
We are in the beginning of a new era for drug safety where protecting public health means that [the Food and Drug Administration’s] responsibility doesn’t end when we grant a product market approval; that is merely the first check point in ensuring safety.
—Dr. Margaret Hamburg, Commissioner, US Food and Drug Administration (FDA, 2011a).
An estimated 48 percent of the US population take at least one prescription drug1 in a given month (Gu et al., 2010). Drugs provide great benefit to society by saving or improving lives. Antibiotics can cure infections, heart medications can decrease the risk of heart attacks, and drugs for multiple sclerosis can decrease the symptoms of the disease and improve patients’ quality of life. At the same time, virtually all drugs have some unintended side effects, some of which are serious and can harm the people who take them. Budnitz et al. (2006) estimate that about 700,000 people are treated in US emergency rooms each year for severe adverse drug reactions, and about 120,000 require hospital admission. In a more recent study using data from 2007 through 2009, Budnitz et al. (2011) estimate that there are about “265,802 emergency department visits (95% confidence interval [CI], 184,040 to 347,563) for adverse drug events annually … among adults 65 years of age or older”, of which 99,628 (95% CI, 55,531 to 143,724) required hospitalization.
The US Food and Drug Administration (FDA) is the agency responsible for ensuring that prescription drugs are safe and effective. FDA’s approval of a drug for use in the United States is the result of a considered judgment based on available data and the agency’s experience with such decisions that overall the potential benefits of the drug outweigh the risks to patients for whom the drug
![]()
1For simplicity, the committee uses the term drugs throughout this report, but similar considerations would apply to biologics. The committee’s charge is related to the US Food and Drug Administration (FDA) regulation of the drug and biologics supply. When discussing FDA’s regulatory authority and mission, therefore, the committee does not address FDA’s roles related to other products, such as tobacco, medical devices, veterinary medicines, food, or animal feed.
is indicated. However, premarketing data used in approval applications are collected from studies that involve small numbers of participants2—often only a few hundred or a few thousand—over a relatively short period of time (IOM, 2007a), so not all risks associated with a drug are known at the time of approval. Warnings or restrictions may be added to the product label, or a drug may be removed from the market because unexpected or greater than expected morbidity or mortality is identified only after a drug enters widespread use. The discovery of new adverse events in the postmarketing setting is part of the normal, natural history of approved drugs. The timely identification of and response to drug-related risks are central to the mission of FDA.
Recent advances in information technology, including electronic health records, and changes in FDA laws, such as the Food and Drug Administration Amendments Act (FDAAA) of 2007,3 provide the opportunity to improve the system for ensuring that drugs are safe and effective. Previous Institute of Medicine (IOM) reports have made recommendations about improving aspects of drug-related patient-safety issues to FDA, other federal agencies, and Congress (IOM, 2002, 2004, 2007a,b). However, no report has focused specifically on the ethical and scientific issues that arise in the postmarketing environment, including how these issues intersect with the authority of FDA to require manufacturers to conduct postmarketing research and how FDA should integrate that authority and evidence, into its regulatory decision-making. The present report addresses those issues in response to the committee’s charge (see Box 1-1) and offers specific recommendations about the ethics and science of FDA required postmarketing research and about the decision-making process about approved drugs when safety issues arise.
THE EVOLUTION OF THE FOOD AND DRUG ADMINISTRATION’S RESPONSIBILITIES IN THE POSTMARKETING SETTING
Food and Drug Administration Authority Before 2007
FDA’s regulatory authority has evolved over the last 100 years, often as a result of serious drug-related adverse events or deaths. Table 1-1 presents some milestones in FDA’s regulatory history. The initial grant of authority to FDA’s predecessor agency began in 1906 with the passage of the Pure Food and Drug Act,4 which, for drugs, focused on misbranding and adulteration. Thirty years
![]()
2Throughout this report, the committee uses the term participants or research participants rather than human subjects. The committee recognizes that both terms have been used in policy discussions on this topic for decades and that neither term perfectly captures the nature of the relationship between the persons who are studied in research (who are both subjects of research and participants in research) and those who are conducting the research.
3Food and Drug Administration Amendments Act of 2007, PL No. 110-85, 121 Stat. 823 (2007).
4Pure Food and Drug Act of 1906, PL 59-384, 34 Stat. 768 (1906).
BOX 1-1
Charge to the Committee
The US Food and Drug Administration (FDA) has requested that the Institute of Medicine convene a committee to evaluate the scientific and ethical issues involved in conducting studies of the safety of approved drugs. Questions to be explored by a committee include
1. What are the ethical and informed consent issues that must be considered when designing randomized clinical trials to evaluate potential safety risks?
2. What are the strengths and weaknesses of various approaches, including observational studies, including patient registries, meta-analyses, including patient-level data meta-analyses, and randomized controlled trials, to generate evidence about safety questions?
3. Considering the speed, cost, and value of studies, what types of follow-up studies are appropriate to investigate different kinds of signals (detected pre-approval or postapproval) and in what temporal order?
4. Under what circumstances should head-to-head randomized clinical trials for safety be required?
5. How should FDA factor in different kinds of safety evidence in considering different kinds of regulatory actions?
later, after more than 100 deaths, many in children, caused by diethylene glycol in an elixir of sulfanilamide, the Food, Drug, and Cosmetic Act (FDCA)5 was enacted (FDA, 2009a). Under the FDCA, a new drug could not enter into interstate commerce unless its sponsor filed a new-drug application (NDA) with FDA that contained convincing evidence from preclinical toxicity testing that the drug was safe for its intended uses (Daemmrich, 2004a; Marks, 1997a). A drug was to be evaluated only with regard to its toxicity; its sponsor was not required to provide FDA with evidence of effectiveness or benefits. FDA could, however, deem a drug misbranded “if its labeling is false or misleading”.6 Under those conditions, FDA had the authority to withdraw its approval of the drug and to prosecute the drug sponsor (Carpenter, 2010a; Daemmrich, 2004a; Grossman et al., 2007; Marks, 1997a).7
![]()
5Food, Drug, and Cosmetic Act, PL No. 75-717, 52 Stat. 1040 (1938).
621 USC § 331(a), (b), (c), (k) (2010).
721 USC § 331 (2010).
|
Table 1-1 Key Milestones in Food and Drug Administration History Related to Drug Safety and Drug Studies |
||
| Year | Milestone | |
| 1906 | Pure Food and Drug Act passed | |
| 1938 | Federal Food, Drug, and Cosmetic Act passed | |
| 1962 | Kefauver–Harris Drug Amendments of 1962 passed | |
| 1981 | Food and Drug Administration (FDA) and Department of Health and Human Services revise and promulgate separate regulations for protection of research participants | |
| 1987 | Investigational-drug regulations revised to expand access to experimental drugs for patients who have serious diseases for which there are no alternative therapies | |
| 1991 | Regulations published to accelerate the review of drugs for life-threatening diseases | |
| 1992 | First Prescription Drug User Fee Act (PDUFA) passed | |
| 1992 | FDA given the authority to require postmarketing trials for accelerated approvals | |
| 1993 | Consolidation of several adverse-reaction reporting systems launched as MedWatch | |
| 1997 | Food and Drug Administration Modernization Act reauthorizes PDUFA (PDUFA II) | |
| 2002 | Public Health Security and Bioterrorism Preparedness and Response Act reauthorizes PDUFA (PDUFA III); some funds allocated for drug-safety activities | |
| 2003 | Pediatric Research Equity Act allows FDA to require clinical research into possible pediatric applications for new drugs and biologic products | |
| 2005 | Drug Safety Board formeda | |
| 2007 | Food and Drug Administration Amendments Act passed; act reauthorizes PDUFA (PDUFA IV) and dedicates a greater portion of funds to drug safetyb | |
aConsisting of FDA staff and representatives of the National Institutes of Health and the Department of Veterans Affairs. The board advises the FDA Center for Drug Evaluation and Research (CDER) on drug-safety issues and works with CDER in communicating safety information to health professionals and patients. The administrative action creating this body occurred after prominent drug-safety controversies (such as controversies about Cox-2 selective agents, such as Bextra and Vioxx). The board was later codified in FDAAA (121 Stat 938).
bPDUFA IV expires in September 2012. PDUFA V is under discussion. Abbreviations: FDA, Food and Drug Administration; FDAAA, Food and Drug Administration Amendments Act; PDUFA, Prescription Drug User Fee Act.
SOURCE: FDA (2009a).
The next major change in FDA’s statutory authority occurred in the early 1960s. Thousands of children in a number of countries other than the United States were born with limb defects to mothers who had been administered thalidomide for morning sickness; FDA had prevented marketing of thalidomide in the United States (Carpenter, 2010b; Hilts, 2003a). Concerns about the implications of this tragedy prompted Congress to pass the Drug Amendments of 1962 (PL 87-781), often referred to as the Kefauver–Harris amendments. That legislation shifted the burden of proof for a drug from FDA proving harm to manufacturers proving safety and efficacy, and represented a major shift in FDA’s role and authority. For the first time, a drug sponsor was required by statute to provide evidence of the effectiveness of a drug, codifying some of FDA’s practices during the 1950s (Carpenter, 2010b). After enactment of the 1962 amendments, the randomized controlled trial (RCT) emerged as the gold standard for the adequate and well-controlled studies required to demonstrate efficacy (Marks, 1997b) and led
to the current drug-approval process summarized in Box 1-2. The 1962 amendments emphasized FDA’s role as the marketplace gatekeeper for new drugs. The Kefauver–Harris amendments also required pharmaceutical companies to keep records of “clinical experience”,8 which were later interpreted as a requirement to report adverse reactions and events. That requirement evolved into today’s MedWatch program, a consolidation of several adverse-reaction reporting systems (available at http://www.fda.gov/Safety/MedWatch/default.htm).
Concerns emerged in the 1980s that the length and complexity of the drug-approval process were delaying the availability of new life-saving drugs, such as those to treat AIDS and cancer (Anderson, 1989; Carpenter, 2010c; Daemmrich, 2004a,b; Hilts, 2003b). Patient groups, regulators, pharmaceutical companies, and others argued that a lack of resources at FDA was slowing the drug-approval process and preventing important drugs from coming to market in a timely matter (FDA, 2011b). In response, Congress passed the Prescription Drug User Fee Act (PDUFA) of 1992,9 whose aim was to increase the pace of drug review by increasing FDA’s resources to expand its drug-review staff and capabilities. In exchange for sponsors paying user fees, FDA agreed to use the funds to meet scheduled review goals, and Congress guaranteed not to reduce FDA’s appropriations to compensate for the user fees (Carpenter, 2010c). FDA also agreed that the increased funds from the PDUFA would “be dedicated towards expediting the drug development process and the process for the review of human drug applications”.10
In an effort to decrease the approval time for selected life-saving drugs, FDA promulgated its accelerated approval regulations in 1992 (21 CFR pt. 314, subpt. H, often referred to simply as “Subpart H”). Under these regulations, FDA may approve new drugs that treat “serious or life threatening illnesses”11 based on clinical trials that used surrogate endpoints12 in assessing the drug’s efficacy.13 In 1997, Congress confirmed FDA’s statutory authority to approve drugs under
![]()
821 USC § 355(k) (2010).
9Prescription Drug User Fee Act of 1992, PL No. 102-571, 106 Stat. 4491 (1992). The PDUFA was written to expire in 5 years, but later laws (PDUFA II through PDUFA IV) have ensured that the user fees continued. The current Congress is expected to approve another extension before October 1, 2012, when PDUFA IV user-fee authority expires.
1021 USC § 379g note (2010).
1121 CFR § 314.500 (2011).
12FDA defines a surrogate endpoint as a “biomarker intended to substitute for a clinical efficacy endpoint. Surrogate endpoints are expected to predict clinical benefit (or harm, or lack of benefit or harm)” (Atkinson et al., 2001; IOM, 2010a). In contrast, a “clinical endpoint is defined as a characteristic or variable that reflects how a patient feels, functions, or survives” (Atkinson et al., 2001; IOM, 2010a). For example, blood pressure might be used as a surrogate endpoint in tests of a drug that decreases the risk of a heart attack or stroke associated with hypertension. As another example, delay in progression to blast crisis—a phase in which immature granulocytes (white blood cells) rapidly proliferate in the chronic phase of chronic myelogenous leukemia—is used as a surrogate endpoint in studies used for evaluation and approval of drugs, but the true clinical benefit of the drug is long-term survival.
1321 CFR § 314.510 (2011).
BOX 1-2
Drug-Approval Process
The development of a candidate drug can begin with preclinical research and short-term animal testing by a drug sponsor. If a candidate drug shows therapeutic potential and low toxicity, the sponsor can submit an investigational new drug (IND) application to the US Food and Drug Administration (FDA), and agency oversight begins.a The IND should contain
• Manufacturing and chemical information about the drug.
• The results of any completed animal tests, toxicology studies, and any other preclinical tests that have been conducted.
• The protocols for Phase 1 human studies.
• The results of any human studies that the sponsor has conducted outside the United States; local institutional review boards must review Phase 1 protocols to ensure protection of reserach participants.
FDA then has 30 days to place a hold on the proposed human trials because of safety concerns. Without a hold, the sponsor can begin to test the compound in humans at 31 days. Long-term animal studies, including carcinogenicity and reproductive-toxicity studies, might occur simultaneously with human studies.
Phase 1 clinical trials test several increasing doses of a drug to assess toxicity and, to some degree, efficacy. In the absence of unacceptable toxicity, Phase 2 clinical investigations (which are conducted on a few dozen to several hundred patients who have the condition for which the drug is being developed) examine both efficacy and safety. Typically, two Phase 3 clinical trials, which can involve fewer than 100 patients or many thousands, are then conducted to evaluate the efficacy of the new product, usually in comparison with a placebo and sometimes in comparison with an already approved drug for the condition. Spon-
these conditions.14 As a condition of the accelerated approval process, Subpart H also requires postmarketing clinical studies15 to confirm the health benefits of the drug that were predicted on the basis of the surrogate endpoints; those trials are typically already underway at the time of approval. FDA also obtained a
![]()
1421 USC § 356(b)(2) (2010).
15Studies conducted after a drug has been approved for marketing are referred to as postmarketing, postapproval, or Phase 4 studies. For consistency the committee refers to such studies as postmarketing studies.
sors develop the study protocols, and trials are conducted under their auspices. FDA provides guidance documents for clinical trials, and the Office of New Drugs review team consults with the sponsor as trial protocols are developed, studies are conducted, and results are obtained. Sponsors are required to notify FDA of serious or unexpected adverse effects that are experienced by trial participants and are potentially attributable to the drug and any findings from animal tests that suggest an important risk for humans, including reports of mutagenicity, teratogenicity, or carcinogenicity.
After the completion of Phase 3 trials, the sponsor can submit a new drug application (NDA) for a chemical drug or a biologic license application for a biologic, which should include
• The chemical composition of the drug.
• The results of pharmacokinetic studies.
• The results of animal tests and clinical trials.
• Details of the manufacturing, processing, labeling, and packaging of the drug.
FDA will either approve the drug and send an “approval” letter to the sponsor with specified labeling and any postmarketing requirements or not approve the drug and send a “complete response” letter to the sponsor explaining why an application is not approved. FDA has a goal, in accordance with the Prescription Drug User Fee Act, of reviewing NDAs for standard approved drugs and for expedited approved drugs determined to be potentially breakthroughs or life-saving within a particular timeframe in accordance with the law.
![]()
aAn investigational new drug application can be opened at any phase of drug testing. Given the global nature of drug development, it is not unusual for the first trial to be submitted to FDA to be a Phase 2 or 3 trial (FDA, 2003).
SOURCE: Modified from FDA (1998).
second authority to require postmarketing studies for approved drugs through the deferred-submission policy under the Pediatric Research Equity Act (PREA) of 2003,16 which allows a drug to be approved for sale if sponsors agree to conduct the required trials in children after a drug enters the market.
![]()
16Pediatric Research Equity Act (PREA) of 2003, PL 108-155, 117 Stat. 1936 (2003). FDA initially published the pediatric rule in the Federal Register in 1998. After that rule was overturned in court because it went beyond FDA’s regulatory authority, Congress passed PREA in 2003, giving FDA the necessary authority.
FDA began receiving more resources for postmarketing activities through the Public Health Security and Bioterrorism Preparedness and Response Act of 2002,17 which included the PDUFA Amendments of 2002 (PDUFA III). Under this act, for the first time some user fees were allocated to postmarketing safety–related activities, for example, to support postmarketing surveillance of already-approved drugs and to allow risk-management oversight of newly approved drugs (2–3 years after approval). Furthermore, pharmaceutical companies could develop and submit a risk-minimization action plan (RiskMAP) with an NDA (FDA, 2005). RiskMAPs were plans intended for a small number of drugs that posed serious risks that warranted additional precautions beyond labeling to manage or limit the risks and ensure that the benefits18 of the drugs outweighed their risks. For example, thalidomide was approved for use in the treatment of multiple myeloma and forms of leprosy and was accompanied by a RiskMAP19 that outlined measures to prevent the risk of fetal exposure. As of February 2007, 30 drugs had been approved with RiskMAPs20; most plans contained only targeted education and outreach requirements (Office of Surveillance and Epidemiology, 2007; Shane, 2009).
The Institute of Medicine’s 2007 Report
Despite those changes, the focus of FDA’s authority and resources remained on the drug-approval process, with little oversight responsibility or authority related to drugs that had entered the market. High-profile withdrawals of some drugs within 5 years of their approval—such as troglitazone (Rezulin®, Resulin®, or Romozin®), cerivastatin (Baycol® or Lipobay®), rofecoxib (Vioxx®), and valdecoxib (Bextra®)—underscored concerns not only about FDA’s ability to recognize and respond to safety signals in the postmarketing setting in a timely fashion but its ability to conduct appropriate oversight of approved drugs and to undertake appropriately targeted regulatory actions short of withdrawal. In 2006, FDA, the Centers for Medicare and Medicaid Services (CMS), the Agency for Healthcare Research and Quality (AHRQ), the National Institutes of Health (NIH), and the Department of Veterans Affairs asked the IOM to examine the US
![]()
17Public Health Security and Bioterrorism Preparedness and Response Act of 2002, PL 107-188, 1165 Stat. 594.
18When discussing the benefits and risks of a drug, like the term risk, the committee uses the term benefits in a probabilistic manner, that is, the potential or probability of benefits. To emphasize the importance of the benefit side of a drug’s benefits and risks, the committee purposefully uses the phrase benefit–risk rather than the more traditional risk–benefit.
19FDA approved thalidomide in 1998 with a System for Thalidomide Education and Prescribing Safety (STEPS) oversight program that contained restrictions on use (see approval letter at http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1998/20785ltr.pdf). The STEPS oversight program predates the establishment of RiskMAPs, but it was considered a RiskMAP after they were established.
20RiskMAPs were replaced with Risk Evaluation and Mitigation Strategies (REMS) following implementation of the FDAAA.
drug-safety system and to make recommendations to improve risk assessment, surveillance, and safe use of drugs (IOM, 2007a).
The 2007 IOM committee’s report, The Future of Drug Safety, focused on how FDA’s structure, organization, and scientific and regulatory activities should change to improve the monitoring and evaluation of drugs. The report concluded that “a transformed drug safety system has at its core a lifecycle approach to drug risk and benefit—not a new concept, but one that has been implemented, at best, in a limited and fragmented manner” (IOM, 2007a). The 2007 IOM committee recognized that changes were needed to implement such a safety system and made a number of recommendations, including (1) changes in the organization and culture of FDA to provide long-term stability and consistent direction for the agency and to increase the role of the Office of Surveillance and Epidemiology (OSE) in drug regulation; (2) an increase in funding for postmarketing activities in FDA; (3) improvements in FDA’s information-technology infrastructure for monitoring drugs once they are approved, including development of public–private partnerships to gain access to and analyze data related to drug safety; (4) development by FDA of “a systematic approach to risk-benefit analysis for use throughout the FDA in the preapproval and postapproval settings”; (5) provision to FDA of new authority to require “postmarketing risk assessment and risk management programs as are needed to monitor and ensure safe use of drug products”21; (6) development and implementation by FDA of a high-quality, flexible system for the evaluation of postmarketing safety issues; and (7) the requirement for industry to register at NIH’s ClinicalTrials.gov all Phase 2 through Phase 4 clinical trials (postmarketing trials) that are intended to be submitted to FDA.22
The Food and Drug Administration Amendments Act: The Food and Drug Administration’s Increased Postmarketing Authority and Responsibility
In 2007, less than a year after that IOM report was published (IOM, 2007a), Congress passed FDAAA, which amended the FDCA to include many of the recommendations presented in the report. In addition to reauthorizing a higher level of prescription-drug user fees (PDUFA IV), Congress earmarked increased resources for postmarketing drug activities and included a number of substantial changes in FDA’s regulatory authority. Described as “the most momentous shift
![]()
21The committee recommended that the risk-assessment and risk-management program could include the authority to require label changes, specific warnings or moratoriums on direct-to-consumer advertising for specific drugs, and restrictions on distribution of a specific drug, such as limiting distribution to particular facilities, pharmacists, or physicians with specific training or only after the performance of specific medical procedures (IOM, 2007a).
22FDA’s responses to IOM’s 2007 report can be found at http://www.fda.gov/Safety/SafetyofSpecificProducts/ucm184598.htm (accessed June 9, 2011).
in drug regulation in half a century” (Evans, 2010), FDAAA gave FDA expanded authority and responsibility in the postmarketing setting.
FDAAA increased requirements for registering clinical trials,23 increased FDA’s authority over the contents of direct-to-consumer advertising,24 increased resources for FDA’s premarketing and postmarketing activities related to drug risks,25 gave FDA the authority to implement safety-labeling changes including class labeling,26 and required that FDA increase the transparency of its information about drugs and improve its risk communication.27 In addition—and most relevant to the present report—FDAAA provided FDA with the authority to require postmarketing studies in some circumstances28 (see Box 1-3 for a discussion of the committee’s terminology for postmarketing studies), provided FDA with the authority to require a risk evaluation and mitigation strategy (REMS),29 and required that FDA develop an active surveillance system.30 Those three elements are described in the subsections below; more details of other parts of FDAAA are presented in Appendix A.
Authority to Require Postmarketing Studies
Before FDAAA, most postmarketing studies were performed under voluntary written agreements between the sponsor and FDA called postmarketing commitments (PMCs), established at the time of drug approval (FDA, 2011c). FDA could require postmarketing studies or clinical trials in only two situations: in 21 CFR 314.510 and 21 CFR 601.4, for products that enter the marketplace as a consequence of accelerated approvals to demonstrate clinical benefit; and in PREA, for products that are approved based only on research with adult participants where postmarketing research is needed to assess their safety and efficacy of the drug in children.31 A number of people have criticized the low completion rate of the postmarketing studies required by FDA through these mechanisms (Avorn, 2005; Carpenter, 2010d; GAO, 2008; Strom, 2006; Wood, 2006).32
FDAAA expanded FDA’s authority to require sponsors of marketed drug and biologic products to conduct and report on postmarketing research studies.33 A
![]()
2342 USC § 282(j) (2010).
2421 USC § 353b (2010).
2521 USC § 379g note (2010).
2621 USC § 355(o)(4) (2010).
2721 USC § 360bbb–6.
2821 USC § 355(o) (2010).
2921 USC § 355(p) (2010).
3021 USC § 355(k)(3) (2010).
31In the remainder of this report, when the committee discusses FDA-required studies, it is referring to studies FDA requires using its authorities in FDAAA, not those it requires using its authorities in 21 CFR 601 Subpart E or in PREA.
32As discussed below, however, at least part of the low completion rate has since been attributed to the tracking system.
3321 USC § 355(o) (2010).
BOX 1-3
Nomenclature for Postmarketing Studies
The Food and Drug Administration Amendments Act of 2007 differentiates between clinical trials and studies.a In response to that differentiation, FDA defines clinical trials and studies as follows (FDA, 2011d):
“Clinical trials are any prospective investigations in which the applicant or investigator determines the method of assigning the drug product(s) or other interventions to one or more human subjects.”
“Studies are all other investigations, such as investigations with humans that are not clinical trials as defined above (e.g., observational epidemiological studies), animal studies, and laboratory experiments.”
In this report, however, the committee uses the term studies in accordance with ordinary usage in the scientific literature as a parent or generic term that encompasses research projects of all types and regardless of design. Thus, as the committee uses the term, studies applies to both clinical trials and non-clinical trial investigations such as observational investigations. When referring specifically to either observational designs or randomized controlled trial (RCT) designs, the committee uses those specific terms.
![]()
a21 USC § 355(o)(3) (2010).
postmarketing requirement (PMR) is an FDA-required research study that a sponsor must conduct after a drug has been approved and is released to the market. Under FDAAA, a PMR can be required to
• Assess a known serious risk related to use of the drug.
• Assess signals of serious risk related to use of the drug.
• Identify an unexpected serious risk when available data indicate the potential of a serious risk.34
Within its definition of a serious risk of an adverse drug experience, FDAAA includes “any failure of expected pharmacological action of the drug” that results in serious medical consequences for the patient.35 On the basis of that definition, Evans (2010) considers FDAAA to provide FDA with the authority to require a postmarketing study when emerging data or results suggest that patients are suffering serious harm because a drug is not performing as effectively as was expected at the time it was approved, either overall or in identifiable patient
![]()
3421 USC § 355(o)(3)(B) (2010).
3521 USC § 355-1(b)(1)(E), (b)(4), (b)(5) (2010).
BOX 1-4
Types of Studies for which the Food and Drug
Administration Considers Postmarketing
Requirements vs Postmarketing Commitments
The FDA recently issued guidance for industry on postmarketing studies that outlines the differences between postmarketing requirements (PMRs) and postmarketing commitments (PMCs) and provides examples of when it would require a PMR or request a PMC. In those guidelines, FDA provides the following examples of the kinds of studies that might be the subject of PMRs:
• “Observational pharmacoepidemiologic studies are generally studies designed to assess a serious risk associated with a drug exposure or to quantify risk or evaluate factors that affect the risk of serious toxicity, such as drug dose, timing of exposure, or patient characteristics. …
• “Meta-analyses may be designed to evaluate a safety endpoint by statistical analysis of data from completed studies or clinical trials. …
• “Clinical trials with a safety endpoint evaluated with prespecified assessments and adequately powered to analyze the serious risk. …
• “Studies or clinical trials designed to evaluate drug interactions or bioavailability when there are scientific data that indicate the potential for a serious safety risk.”
A current example of a PMR is a current clinical trial that is assessing whether there is decreased survival with concomitant use of pani-
subgroups. The present committee considers providing FDA with that authority to be in the interest of the public’s health. When questions arise about the health benefits of a drug, studies to document a drug’s effectiveness may be as critical for ensuring that the benefit–risk profile of a drug remains favorable as studies that investigate its risks.
FDA can require a drug sponsor to conduct an observational study only when the information that FDA needs cannot be obtained from adverse-event or surveillance data,36 including data from the Sentinel system (discussed below).37 FDA’s
![]()
3621 USC § 355(o)(3)(D)(i) (2010).
3721 USC § 355(k)(3) (2010).
tumumab (Vextibix®) and other chemotherapy for metastatic colorectal cancer (FDA, 2011d).
In contrast, the guidelines state that the following studies would generally not meet the statutory conditions for a PMR, and FDA would request them as a PMC:
• “Drug and biologic quality studies, including manufacturing, stability, and immunogenicity studies that do not have a primary safety endpoint. …
• “Pharmacoepidemiologic studies designed to examine the natural history of a disease or to estimate background rates for adverse events in a population not treated with the drug that is the subject of the marketing application. …
• “Studies and clinical trials conducted with vaccines, such as surveillance and observational pharmacoepidemiologic studies when data do not suggest a serious risk or signals of serious risk related to the use of the vaccine and when available data do not indicate the potential for serious risk”, including studies to “evaluate longterm effectiveness or duration of response. …
• “Clinical trials in which the primary endpoint is related to further defining efficacy, designed to
o evaluate long-term effectiveness or duration of response (that are not required under accelerated approval)
o evaluate efficacy using a withdrawal design
o evaluate efficacy in a subgroup”.
![]()
SOURCE: FDA (2011c).
authority to require a clinical trial is further restricted to contexts in which the information that it needs cannot be obtained by requiring observational studies.38
In 2011, FDA issued guidance for industry that provides information on the implementation the section of FDAAA39 that authorizes FDA to require certain postmarketing studies and trials (FDA, 2011c). The guidance outlines the differences between PMRs and PMCs and provides examples of PMRs and PMCs (see Box 1-4) (FDA, 2011c). In contrast to a PMR, a PMC that a sponsor agrees to in writing after approval of a product is typically designed to gather additional
![]()
3821 USC § 355(o)(3)(D)(ii) (2010).
3921 USC § 355(o) (2010).
information about product safety, efficacy, or optimal use. FDA has decided that such information is useful or important but is not a condition of approval.
FDA is required to track and monitor the progress of PMRs and PMCs to ensure that they are completed in a timely manner, and it reviews annual status reports submitted by sponsors. FDA has been criticized for not ensuring the initiation and completion of PMRs and PMCs; recent audits suggest, however, that although FDA system for tracking the progress of the studies was incomplete, most studies either had been completed or were under way. As of September 30, 2011, there were 675 PMRs and 369 PMCs open for drugs. Of those, 590 (87 percent) of the PMRs, and 295 (80 percent) of the PMCs were on schedule (FDA, 2012).
Risk Evaluation and Mitigation Strategies
As discussed above, PDUFA III in 2002 allowed drug sponsors to provide a RiskMAP for products that posed serious risks that outweighed their benefits when specific precautions could be implemented that would result in a favorable benefit–risk profile. Under FDAAA in 2007, REMS was introduced to replace the RiskMAP as part of a risk-management strategy intended to manage a known or serious risk posed by a drug or biologic product.40 The possible elements of a REMS are listed in Box 1-5. FDAAA grants FDA the authority to require sponsors to submit a REMS before approval of a drug if it “determines that a [REMS] is necessary to ensure that the benefits of the drug outweigh the risks”41 or after approval if FDA “becomes aware of new safety information and makes a determination that such a strategy is necessary to ensure that the benefits of the drug outweigh the risks of the drug”.42 As of March 1, 2012, there were 103 pharmaceutical products with active REMSs. Of those, 26 (25 percent) required only a medication guide; 39 (38 percent) required a communication plan, or a medication guide and communication plan; and 38 (37 percent) required elements to ensure safe use and other components (Center for Healthcare Supply Chain Research, 2012).43
Development of a Large-Scale Active Surveillance System
FDAAA mandated that FDA establish an active surveillance system for monitoring drugs by using large electronic databases belonging to health care information holders. An internal FDA task force had previously concluded that FDA’s postmarketing surveillance would be improved by the agency having
![]()
40In general, products previously approved with a RiskMAP that had elements to ensure safe use were “deemed to have an approved REMS” (FDA, 2009b).
4121 USC § 355-1 (a)(2)(A) (2010).
4221 USC § 355-1 (a)(2)(A) (2010).
43The raw data are available on FDA’s website: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm111350.htm (accessed June 9, 2011).
BOX 1-5
Potential Elements of a Risk Evaluation
and Mitigation Strategy (REMS)
In addition to the required timetable for submission of assessments of the strategy, potential elements of the strategy area
1. A medication guideb and, if helpful to mitigate a serious risk posed by the drug, a patient package insert.
2. A communication plan to health care providers that may include
• Sending letters to health care providers.
• “Disseminating information about the elements of the risk evaluation and mitigation strategy to encourage implementation by health care providers of components that apply to such health care providers, or to explain certain safety protocols (such as medical monitoring by periodic laboratory tests)”.
• “Disseminating information to health care providers through professional societies about any serious risks of the drug and any protocol to assure safe use”.
3. Elements as are necessary to ensure safe use of the drug because of its inherent toxicity or potential harmfulness. The elements to ensure safe use shall include one or more goals to mitigate a specific serious risk listed in the labeling of the drug and, to mitigate such risk, may require that
• Health-care providers who prescribe the drug have particular training or experience or are specially certified (the opportunity to obtain such training or certification with respect to the drug shall be available to any willing provider from a frontier area in a widely available training or certification method, including an online course or via mail, as approved by the secretary at reasonable cost to the provider).
• Pharmacies, practitioners, or health care settings that dispense the drug are specially certified (the opportunity to obtain such certification shall be available to any willing provider from a frontier area).
• The drug be dispensed to patients only in specific health care settings, such as hospitals.
• The drug be dispensed to patients with evidence or other documentation of safe-use conditions, such as laboratory test results.
• Each patient using the drug be subject to monitoring.
• Each patient using the drug be enrolled in a registry.
![]()
a21 USC §§ 355-1(e), (f).
bFDA can require a Medication Guide on its own or as part of a REMS. November 2011 FDA guidance states that “depending on the risks involved, FDA may approve a Medication Guide … without requiring a REMS when that alone is adequate to address the serious and significant public health concern” (FDA, 2011e).
“access to external healthcare databases” (FDA, 1999). Congress established goals and a timetable for the system, including a goal of the capacity to access data on 25 million patients by July 1, 2010, and 100 million patients by July 1, 2012.
That national electronic system, developed through the Sentinel Initiative, was launched by FDA in 2008 and has the aim of facilitating the development of active surveillance methods related to “signal detection, strengthening, and validation” (FDA, 2008; HHS, 2010; Platt et al., 2009, 2012; Robb et al., 2012). Sentinel will increase FDA’s ability to track the benefit–risk profiles of drugs, biologics, medical devices, and other FDA regulated products and has the potential to decrease the time needed to identify and evaluate drug-related safety signals. Sentinel will be an active surveillance system and will complement the existing mandatory and voluntary reporting systems that FDA already has in place to track reports of adverse events linked to use of its regulated products. Sentinel will enable FDA to actively query diverse automated health care data holders—such as electronic health-record systems, administrative and insurance-claims databases, and registries—and to obtain the results of specific queries rapidly and securely (FDA, 2008). Sentinel is proceeding in stages, starting with a series of Mini-Sentinel projects to develop the individual parts of the system (HHS, 2010). The electronic data “will be accessed, maintained, and protected by the Sentinel System’s data partners” (FDA, 2008). In such a structure, termed a distributed system, “data remain in their existing secure environments, rather than being consolidated into one database” (FDA, 2008). The Mini-Sentinel initiative also includes development of statistical and epidemiologic methods to improve the use of data from active surveillance (Cook et al., 2012; IOM, 2007a). Concurrently, the Observational Medical Outcomes Partnerships (OMOP), a “public–private partnership among the FDA, academia, data owners, and the pharmaceutical industry” is studying governance, data resource, and methodological issues with a national drug surveillance program (Stang et al., 2010). Those activities will facilitate the development and improvement of postmarketing surveillance systems.
FDA’s Organizational Structure and the Postmarketing Setting
There are two offices within FDA’s Center for Drug Evaluation and Research (CDER) of relevance to the postmarketing setting. The Office of New Drugs (OND) is responsible for “providing regulatory oversight for investigational studies during drug development and making decisions regarding marketing approval for new (innovator or non-generic) drugs” (FDA, 2011f), including decisions governing accelerated approvals. The Office of Surveillance and Epidemiology (OSE) is responsible for “postmarketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug development process” (FDA, 2009c). IOM’s 2007 drug safety report (IOM, 2007a) concluded
that “the Office of Surveillance and Epidemiology … has not had a formal role in drug regulation—neither formal opportunities to learn from and participate in relevant aspects of the review process nor the authority to take action regarding postmarketing safety” (IOM, 2007a). The 2007 report noted that there existed interdisciplinary tension between the two offices, and the negative effect that tension has on anticipating and planning for postmarketing oversight of the benefit–risk profiles of drugs and recommended “that CDER appoint an OSE staff member to each New Drug Application review team and assign joint authority to the Office of New Drugs … and the Office of Surveillance and Epidemiology … for postapproval regulatory actions related to safety” (IOM, 2007a). Others have also described the tensions between OSE and OND (Carpenter, 2010d), and some researchers have commented on the need for independent drug safety review (Avorn, 2005; Strom, 2006; Wood et al., 1998).
Since the publication of the 2007 report (IOM, 2007a) and the passage of FDAAA, FDA has implemented a number of changes to increase the standing of the OSE and improve collaboration between OND and OSE on drug regulatory decisions. In January of 2008 FDA established its Equal Voice Initiative, an “operational philosophy and set of practices to ensure that each professional viewpoint has been fully expressed, understood, and brought into the decision-making process” (FDA, 2010a). In June 2009, OND and OSE signed a Memorandum of Agreement “on the management of significant safety issues associated with pending drug applications and approved drug products”.44 The document is designed to clarify the roles and responsibilities of OND and OSE, and it specifies that “OND and OSE views are to be given equal weight in determining how significant safety issues affecting drug products are resolved” (FDA, 2009d). OND has also established the positions of Deputy Director for Safety and Safety Regulatory Project Manager in each review division.
In 2009, GAO reported that the “OND retains the authority to decide whether to take regulatory action”. It recommended that the FDA commissioner “develop a comprehensive plan to prepare OSE for the transfer of additional regulatory authorities from OND” (GAO, 2009). GAO indicated that such an approach is intended to improve the management and oversight of postmarketing studies. Consequently, an internal Manual of Policies and Procedures was developed
![]()
44“A significant safety issue for purposes of this memorandum of agreement is a safety issue that has the potential to lead to, for example: withdrawal of an approved drug from the market; withdrawal of an approved indication; limitations on a use in a specific population or subpopulation in the post-marketing setting; changes to the warnings, precautions, or contraindication sections of the labeling (including the addition of a boxed warning to the label); the establishment of, or changes to, the proprietary name/container label/labeling/packaging to reduce the likelihood of medication errors; the establishment or modification of a risk evaluation and mitigation strategy (REMS); addition or modification of a Medication Guide or other required Patient Package Insert that addresses a safety issue; the requirement that a sponsor conduct a post-marketing clinical trial; or the conduct of an observational pharmacoepidemiological study by the sponsor or FDA” (FDA, 2009d).
by FDA, effective September 16, 2010, that “provides the general guidance for incorporating the philosophy and practices of EV [Equal Voice] into CDER decision-making processes” (FDA, 2010a). FDA has also hired additional staff responsible for postmarketing monitoring of the benefit–risk profiles of drugs (FDA, 2009d), and increased the staff located within OSE and additional staff within OND responsible for evaluating and interpreting safety signals. The extent to which these changes have improved the communication and collaboration of OSE and OND with regard to postmarketing monitoring is unclear. A recent report by FDA’s Science Board Subcommittee noted that tensions between the two offices remain (FDA Science Board Subcommittee, 2011). These issues have relevance to this report to the extent that they affect the effect or implementation of its recommendations designed to improve the quality of FDA decision-making in the postmarketing context.
The last 5 years have seen a major transformation in drug law and regulation. Before 2007, drug regulation emphasized primarily premarketing oversight. Although FDA could work with drug manufacturers to have drug labels changed, warnings and contraindications added, promotional materials modified, and restrictions added to distributions (Carpenter, 2010d), FDA had little statutory authority to manage the risks posed by drugs that had been approved for use other than to require a manufacturer to withdraw a drug from the market (CRS, 2008). FDAAA gave FDA expanded authorities and additional regulatory tools, such as the ability to require changes in a product label or to require the conduct of clinical trials or other studies in the postmarketing setting, all of which enable FDA to protect the health of the public better.
The authority to require postmarketing studies presents a new set of ethical and scientific questions for FDA to consider. Many of the new questions that FDA faces are illustrated by the Thiazolidinedione Intervention with Vitamin D Evaluation (TIDE) trial, in which the drug sponsor of a diabetes medication, rosiglitazone (Avandia®), was required by FDA to conduct a postmarketing clinical trial. TIDE was to be a “long-term controlled trial to assess the cardiovascular outcomes of patients treated with rosiglitazone, pioglitazone, and placebo (in addition to a other anti-diabetic background treatment)” (Jenkins, 2010). Key details of the research on rosiglitazone, including the TIDE trial, and its regulatory history are summarized in Box 1-6. The committee has identified a number of ethical and scientific questions that arise from the rosiglitazone case, and that might have led FDA to request this study, including
• How should FDA respond when questions about the effectiveness of a drug that was approved on the basis of a surrogate endpoint arise in the postmarketing setting? Rosiglitazone was approved on the basis of two surrogate endpoints—
blood glucose and glycated hemoglobin concentrations in diabetic patients—not on the basis of evidence on the effect of the drug on clinical outcomes.
• How should FDA consider data from different types of studies when making its regulatory decisions? In the case of rosiglitazone, FDA had to evaluate results from numerous studies, including clinical trials that used a variety of regimens, comparators, outcomes and dosing, and meta-analyses of data from these trials, and observational studies using administrative and outcomes data.
• How should FDA’s response to concerns about the safety of an approved drug be affected by the existence of a similar drug that is approved for the same indication? In the setting of new safety concerns about rosiglitazone, FDA had to take into account that physicians and patients could choose to use pioglitazone, which is in the same drug class as rosiglitazone and approved for the same indication.
• In the face of increasing concerns about potentially life-threatening risks, when is it ethically justifiable for FDA to require postmarketing clinical research? Critics argued that enough was known about the risks of rosiglitazone to render TIDE trial unethical and unnecessary.
• What role should FDA play in overseeing the postmarketing studies that it requires? In the case of the TIDE trial, as has been customary, there was no expectation of an interaction between FDA and the trial’s institutional review boards and data and safety monitoring board. The question going forward is whether there should be such interaction when FDA requires postmarketing research.
• Are there relevant ethical differences between postmarketing trials that FDA requires sponsors to conduct and trials that sponsors conduct in anticipation of seeking FDA approval? That so many physicians appeared to be taking their patients off rosiglitazone while other physicians and patients appeared to want to continue with the drug was a consideration that has no analogue in the premarketing context.
• How should FDA make regulatory decisions when scientists disagree about the interpretation of the evidence? There was disagreement among scientists both inside and outside FDA about how to interpret and respond to the evidence on rosiglitazone, including disagreements both between and within FDA’s OND and OSE.
It is precisely those types of questions that are the subject of the present report, which was requested by FDA to help it to answer ethical and scientific challenges that arise from its new postmarketing authorities and responsibilities under FDAAA.
In April 2010, FDA asked IOM to “convene a committee to evaluate the scientific and ethical issues involved in conducting studies of the safety of approved
BOX 1-6
Regulatory and Scientific History of Rosiglitazone
The Food and Drug Administration (FDA) approved rosiglitazone in 1999 on the basis of findings that indicated that it lowered blood glucose and glycated hemoglobin concentrations in diabetic patients. Concerns about adverse effects of rosiglitazone on lipids in the premarketing studies prompted FDA to ask the manufacturer to conduct a clinical trial to compare rosiglitazone with other diabetes drugs in the same class (A Diabetes Outcome Progression Trial) (FDA, 2007a). Major questions about the safety of rosiglitazone arose after a meta-analysis of the results of clinical trials showed an increased risk of cardiovascular events in people who took rosiglitazone (odds ratio [OR] for myocardial infarction, 1.43; 95% confidence interval [CI], 1.03–1.98; P = 0.03; OR for death of cardiovascular causes, 1.64; 95% CI, 0.98–2.74; P = 0.06) (Nissen and Wolski, 2007).
In July 2007, after publication of those results, an FDA advisory committee concluded that “the use of rosiglitazone for the treatment of type 2 diabetes was associated with a greater risk of myocardial is chemic events than placebo” and similar diabetes drugs (Rosen, 2007). Because of some limitations in the meta-analyses, however, the advisory committee voted that the drug should not be withdrawn from the market but “rather that label warnings and extensive educational efforts be instituted immediately”, and it requested further studies (Rosen, 2007). In November 2007, FDA announced its decision that there was not “enough evidence to indicate that the risk of a heart attack or cardiac ischemia is higher for Avandia than other [t]ypes of diabetes treatment” and that it would allow rosiglitazone to remain on the market (FDA, 2007b,c). FDA, however, required the manufacturer to revise rosiglitazone’s package insert to include a “boxed” warning regarding cardiovascular risk, to make a medication guide available to inform patients of the risk, and to conduct a long-term randomized controlled head-to-head clincal trial to evaluate the potential cardiovascular risk associated with rosiglitazone compared with an active control agent (that is, compared with another diabetes drug). Evidence from observational studies continued to appear in the literature as well.
By July 2008, the manufacturer had submitted the protocol for the Thiazolidinedione Intervention with Vitamin D Evaluation (TIDE)a trial to meet the third requirement, and the protocol had been approved by FDA.
Patient enrollment began in May 2009. Researchers not involved in the TIDE study, public health advocates, members of Congress, and some FDA staff, however, declared that there was already sufficient evidence that rosiglitazone was associated with an increased risk of cardiovascular events and questioned the ethics of requiring such a study (Bloomgarden, 2007).
A Senate Committee on Finance report in February 2010b reiterated many of those concerns about FDA’s position on rosiglitazone. The Institute of Medicine committee’s letter report was released on July 9, 2010. FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee held a joint meeting on July 13–14, 2010, at which they voted on several possible approaches to responding to concerns about rosiglitazone. Although the advisory-committee members voted overwhelmingly to take some actions on rosiglitazone—such as to increase warnings, to restrict access, or to remove access—no consensus emerged as to which of those actions FDA should take. On July 21, 2010, FDA put the TIDE trial on partial clinical hold, thus barring the enrollment of new patients. On September 23, 2010, FDA discontinued the TIDE trial and placed severe restrictions on the continued availability of rosiglitazone (FDA, 2011g).c
![]()
aRosiglitazone belongs to a class of drugs called thiazolidinediones that are used to treat diabetes mellitus type 2. The drug sponsor designed the Thiazolidinedione Intervention with Vitamin D Evaluation (TIDE) trial to evaluate “the cardiovascular effects of long-term treatment with rosiglitazone or pioglitazone when used as part of standard of care compared to similar standard of care without rosiglitazone or pioglitazone in patients with type 2 diabetes who have a history of or are at risk for cardiovascular disease” and “the effects of long-term supplementation of vitamin D on death and cancer” (NIH, 2011).
bThe letter, dated February 18, 2010, from Senators Baucus and Grassley—the chair and ranking member, respectively, of the Senate Committee on Finance—is available at http://finance.senate.gov/newsroom/chairman/release/?id=bcf5aef6-9bc5-45ca-9cab-aadf5df135fa.
cFDA required the drug sponsor to issue a risk evaluation and mitigation strategy according to which “the drug will be available to patients not already taking it only if they are unable to achieve glycemic control using other medications and, in consultation with their health care professional, decide not to take pioglitazone [a diabetes medication in the same class of drugs] for medical reasons. Current users of rosiglitazone will be able to continue using the medication if they appear to be benefiting from it and they acknowledge that they understand [the risks associated with its use]. Doctors will have to attest to and document their patients’ eligibility; patients will have to review statements describing the cardiovascular safety concerns” (Woodcock et al., 2011).
drugs”. The five specific questions posed by FDA appear in Box 1-1. In response to FDA’s request, IOM convened a committee of 12 members who have expertise in bioethics, biostatistics, clinical trials, epidemiology, health policy, law, patient safety, pharmacoepidemiology, and regulatory science.
FDA requested two reports: a letter report due in July 201045 and the present report. In its letter report, Ethical Issues in Studying the Safety of Approved Drugs: A Letter Report (see Appendix A), released on July 9, 2010, the committee addressed the first question of the committee’s charge—related to the ethical and informed consent issues that must be considered in designing RCTs—by presenting a conceptual framework for analyzing the ethics of postmarketing RCTs required by FDA (Box 1-7) (IOM, 2010b). In this final report the committee addresses all five specific questions posed to the committee by FDA.
THE COMMITTEE’S APPROACH TO ITS CHARGE
The committee met in person six times, including two open information-gathering sessions at which representatives of FDA, AHRQ, NIH, other stakeholders, and researchers appeared (see Appendix B) to address the committee’s broader charge.
The committee used the conceptual framework in its letter report (Box 1-7) (IOM, 2010b) as a starting point for this final report but conducted further research and deliberations related to its full charge. Several underpinnings of the conceptual framework and additional themes that emerged as the committee deliberated on its full charge shaped this report. These include an understanding of FDA’s public health mission; the importance of adopting a lifecycle approach to drug safety and benefit–risk assessment (see Figure 1-1 for a schematic of a drug’s lifecycle); FDA’s ethical obligations in making regulatory decisions, including the centrality of transparency and communication of the decisions; and a commitment to using best practices in regulatory science46 and high-quality evidence in regulatory decision-making.
Considerations of the Agency’s Mission
FDA is a public health agency; its mission is to protect “public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products” (FDA, 2011h). In making decisions about potential regulatory actions, therefore, FDA should consider their potential public
![]()
45FDA requested that the letter report be completed before a July 13–14, 2010, joint meeting of FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee on rosiglitazone.
46FDA has defined regulatory science as “the development and use of new tools, standards and approaches to more efficiently develop products and to more effectively evaluate product safety, efficacy and quality” (FDA, 2010b).
BOX 1-7
Conceptual Framework from Ethical Issues in Studying the Safety of Approved Drugs: A Letter Report (IOM, 2010a) for Analyzing the Ethics of Postmarketing Randomized Clinical Trials Required by the Food and Drug Administration:
Four Central Classes of Considerations and Recommendations
I. The Public Health Context. The Food and Drug Administration (FDA) should determine that there is a substantial public health question about the nature or acceptability of the risks, or the risk–benefit profile, of a marketed drug—a question that requires a policy decision from FDA.
II. Regulatory Science and Public Accountability. FDA should use regulatory-science principles and practices that include processes of public accountability and transparency to determine the need for a policy decision, the need for new knowledge to support a policy decision, and the policy decision based on the new knowledge.
III. Design Considerations. It is appropriate for FDA to require that a randomized controlled trial be conducted to provide additional evidence about an approved drug’s efficacy and safety only when (i) uncertainty about the risk-benefit balance is such that a responsible policy decision cannot be made based either on the existing evidence or on evidence from new observational studies, and (ii) the trial is properly designed and implemented to reduce uncertainty about the benefit–risk balance sufficiently for a responsible policy decision to be made.
IV. Additional Ethical Obligations to Trial Participants. FDA should ensure that the trial will answer the public health question with a design that minimizes risks to trial participants and involves ongoing monitoring of risks. The risks should be judged to be acceptable by appropriate oversight bodies before and during the trial and by trial participants at enrollment and as appropriate during the trial. Specifically, FDA and appropriate oversight bodies should ensure that the trial includes a comprehensive and meaningful informed consent process that continues during the trial and that takes into account any substantial changes in clinical practice and professional standards and any new research findings relevant to a participant’s willingness to accept the risks associated with the trial. The FDA and appropriate oversight bodies should ensure that those conducting the trial convey such changes to participants in a timely and understandable fashion.
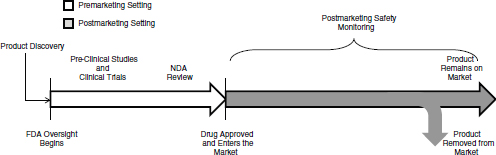
FIGURE 1-1 The lifecycle of a drug for a new molecular entity. After a product is discovered and the sponsor approaches FDA with the product as an investigational new drug, FDA oversight begins. After approved, FDA and the drug sponsor conduct postmarketing safety monitoring of the drug, which could include passive surveillance, active surveillance, observational studies, and randomized controlled studies. A product remains on the market until it is removed from the market either at FDA’s request, FDA’s withdrawal of marketing approval, or the company decides to no longer market it. FDA oversight of the drug continues for as long as the drug is on the market.
health consequences.47 The committee emphasizes FDA’s public health role, and the consequences of not protecting the public’s health, throughout this report (Hamburg and Sharfstein, 2009).
FDA has a complex mission with obligations that sometimes conflict with one another. The committee recognizes that the effort to resolve those conflicts can complicate the agency’s decisions but could only provide general guidance on how FDA may do it. In the premarketing context, FDA has to balance the effort to approve new drugs that could be beneficial to people with the effort to characterize potential harms, as well as benefits, and to make measured judgments about a new drug’s benefit–risk profile with incomplete information. In the postmarketing context, new information requires that FDA continually re-examine its judgment about the benefit–risk profile and make regulatory decisions about the drug while being responsive to advocacy groups that are working from both sides of the issue. From an ethical standpoint, throughout the lifecycle of a drug, FDA has to balance its obligation to protect the public’s health by having strong science on which to base regulatory decisions with its obligation to protect participants in research that it requires.
Such factors as the severity and prevalence of a disease or an adverse event, and the availability of effective alternative treatments necessarily affect FDA’s regulatory decisions, including decisions about what postmarketing studies to require and how to use the information from different studies. Such decisions
![]()
47The committee views FDA’s decisions as having public health consequences regardless of whether they affect a large population or a small group.
need to be made case by case, so the committee cannot provide a prescriptive formula for making them. Instead, the committee discusses principles that should be taken into account in decision-making, provides guidance on how to account for the factors, and offers a general framework for decision-making.
The FDA mission is to allow a drug to enter and remain on the market if, on the basis of its considered judgment, the benefits of the drug outweigh the risks that it poses; it is not charged with establishing, either for initial approval or for continued presence on the market, that a drug has the most favorable benefit–risk profile compared with other drugs for the same indication. FDA is one of many entities that influence the availability and safe use of drugs in the United States. Physicians and other health care providers, professional societies, pharmacies, and hospitals play crucial roles in ensuring that the expected benefits of a prescribed drug outweigh the risks in an individual patient. In addition, payers, such as medical insurance companies, take cost and the availability of alternative treatments into account and decide which drugs they will cover and at what level of reimbursement.
Government agencies are involved in sponsoring drug research, including an increasing focus on translational research in NIH and comparative-effectiveness research in AHRQ, CMS, and the Patient-Centered Outcomes Research Institute. Research sponsored or conducted by those agencies may inform FDA’s evaluations of the benefits and risks associated with drugs. Moreover, the studies required by FDA may inform health care providers, payers, and other agencies and organizations in their drug-related decisions. The intersections of those roles require increased interactions among agencies; a number of initiatives indicate that the agencies recognize the need to coordinate their activities better. For example, FDA and NIH have established a collaborative initiative to move innovations to the public quickly (FDA, 2010b). The fact that the present report was financially supported by NIH and AHRQ, as well as FDA, illustrates the cooperation among the agencies. However, the committee’s charge is related to FDA’s role in regulating drugs, so it does not discuss the roles of other agencies and entities related to drugs.
A Lifecycle Approach to Safety and the Assessment
of a Drug’s Benefits and Risks
The committee starts with the assumption that ensuring the acceptability of the benefit–risk profile of a drug after it is approved for the US market is as important to FDA’s public health mission as ensuring the acceptability of the benefit–risk profile before it is permitted to enter the market. A lifecycle approach to the regulatory oversight of drugs is therefore critical. The new authorities in FDAAA provide FDA with the tools to adopt a more comprehensive lifecycle approach than prior to FDAAA.
FDA assesses drug safety in relation to the drug benefits. For example, cancer chemotherapy drugs can cause serious adverse effects, including death, but
are deemed adequately safe for their intended use because of the greater benefit of reducing cancer mortality risk. This report will use the word “safe” in this same sense, that is, that the magnitude and distribution of benefits and risks is acceptable for the intended use. The word “safety endpoint” will be used to describe the harms associated with a drug. “Risk” is the probability that those harms will be incurred. The committee will use an “efficacy” or “effectiveness” endpoint as the clinical outcome that a drug is intended to improve.
Major regulatory changes in a drug once it is on the market—such as changes in a drug’s label, the addition of a boxed warning, or withdrawal of the drug—may appear to represent failures of the drug regulatory system (IOM, 2007a). It is important to recognize that the discovery of new information about a drug’s adverse events or clinical effectiveness, if ascertained in a timely manner, is a normal and desirable part of the natural history or lifecycle of the drug. It is impossible to know everything about a drug at the point of approval. A responsible public health agency is structured to learn continually about the drugs that it approves with the expectation that what is known about a drug’s benefits and risks will change over time. The timeliness of the identification of and response to new serious adverse events is an indication of a high-quality postmarketing system. The focus of this report thus responds to an expanded understanding of FDA as a public health agency whose approach to its mission is and should be shifting from a premarketing focus on efficacy and short-term harms, and a postmarketing focus on harms, to a continuous and integrated assessment of the benefit–risk profile of a drug throughout its market life.
Ethics and Decision-Making in the Food and Drug Administration
The committee’s letter report focused heavily on FDA’s ethical obligations to research participants. The present report continues that focus but broadens the discussion to include the ethical aspects of study design, how ethical considerations should be taken into account and integrated in FDA’s decision-making framework, and how FDA can incorporate two key components of the ethics of public processes—stakeholder engagement and transparency—into its decision-making practices. The committee views ethical issues as inextricably intertwined with scientific and regulatory issues. Ethical issues are therefore discussed throughout this report.
Public engagement and transparency increase the likelihood that the perspectives of patients and consumers, who have knowledge different from those of technical experts, are included in the making of policy decisions. When including such perspectives, however, FDA should ensure that patient advocacy groups represent the views of patients rather than the views of commercial entities that provide funding to the organizations. The concerns and practical considerations voiced by other stakeholders, such as health care providers, payers, industry, and academe, are also important to include. Transparency and other public account-
ability processes may also increase the likelihood that the public will view regulatory and policy decisions, including a decision by FDA to require a sponsor to conduct postmarketing research or a decision to continue or discontinue a required postmarketing clinical trial, as fair and acceptable.48
Modern tools for risk communication and public engagement ensure that all stakeholders—including physicians, other health professionals, interested patients and their families, and members of the general public—understand the decision the agency is facing, including what is known about the benefits and risks associated with the therapy in question and the pertinent uncertainties. Uncertainties might pertain to the quantity and quality of evidence, the benefit–risk profile, or the effect of policy decisions on the health of the public. Engagement with stakeholders helps to explain the types of uncertainties at issue and how the agency is dealing with uncertainties in making its policy decisions and helps the agency to understand how those affected by its actions weigh benefits and risks.49 Communication to stakeholders will be more important, and in some ways more complex, as FDA moves more toward a lifecycle approach to drug regulation. Education and outreach will help to ensure that the public understands the change in the regulation of a drug as part of the normal natural history of its lifecycle.
Regulatory Science and High-Quality Evidence
The committee was guided by the view that regulatory decision-making, including decisions that require the integration of postmarketing safety information, should be based on the best principles and practices for making policy decisions under conditions of uncertainty, such as appropriate processes for transparency in decision-making and public accountability. Those principles and practices, sometimes referred to as the emerging field of regulatory science, require that policy decisions reflect the best available scientific evidence and analytic techniques drawn from a wide array of disciplines and technical expertise, including decision science, behavioral economics, and cognitive psychology.
Accurately assessing the potential benefits of and risks posed by a drug requires the use of a wide variety of scientific data, including findings from clinical trials; epidemiologic and outcomes research, such as observational studies and meta-analyses; and postmarketing surveillance systems that detect and help to characterize adverse events. All sources of data—not only or primarily those
![]()
48FDA and those advising it should have access to all information relevant to a given public health question, whether or not the information is deemed proprietary or to constitute a trade secret. One source of tension in meeting acceptable standards of transparency with stakeholders is the management of public access to such information.
49The committee acknowledges that there are important challenges to implementing policy making and regulatory processes that balance scientific evidence and stakeholder input appropriately (Lomas, 2005).
obtained from clinical trials—have the potential to contribute to sound regulatory decision-making. The critical factors in determining how much weight to give to various data resources are the quality of the studies that generated them and their relevance to the public health questions at issue, not simply whether the studies were experimental or observational. The committee further recognizes the importance of toxicology studies—including molecular toxicology and animal studies—in both the premarketing and postmarketing setting, especially as genomic sciences progress. The committee, however, considered a review of pharmacologic, metabolic, or toxicologic studies as beyond the scope of its charge.
The transformation in FDA’s authorities and responsibilities over the last five years provides FDA with valuable tools to help ensure that the benefits of a drug outweigh its risks throughout its lifecycle. Despite the challenges that this new era in postmarketing oversight brings, FDA should embrace the opportunities presented by FDAAA to protect the public’s health. FDA has made policy and organizational changes, and has implemented new initiatives that are aimed at improving its oversight in the postmarketing setting. There are also indications in FDA’s strategic plan and in negotiations for PDUFA V that FDA is moving further in the direction of strengthening its assessment of benefits and risks throughout a drug’s lifecycle. The effects of these policies and initiatives, and even the full effect of the sweeping changes precipitated by FDAAA, will take several years to be completely realized. Those changes are promising, and they posed a challenge for the committee, which was dealing with a rapidly evolving FDA role in the postmarketing setting as this report was being written. The committee tried to make general, flexible recommendations that would be relevant in this changing landscape, and that could affect the course of these changes. The committee sees the present report and its recommendations as providing guidance to FDA as part of its evolving approach to drug oversight in which drug safety monitoring and regulatory action after drug approval is seen as increasingly important for protecting the health of the public.
Chapter 2 presents a broad framework for FDA’s regulatory decision-making. The framework addresses the need for a clear explanation of the agency’s decisions and organizational considerations that facilitate decision-making, and the committee recommends a process and formal documentation intended to help FDA assess the benefits and risks associated with a drug throughout its lifecycle. A major challenge for FDA is making decisions in the face of scientific disagreement about available evidence, and the implications of that evidence. Chapter 3 looks more closely at that particular challenge and discusses the nature of evidence and why scientists sometimes disagree about how to interpret and respond to evidence in a regulatory decision. Chapter 4 focuses on one of FDA’s regulatory actions that were highlighted in the committee’s charge (Box 1-1): the
ability to require different types of postmarketing studies. That chapter discusses and makes recommendations about the circumstances under which different types of studies are ethically and scientifically justified. Chapter 5 summarizes the committee’s findings by answering the specific questions in the committee’s charge, and it reiterates the committee’s recommendations.
A summary of key aspects of FDAAA, the agendas of the committee’s public meetings, the committee’s letter report, information on decision conferencing and multicriteria decision analysis, and biographies of the committee members are presented in Appendixes A, B, C, D, and E, respectively.
Anderson, L. F. 1989. Cancer and aids groups push for changes in drug approval process. Journal of the National Cancer Institute 81(11):829-831.
Atkinson, A. J., W. A. Colburn, V. G. DeGruttola, D. L. DeMets, G. J. Downing, D. F. Hoth, J. A. Oates, C. C. Peck, R. T. Schooley, B. A. Spilker, J. Woodcock, and S. L. Zeger. 2001. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clinical Pharmacology & Therapeutics 69(3):89-95.
Avorn, J. 2005. Navigating the third dimension. In Powerful medicines: The benefits, risks, and costs of prescription drugs. New York: Vintage Books.
Bloomgarden, Z. T. 2007. The Avandia debate. Diabetes Care 30(9):2401-2408.
Budnitz, D. S., D. A. Pollock, K. N. Weidenbach, A. B. Mendelsohn, T. J. Schroeder, and J. L. Annest. 2006. National surveillance of emergency department visits for outpatient adverse drug events. JAMA 296(15):1858-1866.
Budnitz, D. S., M. C. Lovegrove, N. Shehab, and C. L. Richards. 2011. Emergency hospitalizations for adverse drug events in older Americans. New England Journal of Medicine 365(21):2002-2012.
Carpenter, D. 2010a. Reputation and gatekeeping authority: The Federal Food, Drug and Cosmetic Act of 1938 and its aftermath. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Princeton, NJ: Princeton University Press. Pp. 73-117.
Carpenter, D. 2010b. Reputation and power crystallized: Thalidomide, Frances Kelsey, and phased experiment, 1961-1966. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Princeton, NJ: Princeton University Press. Pp. 228-297.
Carpenter, D. 2010c. Reputation and power contested: Emboldened audiences in cancer and AIDS, 1977-1992. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Princeton, NJ: Princeton University Press. Pp. 393-464.
Carpenter, D. 2010d. The other side of the gate: Reputation, power, and post-market regulation. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Cambridge, NY: Princeton University Press. Pp. 585-634.
Center for Healthcare Supply Chain Research. 2012. The center publishes new report on Risk Evaluation and Mitigation Strategies (REMS). http://www.hcsupplychainresearch.org/projects/rems.asp (accessed April 2, 2012).
Cook, A. J., R. C. Tiwari, R. D. Wellman, S. R. Heckbert, L. Li, P. Heagerty, T. Marsh, and J. C. Nelson. 2012. Statistical approaches to group sequential monitoring of postmarket safety surveillance data: Current state of the art for use in the Mini-Sentinel pilot. Pharmacoepidemiology Drug Safety 21(Suppl 1):72-81.
CRS (Congressional Research Service). 2008. Report to Congress: FDA Amendments Act of 2007 (P.L. 110-85). Washington, DC: Congressional Research Service.
Daemmrich, A. A. 2004a. Drug laws and therapeutic cultures. In Pharmacopolitics: Drug regulation in the United States and Germany. Chapel Hill, NC: The University of North Carolina Press.
Daemmrich, A. A. 2004b. Clincal trials as test and therapy, 1980-2000. In Pharmacopolitics: Drug regulation in the United States and Germany. Chapel Hill, NC: The University of North Carolina Press. Pp. 81-115.
Evans, B. 2010. Seven pillars of a new evidentiary paradigm: The Food, Drug, and Cosmetic Act enters the genomic era. Notre Dame Law Review 85(2):419-524. Pp. 19-47.
FDA (US Food and Drug Administration). 1998. The CDER handbook. Rockville, MD: Department of Health and Human Services.
FDA. 1999. Managing the risks from medical product use: Creating a risk management framework. Report to the FDA commissioner from the Task Force on Risk Management. Washington, DC: Department of Health and Human Services.
FDA. 2003. Guidance for industry INDs for Phase 2 and Phase 3 studies. Chemistry, manufacturing, and controls information. Washington, DC: Department of Health and Human Services.
FDA. 2005. Guidance for industry: Development and use of Risk Minimization Action Plans. Washington, DC: US Food and Drug Administration.
FDA. 2007a. Advisory committee briefing document: Cardiovascular safety of rosiglitazone. Washington, DC: US Food and Drug Administration.
FDA. 2007b. FDA news release: FDA adds boxed warning for heart-related risks to anti-diabetes drug Avandia. Agency says drug to remain on market, while safety assessment continues. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm109026.htm (accessed August 23, 2011).
FDA. 2007c. Transcript of FDA press conference on the update to the existing box warning on Avandia. November 14, 2007. http://www.fda.gov/NewsEvents/Newsroom/MediaTranscripts/ucm122244.htm (accessed August 23, 2011).
FDA. 2008. The Sentinel Initiative. Washington, DC: Department of Health and Human Services.
FDA. 2009a. The history of drug regulation in the United States. http://www.fda.gov/AboutFDA/WhatWeDo/History/FOrgsHistory/CDER/CenterforDrugEvaluationandResearchBrochureand Chronology/ucm114470.htm (accessed September 9, 2011).
FDA. 2009b. Guidance for industry format and content of proposed Risk Evaluation and Mitigation Strategies (REMS), REMS assessments, and proposed REMS modifications. Washington, DC: Department of Health and Human Services.
FDA. 2009c. Office of Surveillance and Epidemiology (OSE). http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm106491.htm (accessed September 14, 2011).
FDA. 2009d. Memorandum of agreement between the Office of New Drugs and the Office of Surveillance and Epidemiology in the Center for Drug Evaluation and Research. Washington, DC: US Food and Drug Administration.
FDA. 2010a. Initiatives (CDER). http://www.fda.gov/AboutFDA/CentersOffices/CDER/WhatWeDo/Initiatives/default.htm#equalvoice (accessed April 15, 2011).
FDA. 2010b. NIH and FDA announce collaborative initiative to fast-track innovations to the public. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm201706.htm (accessed August 23, 2011).
FDA. 2011a. Strategic Priorities, 2011-2015. Washington, DC: Food and Drug Administration.
FDA. 2011b. FY 2010 performance report to the President and Congress for the Prescription Drug User Fee Act. Washington, DC: Food and Drug Administration.
FDA. 2011c. Guidance for industry postmarketing studies and clinical trials—implementation of section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act. Washington, DC: Department of Health and Human Services.
FDA. 2011d. Postmarket requirements and commitments searchable database: Record for vectibix, NDA # 125147. http://www.accessdata.fda.gov/scripts/cder/pmc/index.cfm?StartRow=6&StepSize=1&Paging=Yes (accessed April 24, 2011).
FDA. 2011e. Guidance: Medication guides—distribution requirements and inclusion in Risk Evaluation and Mitigation Strategies (REMS). Washington, DC: US Food and Drug Administration.
FDA. 2011f. Office of New Drugs. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm184426.htm (accessed April 10, 2011).
FDA. 2011g. FDA significantly restricts access to the diabetes drug Avandia. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm226956.htm (accessed January 14, 2011).
FDA. 2011h. FDA fundamentals. Do you know what FDA does? http://www.fda.gov/aboutfda/transparency/basics/ucm192695.htm (accessed April 24, 2011).
FDA. 2012. Report on the performance of drug and biologics firms in conducting postmarketing requirements and commitments; availability. Federal Register 77(44):13339-13343.
FDA Science Board Subcommittee. 2011. Review of the FDA/CDER pharmacovigilance program. Washington, DC: US Food and Drug Administration.
GAO (Government Accountability Office). 2008. FDA has begun to enhance its oversight of drugs approved on the basis of surrogate endpoints. Washington, DC: Government Accountability Office. GAO. 2009. Drug safety: FDA has begun efforts to enhance postmarket safety, but additional actions are needed. Washington, DC: Government Accountability Office.
Grossman, L. A., R. A. Merrill, and P. B. Hutt. 2007. FDA jurisdiction: A matter of definition. Washington College of Law Research Paper No. 2008-38. American University Washington College of Law.
Gu, Q., C. D. Dillon, and B. V. L. 2010. Prescription drug use continues to increase: U.S. prescription drug data for 2007-2008. Atlanta, GA: Centers for Disease Control and Prevention.
Hamburg, M. A., and J. M. Sharfstein. 2009. The FDA as a public health agency. New England Journal of Medicine 360(24):2493-2495.
HHS (US Department of Health and Human Services). 2010. HIPAA Mini-Sentinel and Sentinel. Washington, DC: Department of Health and Human Services.
Hilts, P. J. 2003a. Thalidomide. In Protecting America’s health. New York: Alfred A. Knoff. Pp. 144-165.
Hilts, P. J. 2003b. The modern plague. In Protecting America’s health. New York: Alfred A. Knoff. Pp. 236-254.
IOM (Institute of Medicine). 2002. Responsible research: A systems approach to protecting research participants. Washington, DC: The National Academies Press.
IOM. 2004. Patient safety: Achieving a new standard for care. Washington, DC: The National Academies Press.
IOM. 2007a. The future of drug safety: Promoting and protecting the health of the public. Washington, DC: The National Academies Press.
IOM. 2007b. Understanding the benefits and risks of pharmaceuticals: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2010a. Evaluation of biomarkers and surrogate endpoints in chronic disease. Washington, DC: The National Academies Press.
IOM. 2010b. Ethical issues in studying the safety of approved drugs: A letter report. Washington, DC: The National Academies Press.
Jenkins, J. K. 2010. Memorandum from John Jenkins to Janet Woodcock (dated September, 2010). Re: Recommendations for regulatory actions—rosiglitazone. Washington, DC: Food and Drug Administration.
Lomas, J. 2005. Using research to inform healthcare managers’ and policy makers’ questions: From summative to interpretive synthesis. Health Policy 1(1):55-71.
Marks, H. M. 1997a. Playing it safe: The Federal Food, Drug and Cosmetic Act of 1938. In The progress of experiment: Science and therapeutic reform in the United States, 1900-1990. Cambridge, NY: Cambridge University Press. Pp. 136-163.
Marks, H. M. 1997b. Managing chance: Statistics and therapeutic experiments, 1950-1960. The progress of experiment: Science and therapeutic reform in the United States, 1900-1990. Cambridge, NY: Cambridge University Press. Pp. 71-97.
NIH (National Institutes of Health). 2011. Thiazolidinedione Intervention with Vitamin D Evaluation (TIDE). http://clinicaltrials.gov/ct2/show/NCT00879970 (accessed April 25, 2011).
Nissen, S. E., and K. Wolski. 2007. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356(24):2457-2471.
Office of Surveillance and Epidemiology. 2007. An overview of RiskMAPs. Washington, DC: US Food and Drug Administration.
Platt, R., M. Wilson, K. A. Chan, J. S. Benner, J. Marchibroda, and M. McClellan. 2009. The new Sentinel network—improving the evidence of medical-product safety. New England Journal of Medicine 361(7):645-647.
Platt, R., R. M. Carnahan, J. S. Brown, E. Chrischilles, L. H. Curtis, S. Hennessy, J. C. Nelson, J. A. Racoosin, M. Robb, S. Schneeweiss, S. Toh, and M. G. Weiner. 2012. The U.S. Food and Drug Administration’s Mini-Sentinel program: Status and direction. Pharmacoepidemiology and Drug Safety 21(Suppl 1):1-8.
Robb, M. A., J. A. Racoosin, R. E. Sherman, T. P. Gross, R. Ball, M. E. Reichman, K. Midthun, and J. Woodcock. 2012. The U.S. Food and Drug Administration’s Sentinel initiative: Expanding the horizons of medical product safety. Pharmacoepidemiology and Drug Safety 21(Suppl 1):9-11. Rosen, C. J. 2007. The rosiglitazone story—lessons from an FDA advisory committee meeting. New England Journal of Medincine 357(9):844-846.
Shane, R. 2009. Risk Evaluation and Mitigation Strategies: Impact on patients, health care providers, and health systems. American Journal of Health-System Pharmacy 66(24 Suppl. 7):S6-S12.
Stang, P. E., P. B. Ryan, J. A. Racoosin, J. M. Overhage, A. G. Hartzema, C. Reich, E. Welebob, T. Scarnecchia, and J. Woodcock. 2010. Advancing the science for active surveillance: Rationale and design for the Observational Medical Outcomes Partnership. Annals of Internal Medicine 153(9):600-606.
Strom, B. L. 2006. How the U.S. drug safety system should be changed. JAMA 295(17):2072-2075. Wood, A. J. 2006. A proposal for radical changes in the drug-approval process. New England Journal of Medicine 355(6):618-623.
Wood, A. J. J., C. M. Stein, and R. Woosley. 1998. Making medicines safer—the need for an independent drug safety board. New England Journal of Medicine 339(25):1851-1854.
Woodcock, J., R. E. Behrman, and G. J. Dal Pan. 2011. Role of postmarketing surveillance in contemporary medicine. Annual Review of Medicine 62(Feb):1-10.
































