative study. Food and Drug Law Journal 55(1):57-80.
Davis, S., E. Gilbertson, and S. Goodall. 2011. EU medical device approval safety assessment: A comparative analysis of medical device recall 2005-2009. Boston Consulting Group.
Defend, B., M. Salajegheh, K. Fu, and S. Inoue. 2008. Protecting global medical telemetry infrastructure. Amherst, MA: I3P Technical Report.
Ergina, P. L., J. A. Cook, J. M. Blazeby, I. Boutron, P. A. Clavien, B. C. Reeves, C. M. Seiler, D. G. Altman, J. K. Aronson, J. S. Barkun, W. B. Campbell, L. S. Feldman, D. R. Flum, P. Glasziou, G. J. Maddern, J. C. Marshall, P. McCulloch, J. Nicholl, S. M. Strasberg, J. L. Meakins, D. Ashby, N. Black, J. Bunker, M. Burton, M. Campbell, K. Chalkidou, I. Chalmers, M. de Leval, J. Deeks, A. Grant, M. Gray, R. Greenhalgh, M. Jenicek, S. Kehoe, R. Lilford, P. Littlejohns, Y. Loke, R. Madhock, K. McPherson, P. Rothwell, B. Summerskill, D. Taggart, P. Tekkis, M. Thompson, T. Treasure, U. Trohler, and J. Vandenbroucke. 2009. Challenges in evaluating surgical innovation. Lancet 374(9695):1097-1104.
Ernst and Young. 2009. Pulse of the industry: Medical technology report.
European Commission. 1998. Commission communication on the application of transitional provision of directive 93/42/EEC relating to medical devices (98/c 242/05).
_______. 2001. Interface with other directives: Guidelines relating to the application of: The council directive 90/385/EEC on active implantable medical devices the council directive 93/42/EEC on medical devices.
_______. 2010a. Classification of medical devices. http://ec.europa.eu/consumers/sectors/medical-devices/files/meddev/2_4_1_rev_9_classification_en.pdf (accessed 02/07/2010).
_______. 2010b. Consultation on the general product safety legislative initiative. http://ec.europa.eu/consumers/safety/prod_legis/GPSD_consultation/index_en.htm (accessed 02/07/2010).
_______. 2010c. Exploratory process on the future of the medical devices: Potential themes for further reflection at the European level and issues identified by the members. http://ec.europa.eu/consumers/sectors/medical-devices/files/exploratory_process/final_report_en.pdf. (accessed 02/07/2010).
_______. 2010d. Roadmap: Revision of the general product safety directive (2001/95/ec). http://ec.europa.eu/consumers/safety/prod_legis/GPSD_consultation/docs/GPSD_roadmap_en.pdf (accessed 02/07/2011).
FDA (Food and Drug Administration). 1993. The Temple report: Final report of the committee for clinical review. Based on a review of selected medical device applications. Silver Spring, MD: Food and Drug Administration.
_______. 1997. Deciding when to submit a 510(k) for a change to an existing device. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080235.htm (accessed 08/20/2010).
_______. 2002. General principles of software validation: Final guidance for industry and FDA staff. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm085371.pdf (accessed 05/20/2011).
_______. 2005a. Guidance for the content of premarket submissions for software contained in medical devices. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089543.htm (accessed 05/20/2011).
_______. 2005b. A pilot program to evaluate a proposed globally harmonized alternative for premarket procedures. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071520.htm (accessed 06/2/2011).
_______. 2008. FY 2008 performance report to Congress for the Office of Combination Products. Silver Spring, MD: Food and Drug Administration.
_______. 2009a. Guidance for industry: New contrast imaging indication considerations for devices and approved drug and biological products. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126051.pdf (accessed 02/07/2011).
_______. 2009b. Medtronic announces nationwide, voluntary recall of model 8870 software application card. http://www.fda.gov/MedicalDevices/Safety/RecallsCorrectionsRemovals/ListofRecalls/ucm133126.htm (accessed 01/03/2011).
_______. 2009c. Registered foreign establishments as of June 15, 2008. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/InternationalInformation/ucm134816.htm (accessed 02/07/2011).
_______. 2010a. CDRH preliminary internal evaluations—Volume I: 510(k) working group preliminary report and recommendations. Silver Spring, MD: Food and Drug Administration.
_______. 2010b. CDRH preliminary internal evaluations—Volume II: Task force on the utilization of science in regulatory decision making preliminary report and recommendations. Silver Spring, MD: Food and Drug Administration.
_______. 2010c. Innovation or stagnation: Challenge and opportunity on the critical path to new medical products. http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm077262.htm (accessed 02/28/2011).
_______. 2010d. What we do. http://www.fda.gov/aboutfda/whatwedo/default.htm (accessed 05/26/2011).
_______. 2011a. 510(k) and science report recommendations: Summary and overview of comments and next steps. Silver Spring, MD: Food and Drug Administration.
_______. 2011b. CDRH innovation initiative. http://www.fda.gov/AboutFDA/CentersOffices/CDRH/CDRHInnovation/default.htm (accessed 02/28/2011).
Foote, S. B. 2005. Frontiers of medical technology: Reflections on the intersection of innovation and the health care system. Minnesota Journal of Law, Science, and Technology 7(1):79-88.
GHTF (Global Harmonization Task Force). 2008. Summary technical documentation for demonstrating conformity to the essential principles of safety and performance of medical devices (STED) (SG1-N11).
_______. 2009. GHTF documents. http://www.ghtf.org/documents/ (accessed 02/07/2011).
_______. 2011. Statement from GHTF chair: Update on future directions of GHTF. http://www.ghtf.org/documents/statement-future-2011.pdf (accessed 04/25/2011).
Grubb, A., J. Laing, J. McHale, and I. Kennedy, eds. 2011. Principles of Medical Law. Third ed. Oxford, UK: Oxford University Press.
Halperin, D., T. Heydt-Benjamin, K. Fu, T. Kohno, and W. H. Maisel. 2008. Security and privacy for implantable medical devices. IEEE Pervasive Computing 7(1):30-39.
IOM (Institute of Medicine). 2010. Public health effectiveness of the FDA 510(k) clearance process: Balancing patient safety and innovation. Washington, DC: The National Academies Press.
_______. 2011. Public health effectiveness of the FDA 510(k) clearance process: Measuring postmarket performance and other select topics. Washington, DC: The National Academies Press.
ITA (International Trade Administration). 2010. Medical device industry assessment updated March 24, 2010. Washington, DC: United States Department of Commerce.
Jorgensen, M. 2010. Identification of more risks can lead to increased over-optimism of and over-confidence in software development effort estimates. Information and Software Technology 52(5):506-516.
Kalorama Information. 2010. The global market for medical devices. Market Research Group, LLC.
Kaplan, A. V., D. S. Baim, J. J. Smith, D. A. Feigal, M. Simons, D. Jefferys, T. J. Fogarty, R. E. Kuntz, and M. B. Leon. 2004. Medical device development: From prototype to regulatory approval. Circulation 109(25):3068-3072.
Lee, S., B. Ransford, K. Fu, T. Kohno, and W. H. Maisel. 2008. Electromagnetic interference (EMI) of implanted cardiac devices by MP3 player headphones. Circulation 118:S596.
Linehan, J. H., and A. Chaney. 2010. Academic/industry challenges for medical device development. Science Translational Medicine 2(63):63mr6.
Long, D. E. 2008. Crossing the innovation divide. Temple Law Review 81(2):507-554.
Makower, J., A. Meer, and L. Denend. 2010. FDA impact on U.S. medical technology innovation: A survey of over 200 medical technology companies. Mountain View, CA.
McCulloch, P., D. G. Altman, W. B. Campbell, D. R. Flum, P. Glasziou, J. C. Marshall, J. Nicholl, J. K. Aronson, J. S. Barkun, J. M. Blazeby, I. C. Boutron, P. A. Clavien, J. A. Cook, P. L. Ergina, L. S. Feldman, G. J. Maddern, B. C. Reeves, C. M. Seiler, S. M. Strasberg, J. L. Meakins, D. Ashby, N. Black, J. Bunker, M. Burton, M. Campbell, K. Chalkidou, I. Chalmers, M. de Leval, J. Deeks, A. Grant, M. Gray, R. Greenhalgh, M. Jenicek, S. Kehoe, R. Lilford, P. Littlejohns, Y. Loke, R. Madhock, K. McPherson, J. Meakins, P. Rothwell, B. Summerskill, D. Taggart, P. Tekkis, M. Thompson, T. Treasure, U. Trohler, and J. Vandenbroucke. 2009. No surgical innovation without evaluation: The ideal recommendations. Lancet 374(9695):1105-1112.
MDMA (Medical Device Manufacturers Association) and NVCA (National Venture Capital Association). 2009. Medical technology and venture capital: A fruitful yet fragile ecosystem. Washington, DC
Merriam Webster. 2011. http://www.merriam-webster.com/dictionary/innovation (accessed 05/26/2011).
NANDO (New Approach Notified and Designated Organisations). 2011. European Commission: Enterprise and industry: NANDO (new approach notified and designated organisations) information system. http://ec.europa.eu/enterprise/newapproach/nando/ (accessed 02/07/2011).
NRC (National Research Council). 1999. Trust in cyberspace. Washington, DC: National Academy Press.
Oracle. 2010. Oracle solaris patching strategy and best practices. Presented at Oracle Open World. September 29
OTA (Office of Technology Assessment). 1984. Federal policies and the medical devices industry. Washington, DC: Congress of the United States, Office of Technology Assessment.
Oxford Dictionaries. 2011. http://oxforddictionaries.com/view/entry/m_en_us1258230#m_en_us1258230 (accessed 05/26/2011).
Pfleeger, S. L. 2005. Soup or art? The role of evidential force in empirical software engineering. IEEE Software January/February 66-73.
Pfleeger, S. L., L. Hatton, and C. Howell. 2002. Solid software. Upper Saddle River, NJ: Prentice Hall.
Platzer, M. 2006. Patient capital: How venture capital investment drives revolutionary medical innovation. Washington, DC: National Venture Capital Association.
Privitra, M. B., M. Design, and J. Johnson. 2009. Interconnections of basic science research and product development in medical device design. Presented at 31st Annual Conference of the IEEE EMBS, Minneapolis, MN.
Rogers, E. M. 2003. Diffusion of innovations. New York: Simon & Schuster, Inc.
Schum, D. 1994. Evidential foundations of probabilistic reasoning. New York: Wiley-Interscience.
Shuren, J. 2011. On the impact of medical device regulation on jobs and patients. Testimony before the Subcommittee on Health, Committee on Energy and Commerce, House of Representatives. February 17, 2011. Washington, DC
Teece, D. J. 1986. Profiting from technological innovation: Implications for integration, collaboration, licensing, and public policy. Research Policy 15:285-305.







































