
As part of its charge the committee was asked how the US Food and Drug Administration (FDA) should “factor in different kinds of safety evidence in considering different kinds of regulatory action”. To respond to that question, the committee considered the relevance of different evidence to decisions about different potential regulatory actions, and how FDA should apply that evidence across the lifecycle of a drug. The committee concluded that there is no one answer to that question, because the evidence and circumstances surrounding each regulatory decision are different. Although decisions as to how to weigh evidence will always have to be made case by case, the committee provides a broad overall approach to guide the assessment of safety evidence and FDA decision-making. In this chapter, the committee highlights the importance of a lifecycle approach to FDA’s regulatory decisions and proposes two mechanisms to facilitate adopting such an approach: a framework for decision-making and a document, referred to as a Benefit and Risk Assessment and Management Plan (BRAMP), to formalize the implementation of the lifecycle approach discussed in Chapter 1, and make FDA’s decisions about each drug transparent. The framework is not intended as a one-time activity, but rather an activity that recurs when questions about the benefits or risks associated with a drug arise. The document is a record that tracks the experience of a drug across its lifecycle, to be updated whenever the framework is used to evaluate the benefits and risks associated with a drug.
EVALUATING BENEFIT AND RISK OVER A DRUG’S LIFECYCLE
FDA’s decision to approve a drug for sale in the United States is based on a judgment that in view of the evidence from premarketing studies and clinical
needs, it is, all things considered, in the interest of the public’s health for the drug to enter the marketplace. In other words, the benefits of the drug outweigh its risks for the intended use and population. Although at the time of approval knowledge about efficacy from small, short-term clinical-trial populations is limited, far less is known about the drug’s risks. Some adverse effects may be too rare to be identified in the small numbers of people who participate in premarketing studies. For example, although the premarketing clinical trials for a second-generation rotavirus vaccine involved relatively large numbers of research participants, the small, increased risk of intussusceptions with rotavirus vaccines was only identified in post-licensure safety monitoring (approximately 1 of every 51,000 to 68,000 vaccinated infants) (Greenberg, 2011; Patel et al., 2011). Other adverse events may have a latent period longer than the duration of premarketing trials or may occur in people who are unlike those who participated in the premarketing trials in relevant respects. For example, they may be less healthy, take other medications, or have comorbidities. Such patients are often excluded from or enrolled in small numbers in premarketing trials (Fung, 2001).
For several reasons, questions about the effectiveness of a drug in actual clinical practice may also remain at the time of approval (Borer et al., 2007; Hiatt, 2006; IOM, 2007a; Ray and Stein, 2006). Long drug exposure during the postmarketing period could lead to a loss of effectiveness as tolerance or resistance to the drug develops. The population taking an approved drug is likely to be more heterogeneous than the people who participated in premarketing clinical trials. The drug may not be as effective in the postmarketing general population as it was in the premarketing test population. Many factors can account for those differences, including differences in environmental factors, genetics, age, race, ethnicity, or sex; interactions with other drugs; comorbidities; and problems with drug adherence. For example, a person who has liver disease might not fully metabolize and activate a drug, leading to decreased clinical effectiveness. A drug approved on the basis of a surrogate endpoint might not be as effective in improving a clinical endpoint, for example tumor shrinkage may not correlate strongly with survival. Once a drug is allowed to enter the market, physicians are free to use it, on-label or off-label, for any indication, including those of which there may be little or no scientific evidence of effectiveness from premarketing trials.
In the remainder of this chapter, the committee outlines a three-stage framework for making regulatory decisions and how FDA could apply the framework as part of the lifecycle approach to drug safety discussed in Chapter 1. (See Box 2-1 for definitions of key terms used in this chapter.) The committee then proposes a BRAMP document as a mechanism for implementing a lifecycle approach to drug regulation and for making FDA’s decisions transparent. Figure 2-1 shows how FDA can incorporate the framework and the BRAMP into a lifecycle approach to drug oversight. The chapter concludes by addressing the circumstance under which regulatory decisions should include requiring manufacturers to conduct postmarketing studies, a focus of the committee’s charge (see
BOX 2-1
Key Definitions
Benefit assessment and risk assessment: The gathering and analyzing of information on the nature and magnitude of potential benefits and potential harms (risks) associated with a drug and the determination of the likelihood that those benefits and harms will occur.
Benefit–risk profile: An overall evaluation of the benefits and risks associated with a drug.
Benefit–risk management: The process of identifying, evaluating, selecting, and implementing actions to increase benefits and reduce risk to human health. The goal of benefit–risk management is scientifically sound, integrated actions that increase or maintain benefits and reduce or prevent risks while taking into account social, cultural, ethical, political, and legal considerations. “A good risk management decision emerges from a decision-making process that elicits the views of those affected by the decision, so that differing technical assessments, public values, knowledge, and perceptions are considered” (Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). The process of benefit–risk management should include not only information about current regulatory actions but plans for future evaluations and regulatory actions as part of the lifecycle process.
Uncertainty: Lack or incompleteness of information. Quantitative uncertainty analysis attempts to analyze and describe the degree to which a calculated value may differ from the true value; it sometimes uses probability distributions. Uncertainty depends on the quality, quantity, and relevance of data and on the reliability and relevance of models and assumptions.
Box 1-1; the question of which study designs FDA should require is addressed in Chapter 4).
THREE-STAGE FRAMEWORK FOR REGULATORY DECISION-MAKING
Overview and Rationale
Responding in a timely and appropriate way to safety signals from already-approved drugs is among the most important and challenging public health jobs that FDA must accomplish. Permitting a drug to stay on the market that is on
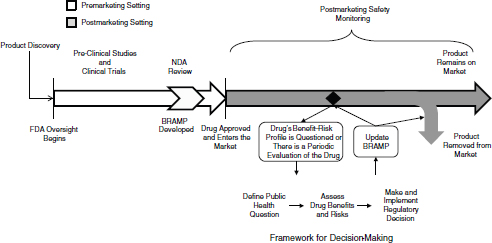
FIGURE 2-1 The lifecycle of a drug for a new molecular entity. After a product is discovered and the sponsor approaches FDA with the product as an investigational new drug, FDA oversight begins. During approval, the company submits information about the benefits, risks, and benefit–risk profile of the drug, and FDA develops a BRAMP. After approved, FDA and the drug sponsor conduct postmarketing safety monitoring of the drug, which could include passive surveillance, active surveillance, observational studies, and randomized controlled studies. If evidence arises that calls into question the benefit–risk profile of the drug, FDA uses the decision-making framework to review the new evidence in the context of existing evidence and the public health context of the drug, to make a regulatory decision about the drug. Depending on the decision, the drug will remain on the market, either with the same or different restrictions and conditions, or will be removed from the market. FDA updates the BRAMP document when it considers a regulatory decision for the drug, and when periodic evaluations occur over the drug’s lifecycle. FDA oversight of the drug continues for as long as the drug is on the market.
balance harmful threatens public well-being,1 but so too does limiting access to a drug whose benefits outweigh its harms. As discussed in Chapter 1, the Food and Drug Administration Amendments Act (FDAAA) of 20072 provides FDA with new tools and authorities to adopt a lifecycle approach to regulatory decision-making—an approach that FDA has endorsed (FDA, 2004). However, FDA has not yet taken full advantage of its new tools and authorities to implement a lifecycle approach in a systematic or comprehensive manner.
![]()
1There are instances where a drug is, on balance, harmful to the overall population but nevertheless provides a net benefit to a specific subgroup within the population. In those instances, the drug could remain on the market with restrictions to limit its use to those subgroups for whom the drug has a favorable benefit–risk profile.
2Food and Drug Administration Amendments Act of 2007, PL No. 110-85, 121 Stat. 823 (2007).
The assessment of benefits and risks and making management decisions in response to the assessment are not new challenges for FDA. The agency has a process in place for reviewing premarketing data on efficacy and risks, and making regulatory decisions about approving a drug on the basis of those data and other considerations. Similarly mature processes do not exist for evaluating a drug’s benefits and risks in the postmarketing setting using FDA’s authority in FDAAA, although in April, 2011 FDA issued guidance providing information for industry on how it would implement the section of FDAAA3 that authorizes FDA to require postmarketing research. The link between benefit assessment, risk assessment, and FDA’s regulatory decision-making has been criticized for failing to be explicit and transparent to external stakeholders (Asamoah and Sharfstein, 2010; Transparency Task Force et al., 2010).
Other US government agencies and organizations have a history of making decisions on the basis of formal assessments of risks. The US Environmental Protection Agency (EPA), for example, conducts formal chemical risk assessments to guide its decisions on allowable concentrations of chemicals in the environment (see for example EPA, 2005, 2009). The process used by EPA has evolved after publication of a number of reports outlining best practices for risk assessment and regulatory decision-making (NRC, 1983, 1989, 1996, 2009; Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). Characteristics of those best practices include the use of the best available scientific evidence, the involvement of parties that would be affected by the decision in the decision-making process, especially to incorporate the perspectives of patients and consumers in the process, and transparency in the process (NRC, 1989, 1996, 2009; Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). The 2009 National Research Council report Science and Decisions: Advancing Risk Assessment proposed that EPA use a formal three-phase framework when making its regulatory decisions (NRC, 2009, 2011). The framework includes a problem-formulation phase, a phase for the planning and conduct of the risk assessment, and a risk-management phase. A recent National Research Council report, A Risk-Characterization Framework for Decision-Making at the Food and Drug Administration, highlighted the 2009 framework and its general usefulness for FDA in its regulatory purview (NRC, 2011). It proposed a similar, three-step process for decision-making that involved identifying and defining the decision context, estimating or characterizing the public health consequences of each decision option, and using the completed characterization to compare decision options and to communicate their public health consequences within the agency, to decision-makers, and to the public. The report highlighted factors that are considered in FDA’s decision-making, including scientific, social, and political factors, as well as the importance of the context of the decision to all steps of the decision-making process.
![]()
321 USC § 355(o) (2010).
The need for a systematic process for drug-regulatory decisions has been discussed previously. The Pharmaceutical Research and Manufacturers of America’s Benefit–Risk Action Team (BRAT)4 discussed the need for a consistent framework for “transparent, rational and defensible decision-making that benefits patients, drug developers, and decision makers” (Coplan et al., 2011). The BRAT proposed a six-step framework for decision-making (Coplan et al., 2011). Health Canada and the European Medicines Agency have also discussed the need for a benefit–risk framework (CHMP, 2008; Health Canada, 2000).
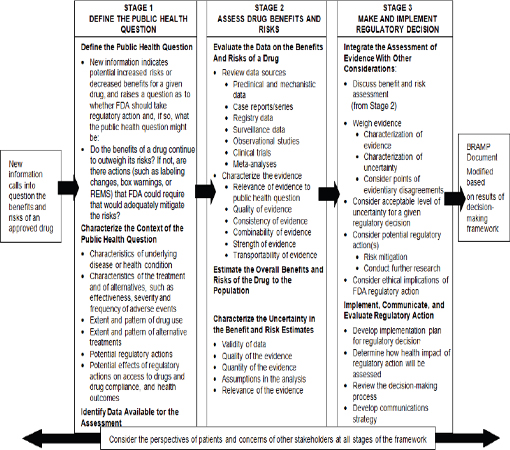
In the present report, the committee adapts the framework from Science and Decisions: Advancing Risk Assessment specifically to FDA’s postmarketing drug-regulatory setting to facilitate managing the benefits and risks associated with a drug throughout its lifecycle (see Figure 2-2). The framework should be used whenever FDA needs to make a regulatory decision about a drug; given its charge, the committee focuses on the use of the framework in the postmarketing setting where it could be used, for example, in choosing regulatory actions when the presence of a serious safety signal may precipitate or require consideration of a regulatory action. The adapted framework has three stages: define the public health question, assess the drug’s benefits and risks, and make and implement the regulatory decision. Central to the framework is the need to elicit and incorporate the perspective of the patient.5
The three-stage framework is designed to be broadly applicable and assist the decision-maker in the exercise of sound judgment. It is intended to place reasonable demands on the limited resources of FDA given the volume of approved drugs, but to ensure that comprehensive evaluations of benefit and risk can be conducted when disagreements arise or when the public health effects may be substantial. FDA’s decisions vary in their complexity (see Box 2-2). A recent NRC report noted that FDA’s decision-making framework should be flexible enough to be applicable to the broad array of decisions FDA faces for its different regulatory purviews (NRC, 2011). That need for flexibility is equally true within the drug-regulatory setting. Although all three stages are necessary regardless of the complexity of the regulatory decision under consideration, the scope of each stage required to support sound policy decision-making will depend on the circumstances and available evidence. Many regulatory decisions will not require comprehensive evaluations at every stage, and efforts should be scaled accordingly.
A number of methods have been proposed for assessing benefits and risks, and for making regulatory decisions in response to these assessments. Some
![]()
4The Pharmaceutical Research and Manufacturers of America has transferred its Benefit–Risk Action Team (BRAT) framework to the Centre for Innovation in Regulatory Science “to further the program’s technical development and broaden input from the scientific community” (PhRMA, 2012).
5FDA also recognizes the importance of the participation of patients, patient advocates, and health professional organizations in its regulatory decisions, and has established an office to facilitate such interactions (FDA, 2011a).
BOX 2-2
Two Examples of the Diversity and Complexity of Food and Drug Administration (FDA) Decisions
FDA’s decisions on drugs range from relatively easy decisions for which the science and the appropriate regulatory action are clear to ones for which the scientific evidence can be complex or contradictory and determining the appropriate regulatory action would benefit from input from many experts.
1. As an example of the former, the scientific evidence on the risk of liver problems associated with trovafloxacin (Trovan®), an antibiotic used for treatment for various infections, was clear soon after it was approved. Trovafloxacin was approved by FDA in December 1997 and became available to patients two months later (FDA, 1999). None of the 7,000 patients in the premarketing clinical trials experienced serious liver problems (hepatic failure, sometimes requiring liver transplantation, or death), but soon after entered the market, FDA began to receive reports of adverse events as early as two days after treatment; more serious adverse events (acute liver failure) occurred in patients after more than two weeks of treatment. Within seven months of approval, FDA had received more than 100 reports of patients’ experiencing symptomatic and asymptomatic hepatic toxicity; some who sustained hepatic damage had to have transplants or died (FDA, 1999). In July of 1998, FDA worked with the drug sponsor to add further toxicity information on the medication label and package insert, informing physicians of the potential for hepatic toxicity. In addition, distribution of trovafloxacin was limited to inpatient facilities, patients receiving trovafloxacin had to have life-threatening or limb-threatening disease, and a physician must believe that the drug’s benefits outweighed the risks it posed for a patient (FDA, 1999). In this example, once the drug was marketed, there was evidence of an association between trovafloxacin and severe, sometimes fatal adverse events. Other drugs that could be used effectively to treat for most infections were on the market. Given that evidence, FDA placed severe restrictions on the use of Trovan.
2. In contrast, when deciding about postmarketing regulatory decisions about aprotinin (Trasylol®), FDA was faced with conflicting scientific evidence about the risks associated with the drug. Aprotinin is a bovine-derived natural protease inhibitor that was approved by FDA in 1993 for use during coronary arterial bypass surgery to reduce blood loss and diminish the need for blood transfusions in surgical patients. From the time of its FDA approval through 2005, several studies and meta-analysis of results of randomized controlled trials supported
the efficacy of aprotinin for reducing the inflammatory response, the need for transfusions, and the risk of stroke, and it showed either no effect or a reduction in mortality, myocardial infarction, or renal failure risk (Henry et al., 2001; Levi et al., 1999; Sedrakyan et al., 2004). In early 2006, however, two observational studies further raised concerns about aprotinin’s safety. Mangano et al. (2006) compared health outcomes related to the use of aprotinin (1,295 patients) with outcomes related to the use of two other hemostatic agents—aminocaproic acid (883 patients) and tranexamic acid (822 patients)—and results in 1,374 patients who did not receive a hemostatic agent. The study found that use of aprotinin doubled the risk of renal failure and was associated with higher rates of heart attacks and stroke than the use of other medications or no treatment. The study by Karkouti et al. (2006) used propensity scores and compared 449 of 586 patients who had received aprotinin during high–transfusion-risk surgery with 449 patents who received tranexamic acid; it determined that aprotinin may be associated with renal dysfunction. On the basis of the results of those two studies, FDA released a public health advisory for aprotinin in February 2006, detailing the results of the two observational studies and cautioning physicians to “consider limiting [aprotinin] use to those situations in which the clinical benefit of reduced blood loss is essential to medical management of the patient and outweighs the potential risks”.
In late September 2006, after FDA held a public meeting of the Cardiovascular and Renal Drugs Advisory Committee, the drug sponsor disclosed preliminary findings from a new observational study that confirmed the findings of the previous observational studies. The new study, which was commissioned by the drug sponsor, reviewed hospital records of 67,000 patients who had undergone coronary bypass graft surgery. Preliminary study results found that the 30,000 patients who received aprotinin during surgery had an increased risk of death, renal failure, congestive heart failure, and stroke. Final study results, published in 2008, concluded that patients who received aprotinin had an estimated mortality 64% higher than patients who received aminocaproic acid (relative risk [RR], 1.64; 95% confidence interval [CI], 1.56–2.02) (Schneeweiss et al., 2008). Another advisory committee meeting was held, but the committee did not find the evidence compelling enough to recommend withdrawal of the product from the market, but did find it compelling enough to recommend a label change and that an RCT be
Continued
BOX 2-2 Continued
conducted (FDA, 2007a). Taking the preliminary data into account, FDA issued a new statement in September 2006, reiterating the cautions from the earlier health advisory and asking physicians to monitor patients for the occurrence of toxicity. In December 2006, FDA strengthened the safety warnings regarding aprotinin and added a warning that the drug increases the possible risk of renal damage. The advisory also included guidance for minimizing the risk.
In the meantime, the Blood Conservation Using Antifibrinolytics in a Randomized Trial (BART)—a multicenter, blinded, randomized, controlled study comparing aprotinin with two other antifibrinolytic agents (aminocaproic acid and tranexamic acid)—had begun to enroll patients in 2002. In October 2007, the study was halted early when preliminary results indicated a higher death rate seen in aprotinin-treated patients (RR, 1.53; 95% CI, 1.06–2.22) (Fergusson et al., 2008). In November, 2007, FDA announced that the sponsor agreed to an FDA-requested marketing suspension of aprotinin in February 2007, after preliminary results from the BART were released.
In May 2008, FDA announced that the drug sponsor would remove remaining stock of aprotinin from the market and limit access to aprotinin to investigational use. The special protocol allows the use of aprotinin for “certain patients who are at increased risk of blood loss or transfusions during coronary artery bypass surgery and who have no acceptable alternative therapy” (FDA, 2008a).
The differences in the complexity of these two examples illustrate FDA’s need for a scalable framework for decision-making. Where the evidence is somewhat more clear-cut, such as the case of trovafloxacin, FDA could use the three stages of the framework, but the decision might not require as extensive weighing of the evidence, or engagement of stakeholders and external experts. Where the evidence is not as clear, where scientists might disagree about the value of different sources of evidence, or where requiring a randomized controlled trial is likely to be ethically controversial, such as in the case of aprotinin, each of the three stages might be more involved, with FDA eliciting external scientific advice, the perspectives of patients, and the concerns of other stakeholders.
![]()
aThe committee uses these drugs as examples of the variability in FDA’s decisions and its approach to safety signals, evidence, and regulatory decision-making. The committee is not commenting on, or drawing any conclusions about, the timing or nature of the regulatory decisions.
researchers and decision scientists have proposed the use of either decision-conferencing or multicriteria decision analysis for benefit-and-risk–based regulatory decision-making (see Appendix D for discussion) (NRC, 2011). The process of decision-conferencing or multicriteria decision analysis can increase transparency in regulatory decision-making, provide formal opportunities for input from stakeholders, and delineate the sources of disagreements among participants. Some of the methods, however, rely heavily on using a common metric, such as dollars saved, lives saved or quality-adjusted life-years, to quantify benefits and risks related to different endpoints and assigning numerical values to a number of subjective considerations, such as the importance of a given adverse event or a specific improvement in quality of life. Appendix C discusses the use of those processes as tools to elicit input into the decision-making process. They can be useful, and, in some cases, can provide informative results, but the committee emphasizes, as have others, that reducing benefits and risks to a common metric as the only output considered in a decision can sometimes lead to oversimplifying complex decisions, misunderstanding, and a lack of trust (NRC, 1989, 2011).
Consistent use by FDA of the proposed framework for decision-making would be valuable for several reasons. First, it would allow stakeholders to understand and anticipate key components of the process by which decisions are made. Second, it would emphasize the dynamic nature of benefit and risk assessments, particularly in the postmarketing setting, and the need for continual re-evaluation of decisions by an organization dedicated to protecting and promoting the public health. Third, it would provide an opportunity to consider the value of additional postmarketing studies (for example, through postmarketing requirements), to explore scientific and ethical issues with regard to the type of postmarketing studies under consideration, and to evaluate the potential effects of future regulatory decision-making. Fourth, use of a systematic approach for the routine re-evaluation of benefits, risks, and regulatory decisions could minimize long delays in decision-making or lack of transparency in the rationale for regulatory decisions may be minimized. It is important to note that while the need to change or modify a regulatory decision about a drug in the postmarketing context can sometimes be traced to errors in premarketing regulatory decision-making, often that is not the case. Rather, the ability to respond to the changing knowledge base regarding the benefit–risk balance is a valued characteristic of the agency that seeks to secure population benefits while mitigating harms, and modifications of regulatory decisions should be expected in the postmarketing phase and not regarded as reflecting a failure.
The following sections describe the three stages and the key elements for consideration in each stage. The three stages should not be interpreted as isolated activities, but as interconnected activities that inform each other and help to ensure that the characterization of risks or, in this case benefits and risks, is decision-driven, recognizes all significant concerns, includes both analysis and
deliberations with input from the interested and affected parties, and is appropriate to the decision (NRC, 1996, 2011).
Stage I—Define the Public Health Question
In their 2009 commentary, Hamburg and Sharfstein defined in a broad outline the public health question that faces the agency (Hamburg and Sharfstein, 2009):
A public health approach recognizes that the potential good of a new medical product or policy must be balanced against the potential harm. Some benefits are not worth the risk; some risks are worth taking. Key considerations are the severity of the illness at issue, the availability of alternative treatments or preventive interventions, and the current state of knowledge about individual responses.
In the postmarketing context, each regulatory decision is usually triggered by the receipt of new information about a drug’s benefits, risks, or both. The new information may come to FDA in many ways, including routine surveillance initiated by FDA, reports of adverse events from physicians and manufacturers, and scientific studies published in the professional literature. Continuous monitoring of those sources of information is part of the lifecycle approach to FDA oversight. Stage I of the regulatory decision-making framework involves defining—each time that relevant new information about a drug emerges—the public health question that the new information raises. The broad public health question raised by new evidence of harms associated with a drug in the postmarketing setting, for instance, is whether any regulatory action is needed to ensure that the public’s health is enhanced, and not unduly jeopardized, by a drug currently on the market.
The primary framing for this question is FDA’s public health mission and responsibilities. Policy makers, regulators, and advisory-committee members need to determine the consequences of alternative regulatory actions on population health and target their efforts accordingly. All FDA regulatory decisions involve complex relationships between scientific evidence, regulatory authority, ethical values, and practical considerations. The public health question and the context within which it is being asked need to be clearly defined to ensure that all those relationships and the potential public health consequences of alternative actions, are properly considered from the onset of the decision-making process.
A first step in the decision-making framework is identifying the general public health question at issue and then making it more particular in response to specific characteristics of the drug and health problem at issue and the new information that has emerged. To make sure that the public health question defined in Stage I reflects a broad understanding of the public health interests at stake, it is important to elicit and take into account the perspectives of patients and the experiences of health-care providers, pharmacists, industry, and other stakeholders. A number of reports discuss the financial ties between some patient groups and the
pharmaceutical industry (Abraham, 2010; Hemminki et al., 2010; Jones, 2008; Lofgren, 2004; Rothman et al., 2011). FDA should ensure that patient advocacy groups represent the views of patients rather than the views of commercial entities that provide funding to the organizations.
The major considerations that should be taken into account when specifying the public health question that underlies a regulatory decision are discussed below. The goal of the “public health question” is to ascertain what the public health impact would be of different regulatory responses triggered by new data pertaining to a drug’s benefit–risk profile, not simply whether the use of the drug incurs unacceptable risk. This public health question contains all the components of a completely specified research question, which can be summarized under the acronym PICOTS: Population, Intervention, Comparison, Outcomes, Timing, and Setting (Brian Haynes, 2006; IOM, 2011a; Richardson et al., 1995; Straus et al., 2010). In the drug safety policy context, the population is those persons with a specified condition who currently take or might be eligible to take the drug, including information about relevant subgroups; the intervention is a regulatory action, with the predicted mix of treatments in the population induced by that action; the comparison is an alternative action (typically, the maintenance of current policies), with a projected population prevalence and pattern of treatments for the condition predicted to exist after that action; the outcomes are all the health outcomes deemed relevant to the policy decision, considered on the population level, due to both the drug and to the disease; the timing includes any time factors relevant to treatment (for example, chronic versus time-limited) or outcomes (immediate versus delayed); and the setting represents those contexts in which treatments for the condition are prescribed or administered.
It is important to note the difference between the elements of the question above and those concerning a simple drug treatment. For a single drug treatment, its effects can often be tested in a randomized controlled trial, where some research participants get the treatment and others do not. But there in fact is no single experiment that can be conducted to test directly the health effects of a given policy change, even if that involves market removal of a drug. Instead, the public health question must be addressed indirectly, piecing together a wide variety of facts and factors—both scientific and social—to estimate the likely population health impact of a policy intervention. Those factors are outlined in the following sections.
Characteristics of the Underlying Disease or Condition and of the Affected Population
Characteristics of the underlying disease or condition—such as its severity, duration, and natural history—are critical for specifying the public health question. A drug that improves quality of life or life expectancy of people who have advanced pancreatic cancer might continue to be acceptable even if new
information suggests a high risk of a new, severe adverse event. Conversely, new information suggesting that a migraine drug poses a high risk of a severe adverse event would raise serious concerns about the drug’s benefit–risk balance even if the drug were effective in controlling migraine attacks in many patients. Patient input may be crucial at this point (NRC, 2011). The value they put on the prevention of migraine attacks or other symptomatic conditions and how much risk of what type they are willing to accept for improvements in quality of life might be quite different than the values put explicitly or implicitly on outcomes by regulators. It is also important to keep in mind that patients will not all have the same values. Sicker patients, for example, might accept more frequent or severe side effects than patients whose illness or condition is well-controlled, and even patients with similar disease severity and therapeutic response might differ in their willingness to risk side effects.
The public health question cannot be properly specified without careful characterization of the population that is taking the drug and could take it in the future. Key considerations include the size of the population; whether it includes people who are in need of special protections, such as children and those with serious cognitive disabilities; whether it is made up largely of communities where there are substantial health disparities; and whether it is a population of mainly well or ill people.
Potential benefits beyond those to the people taking the drug should also be identified. Family and friends of patients benefit when drugs enhance the well being and functioning of those they love. For example, a marketed drug that improves the symptoms of dementia, other cognitive disabilities, or severe mental illnesses may have a direct effect on the quality of life of family members and other caregivers. Similarly, the benefits of a vaccine can extend far beyond the person vaccinated to others who might not be able to be vaccinated and more broadly to society.
Also relevant is whether the aim is prevention or treatment. Judgments about the acceptability of risks posed by such products as vaccines or drug therapies administered to healthy people to prevent them from becoming ill will be different from judgments about the acceptability of risks posed by drugs for treating those who are already ill.
Available Information About the Drug
To prepare for the formal assessment in Stage II of the quality of new and existing evidence about a drug’s benefits and risks, it is important in Stage I to broadly identify what is understood about why the drug is given, how it is given, at what dose and how long, the variation in administration seen in the community with concomitant reasons, what monitoring or oversight is required for proper use, where it is given, and how the drug is experienced by patients,
including benefits, harms and typical adherence. That understanding should be incorporated into the framing of the public health question. Additional characteristics related to safety include the severity, duration and reversibility of potential adverse events associated with the drug, whether it would be easy to mitigate or improve outcomes resulting from adverse events with close monitoring or other treatments, and whether estimates of efficacy are based on clinical endpoints or surrogate markers.
Availability of Alternative Treatments for the Disease or Condition
When deciding how to specify the public health question raised by new information about an approved drug, FDA should consider whether the drug is “first-in-class”, and the benefits and risks of any alternative treatments, including whether there is a subgroup of patients in whom the other treatments do not appear to be effective. The willingness to restrict the use of rosiglitazone (Avandia®) in light of concerns about its risks was prompted in part by the availability in the market of a similar drug, pioglitazone, for the same indication that appeared to have a more favorable benefit–risk profile, although pioglitazone had not been on the market for as long as rosiglitazone.
Plausible Regulatory Actions and the Potential Effects of Alternative Regulatory Actions
It can be helpful in Stage I to narrow the public health question by specifying which regulatory actions are plausible candidates for a given situation. In some cases, a drug may already be subject to one or more of the regulatory actions, or it may already be clear that some options, such as withdrawal of the drug from the market, are inappropriate in a given context. Insofar as it is possible to narrow the range of plausible regulatory actions and thus the scope of the public health question, efforts in Stages II and III can be more focused. Potential options for postmarketing FDA regulatory action are defined by statute, implementing regulations, and judicial decisions. FDA’s potential regulatory actions in the postmarketing setting expanded considerably with the passage of FDAAA (Kessler and Vladeck, 2008). Those regulatory actions are described in Chapter 1 and summarized in Box 2-3. The actions are not mutually exclusive; FDA can choose to use a single action or a mix of actions. For example, FDA could require both a Risk Evaluation and Mitigation Strategy (REMS) and a postmarketing study of a drug.
One important consideration is the extent to which alternative regulatory actions affect access to a drug, and whether any resultant restrictions in access would be desirable and fair. For example, the implementation or expansion of a REMS that only permits specially trained physicians to prescribe a drug or
BOX 2-3
Summary of Food and Drug Administration (FDA) Postmarketing Regulatory Actions and Authorities
No Change in Regulatory Action
FDA may determine that available evidence does not justify any change in regulatory action.
Changes in Labeling and Letters to Health Care Professionals
FDA may order changes in the labeling of approved prescription drugs and biologics to make patients and medical professionals aware of new safety information and recommendations concerning their safe use.a Manufacturers must submit their proposed labeling changes for FDA review. FDA has new enforcement tools to ensure timely and appropriate safety-labeling changes,b and may request that the manufacturer write an informational letter to health professionals indicating new label changes. FDA may also require boxed warnings if special problems are associated with a drug, particularly problems that may lead to death or serious injury.c Boxed warnings are intended to call special attention to the risks involved with a marketed drug.
Request that the Drug Sponsor Conduct a Postmarketing Study
FDA may request that a manufacturer enter into a postmarketing commitment (PMC) to conduct postmarketing studies (that is, Phase 4 trials) to characterize the safety and clinical effectiveness of an approved drug better. PMCs are not required.
Requirement that the Drug Sponsor Conduct a Postmarketing Study
FDA may require a manufacturer to conduct postmarketing studies (postmarketing requirements, PMRs). FDA may impose a PMR at any time in the lifecycle of a drug when (1) the need for a study is based on appropriate scientific data and (2) the adverse-event reporting and pharmacovigilance systems will not be sufficient to assess known or new signals of serious risk or the available data indicate the potential for unexpected serious risks.d
specially trained pharmacists to dispense it in rural communities where a short supply of such specially trained health professionals could have the effect of limiting drug availability in ways that are not intended.6 In addition, removal of a warning label or of a REMS requirement could result in increased prescribing and
![]()
6Congress recognized this and tried to make it easier for “frontier” providers to participate in REMS training (21 USC § 355-1[a][f][3]).
Establishment or Modification of Risk Evaluation and Mitigation Strategies (REMS)
FDA may require, if new safety information concerning a drug arises, that the manufacturer submit a proposed REM that will ensure that the benefits of the drug will outweigh its risks.e FDA reviews and approves proposed REMSs to ensure their adequacy and compliance with statutory criteria. Failure to comply with an approved REMS can result in civil penalties and a removal of the drug from the market. Potential components of a REMS may be the inclusion of a medicine guide or patient package insert and limitation of an approved drug to a specific population, or a particular indication.
Withdrawal of an Approved Drug
FDA has the authority to withdraw approval of a marketed drug using several procedures, after due notice and opportunity for hearing to the manufacturer, if, among other things, new information shows that the drug has not been demonstrated to be either safe or effective.f FDA may also withdraw approval of a marketed drug if the new drug application contained false and misleading statements of material fact or if it is misbranded. It is relatively rare for FDA to remove an approved drug from the market entirely (McClellan, 2007). If a drug was granted accelerated approval for a serious or life-threatening disease, FDA has the authority to use an expedited procedure to remove it from the market if later clinical trials fail to confirm its expected clinical benefit or if the manufacturer did not satisfy its obligations for additional postmarketing studies.g
![]()
a21 USC § 355(o)(4) (2010).
b21 USC § 355(o)(4)(G) (2010).
c21 CFR 201.57(e).
d21 USC §§ 355(k), (o)(3) (2010).
e21 USC § 355-1 (2010).
f21 USC § 355(e) (2010).
g21 USC § 356(b)(3); 21 CFR 314.500 et seq.
use of a drug, whereas a major label change could decrease the use of or compliance with a drug (see Box 2-4). For FDA to take proper account of unintended consequences of different regulatory actions in Stage III, it is important that, to the extent possible, FDA identify in Stage I of the decision-making process the potential consequences of a decision on the availability and utilization of a drug. Identifying those consequences at the onset of the process will help ensure that the affects of those consequences on the benefits and risks associated with dif-
BOX 2-4
Antiepileptic Drugs: An Example of Unintended
Consequences Playing a Role in a Regulatory Decisiona
Case reports of suicides or ideas of committing suicide, referred to as suicidality, resulting from the use of antiepileptic drugs triggered the US Food and Drug Administration (FDA) to ask drug sponsors to submit data from placebo-controlled trials for meta-analysis. FDA sent letters to sponsors requesting submission of clinical data on 11 antiepileptic drugsb from March 2005–January 2007 (FDA, 2008b). In all, FDA reviewed the data from 210 trials—199 placebo-controlled and 11 trials that used low-doses as the control. In total, the drug arms contained 27,863 patients and the placebo arms 16,029 patients (FDA, 2008c). FDA analyzed the data on patients who had epilepsy, psychiatric disorders, or other indications for four primary end points: completed suicide, attempted suicide, preparation toward suicide behavior, and suicidal ideation. Results from the meta-analysis, published in May 2008, indicated that a higher risk of suicidality occurred as early as 1 week after starting on antiepileptic treatment and continued for at least 24 weeks (FDA, 2008c). Overall, patients on treatments had a higher risk of experiencing a suicidal behavior or ideation event than placebo patients (odds ratio, 1.80; 95% confidence interval, 1.24, 2.66).
FDA issued an alert to physicians and other health care professionals of the increased risk of suicidal behavior or thoughts in patients taking antiepileptic drugs “to treat epilepsy, bipolar disorder, migraine headaches, and other conditions” (FDA, 2009a).
At a July 2008 joint meeting of FDA’s Peripheral and Central Nervous System Drugs Advisory Committee and Psychopharmacologic Drugs Advisory Committee, the committees voted in favor of adding warnings to
ferent regulatory actions are assessed in Stage II of the process, and considered in the decisions made in Stage III of the process.
At the end of Stage I, FDA should have a clearly stated and carefully specified public health question that identifies the relevant regulatory actions under consideration. For example, as noted previously, when the decision-making process is initiated because of new evidence of harms associated with a drug, the general public health question is whether any regulatory action is needed to ensure that the approved drug or class of drug still has a favorable benefit–risk profile, and, if so, what those actions should be. By the end of the Stage I, the question should carefully articulate which regulatory actions might be needed to ensure that a given drug is still acceptable for a specified population with a
prescribing information and requiring a medication guide for antiepileptic drugs but voted against adding a boxed warning to the drugs. The meeting included discussion of the potential for a decrease in prescriptions for and use of the drugs in the wake of a boxed warning, and the adverse effect on patient care such decreases might cause (FDA, 2008d).
On the basis of the meta-analysis results and the recommendations from the advisory committees, FDA required all sponsors of the class of antiepileptic drugs to include a warning label, not a boxed warning, and develop a medication guide informing patients of the possible increased risk of suicidal thoughts and behavior with the initiation of this class of drug. Health-care professionals were informed of the updated warning label “for antiepileptic drugs used to treat epilepsy, psychiatric disorders, and other conditions (e.g. migraine and neuropathic pain syndromes)” (FDA, 2008c). FDA also warned patients that suddenly stopping the use of antiepileptic drugs could cause serious problems.
The regulatory actions in connection with the antiepileptic class of drugs constitute an example of a complex regulatory decision that required balancing the risks posed by a drug and the unintended risks of imposing more stringent regulatory requirements, such as inclusion of a boxed warning about the association between the drugs and suicidality, were to be imposed.
![]()
aThe committee uses this examples to illustrate the importance of considering all potential consequences of a regulatory decision. The committee is not commenting on, or drawing any conclusions about, the timing or nature of the regulatory decisions.
bThe Food and Drug Administration requested clinical data on the following drugs: carbamazepine, felbamate, gabapentin, lamotrigine, levetiracetam, oxcarbazepine, pregabalin, tiagabine, topiramate, valproate, and zonisamide (FDA, 2008b). In 2009, clonazepam was added to the list of antiepileptic drugs associated with an increased risk of suicidality.
disease that has a characterized public health impact for which alternative interventions are or are not available. The specification of the public health question should have engaged patients and other stakeholders including clinicians, industry, and family members, and incorporate their perspectives when planning the benefit and risk assessments, and to identify their concerns, including practical considerations related to different regulatory actions.
Stage II—Assess Drug Benefits and Risks
In Stage II, scientific and technical experts—in conjunction with risk managers, policymakers, and regulators—evaluate the quality of evidence on both the benefits and the risks associated with an approved drug, including any new
information that has triggered the need to consider regulatory action. The output of this stage includes estimates of the likelihood and magnitude of a drug’s benefits and risks, and a characterization of the scientific evidence on which the estimates are based.
Evaluate the Data on the Benefits and Risks of a Drug
Evaluating a drug’s benefits or harms on the population level is a two-step process. The first step involves scientific studies—usually controlled experiments—that aim to estimate the degree of difference in health outcomes under one treatment regimen versus another in a defined population. The second step is to use this and additional information to estimate the population impact of the drug’s use in a given condition. This requires understanding population patterns of use, including dosage and co-treatments, and impact in more heterogeneous populations than might have been studied. This sometimes can be assessed in community-based studies or trials, but often must be evaluated by modeling the impact of a variety of disparate data sources pertaining to the above factors. These include surveillance data, observational studies, registry data, published and unpublished clinical trial data, and relevant case reports or series as appropriate. Information on structurally similar drugs and drugs in the same class may also be relevant and should be considered for inclusion.
The goal of the benefit assessment and risk assessment is to support FDA’s decision-making. The assessments therefore should be designed with the public health question that needs to be answered and the array of regulatory actions in mind. The available evidence relevant to the public health question at issue, the expertise required to assess that evidence, and the additional evidentiary needs should be identified at the onset of the assessments. The evidence considered in benefit and risk assessments, at least initially, should be inclusive to avoid biasing their outcome.
The benefit-assessment and risk-assessment stage typically includes scientific judgments. For example, judgments are made about the strength of evidence on the basis of interpretations of the quality and applicability of individual studies, and about what constitutes a benefit or an adverse event. Those judgments can depend on a person’s professional training, ethical values, and personal preferences. Such judgments are unavoidable but should be explicitly discussed among the participants in the assessment and documented to ensure transparency regarding the sources of the differences. Chapter 3 discusses the potential sources of disagreements in detail.
The expertise necessary for evaluating the quality of evidence and characterizing the evidence can differ substantially between the premarketing and postmarketing settings (GAO, 2009a). Given the rapidly evolving types of data sources, study designs, and analytic approaches used in the postmarketing setting (as described in Chapter 3), expertise in many fields is necessary for evaluating the quality of evidence for assumptions used in benefit and risk assessment.
Participants in the benefit-assessment and risk-assessment process should include, as appropriate, FDA policymakers, regulators, and scientists and external experts who have sufficient experience and expertise—including expertise in observational studies and clinical trials and in causal-inference methods—to evaluate for each study the quality of data sources, study-design elements, study conduct, data analyses, and interpretations of the results. The participants should also include persons with training and experience in clinical medicine, particularly in the specific specialties relevant to the disease in question and its potential adverse events, so that they can understand the clinical context within which medical care takes place. The people assessing the benefits and risks should have a comprehensive understanding of the public health question under consideration and of the potential implications of the assessments for regulatory decision-making.
To that end, there may be a need for capacity-building in FDA for it to be able to conduct, develop, and implement the benefit-assessment and risk-assessment process, including the elicitation of individual values and understanding of regulatory judgments about the different dimension of the decision-making process, and to have shared responsibility with industry for the development and oversight of benefit and risk management and planning documents of the sort described later in this chapter. If such resources are not yet fully available in FDA, FDA could build on its partnerships through federal collaborative initiatives, and public–private–nonprofit–academic partnerships. It could also build capacity through the centers-of-excellence models discussed in the Institute of Medicine regulatory science workshop summary (IOM, 2011b). Interactions and models of this kind can mitigate the issue of workforce constraints while providing FDA with access to requisite expertise. Appropriately managed interactions can also enhance transparency, minimize conflict of interest, and facilitate peer review. The overall decision, however, should rest with FDA.
Characterization of the Strength of Evidence
The estimates of the benefits and risks associated with a drug need to be characterized for regulatory decision-makers and stakeholders. The characterization should include a discussion of the nature of the evidence, including the types of studies that have been conducted (for example, observational studies and clinical trials), the quality of the studies, and the consistency of the findings among studies. Such a characterization will provide FDA’s decision-makers and stakeholders with an indication of the confidence that they should place in the overall body of literature and in the benefit and risk estimates that are based on it. The characterization can be qualitative or, if sufficient data are available and the precision is warranted, the uncertainty in the benefit and risk assessments can be quantified.
The extent of the analysis of uncertainty and the complexity of the process used to incorporate expertise and perspectives will depend on the available evidence. If, for example, the available studies are of high quality and the results
are consistent among studies, various experts are more likely to agree on the assessment of the risks and benefits and on the characterization of the evidence on which the assessment is based. As the quality and consistency of evidence decrease, disagreements are more likely, and more complex and formal processes may be helpful in clarifying and helping to resolve them.
A variety of approaches to characterization of the strength of evidence are possible and used by other US government agencies, foreign governments, and organizations (AHRQ, 2002; Miksad et al., 2009). Different approaches have their own strengths and limitations; some are simply qualitative assessments that provide categories of evidence, and others involve complex, quantitative methods for combining and statistically analyzing data from different studies. Chapter 3 further discusses the dimensions that contribute to the strength of evidence.
At the end of Stage II, FDA decision-makers should be provided with estimates of the likelihood and magnitude of the benefits and the risks of a drug, and a characterization of the scientific evidence on which those estimates are based. That characterization should include a summary of the data on which those estimates are based, the strengths and weaknesses of the data, the confidence in the evidence base, and any disagreements in the evaluation of the quality of the evidence (NRC, 2009, 2011).
Stage III—Make and Implement Regulatory Decisions
In the third stage of the framework, regulatory decisions are made and implemented. This stage involves synthesizing and integrating the estimates of benefits and risks and the quality of the evidence on which these are based (from Stage II) with the public health question (as specified in Stage I); deciding on the appropriate regulatory actions, including whether further study should be required; implementing the regulatory actions; and evaluating the effects of the regulatory actions.
Integrating the Assessment of the Evidence with Other Considerations
When making a regulatory decision, FDA should take into account not only the best available scientific evidence on a drug’s benefits and risks but also a variety of legal, ethical, and practical considerations. A key benefit of distinguishing between Stage II from Stage III is the ability to disentangle technical scientific considerations from other factors relevant to decision-making (NRC, 2009, 2011; Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). That separation helps to ensure, for example, that there is clarity about the reasons behind a decision that is taken and about the sources of disagreements regarding the decision. For example, if the decision is that no new regulatory action is needed, it is important to know whether the decision reflects the scientific conclusion that the evidence associating the drug with a new adverse
event is of poor quality or whether, on the basis of stakeholder input, FDA has judged the benefits of the drug to be so important that the newly discovered risk is acceptable. The various factors and their role in regulatory decision-making are discussed below.
Scientific Considerations
In Stage II of the framework, scientific and technical experts provide to regulators estimates of the benefits and risks associated with a drug and characterize the uncertainty in the estimates (NRC, 2009). Regulators should be given other relevant information that was identified in Stage I that also engages scientific assessments, in this case assessments of the public health and clinical effects of the drug and alternative regulatory actions. The information should include characteristics of the disease and of the adverse events, the availability of alternative treatments, and some features of the population affected (NRC, 2009).
Legal Considerations
Each postmarketing regulatory action has legal requirements that must be met before FDA may take it. Statutes and implementing regulations, as interpreted by US courts, define the requirements. Boxed warnings, for example, are “most likely to be based on observed serious reactions” (FDA, 2011b), but FDA can order a postmarketing requirement based on an indication of a potential serious health risk.7 Most of the listed postmarketing regulatory actions also have procedural requirements—such as notice to the manufacturer and an administrative hearing with the right of judicial appeal8—that generally increase with the consequences of the regulatory action for stakeholders (Evans, 2010; FDA, 2012a).
FDA can be further constrained in choosing among regulatory actions by its precedent policies, and nonbinding guidance documents. There are many good reasons for FDA to maintain consistency and predictability in its regulatory actions. The perception of arbitrariness contributes to public mistrust and undermines compliance with decisions (FDA, 2011b; Transparency Task Force et al., 2010). FDA regulatory actions can be the subject of litigation because of their economic, political, and social consequences (Carpenter, 2010a; O’Reilly, 2008). Courts will largely defer to FDA’s judgments about how to interpret the statutes that it is charged with administering. FDA decisions, however, can be overturned by the courts (O’Reilly, 2008).
Given the variety of factors involved, there is no single method for determining whether the relevant threshold for a particular regulatory action is satisfied. In some cases, there may be insufficient evidentiary certainty to support some kinds of regulatory actions. In others, the participants in the benefit–risk management
![]()
721 USC § 355(o)(3)(B) (2010).
8For example, see 21 USC § 355(o)(3)(E), (F), and 355(o)(4)(B), (C), (D), (E), (F) (2010).
process may agree on the quality and degree of certainty of the overall evidence but disagree on whether that evidence satisfies legal or precedent thresholds for particular regulatory actions. Being clear on where the discrepancy originates is important for determining whether to pursue additional study through a postmarketing requirement and whether the evidence generated from the study would meet the threshold requirements for a particular regulatory action and result in a change in policy.
Ethical Considerations
Regulatory decision-making is an exercise in judgment by a person or leadership team in a position of authority, but the judgment is best made with the fullest possible understanding of the values and views of the relevant parties (NRC, 1994, 1996, 2009, 2011; Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). As has been discussed in the environmental regulatory context (NRC, 1996), people’s values affect their preferences for health states, their perceptions of the benefit–risk balance of a drug, and their views regarding appropriate drug regulatory decisions. That is as true for policy makers, scientists, regulators, and industry executives as it is for patients, physicians, and health care managers. Among the key values relevant to drug decision-making are views about quality of life, risk tolerance, the appropriate role of government, and the overall objective of regulatory actions (NRC, 1996).
Patients’ and physicians’ preferences for health states reflect how they think about the value of prolonging life and about potential tradeoffs between prolonging life and preserving or maximizing quality of life. Some cancer patients and oncologists may feel disinclined to pursue chemotherapy regimens that hold out the prospect of extending life for a few weeks but involve burdensome side effects; others may wish to have access to such drugs notwithstanding their known harms and risks. Such differences in values are not restricted to life-threatening diseases. In February 2005, at an advisory-committee meeting on Vioxx®, a number of doctors representing arthritis patients urged FDA to leave Vioxx on the market because their patients, aware of the risks associated with the drug, were willing to accept the risks given the benefit of relieving the pain of arthritis (FDA, 2005a).
People’s degree of risk tolerance affects not only their willingness to take a drug or have it on the market but also their willingness to tolerate uncertainty about the bases of regulatory decisions. More risk-averse people, for instance, might support regulatory action to remove a drug from the market on the basis of fairly modest evidence of a safety problem, whereas people with a higher risk tolerance would require a greater level of certainty about a safety problem before deeming such action acceptable.
Another important value is people’s view of the appropriate exercise of regulatory authority in the limiting of access to therapies and the constraining
of professional judgment and patient choice. Some stakeholders may emphasize the government’s public-protection role, whereas others may feel that the free market and private decision-making by patients and their physicians should play a more prominent role in determining who has access to which drugs. Those views will drive stakeholders’ perceptions of what postmarketing regulatory actions are appropriate in response to new information that emerges about a marketed drug.
An additional value concerns the overall purposes or objectives of regulatory decisions. Should decisions be made with the sole goal of maximizing the health and welfare of patients for whom the drug is indicated, or in some circumstances should decisions take into account the drug’s effect on the well-being of family and other caretakers and on the health of others? Should the benefits and risks of a drug be weighted differently if they are experienced by particular population groups with special or unusual needs or whose health interests have not been well served historically?
The judgments of regulators and experts about alternative regulatory actions will necessarily reflect their own values about those issues, but they should also take into account the values of others, especially those most affected by their decisions, including principally patients and their families. It is important to recognize that there may be a wide array of values about, for example, quality of life and risk tolerance among patients and families. Particularly in contentious and high-stakes contexts, care should be taken throughout the decision-making process, especially in Stages I and III, to ensure that the full array of views is elicited (NRC, 1996). The values of patients and family members can give meaning to the scientific determinations that emerge from consideration of the evidence concerning a drug’s benefit–risk balance. They may be helpful in pointing regulators toward a course of action in which the scientific assessment does not suggest a clear direction—for instance, when a drug is found to have both substantial benefits and substantial risks, when the benefits and risks are very different, or when there is considerable uncertainty about the benefit–risk balance. Explicitly describing the values that played a role in the decision-making process, whose values they were, and how they were elicited is critical for facilitating public understanding of how regulators reached a given decision (NRC, 1996).
The importance of seeking stakeholder participation in regulatory decision-making has been highlighted in many reports (NRC, 1996, 2009; Presidential/ Congressional Commission on Risk Assessment and Risk Management, 1997). Recent examples of FDA decisions about regulatory actions in the postmarketing period also underscore how different the values and preferences of various stakeholder groups may be from one another and from those of FDA officials. Box 2-5 illustrates the importance of patient preferences in FDA decision-making using the examples of Tysabri® for patients with multiple sclerosis and Lotronex for patients with irritable bowel syndrome.
BOX 2-5
Natalizumab and Alosetron:
The Importance of Patient Perspectivea
The cases of natalizumab (Tysabri®) and alosetron (Lotronex®) highlight the importance of patient preferences in FDA decision-making.
The US Food and Drug Administration (FDA) approved natalizumab—an intravenous monoclonal antibody approved for treatment for relapsing multiple sclerosis (MS)—for marketing in November 2004, under an accelerated approval, on the basis of “positive results to patients after one year of treatment” in two randomized, double-blind, placebo-controlled clinical trials (FDA, 2010a). Uncommon serious adverse events (such as pneumonia, rash, fever, depression, and gallstones) and common adverse events (such as urinary tract infections, headaches, and menstrual disorders) were seen in study participants. FDA approval was conditioned on the manufacturer’s continuing clinical trials for one year after approval. Progressive multifocal leukoencephalopathy (PML), a serious adverse event, was reported in three clinical-trial participants three months after approval (February 2005)—nonfatal in two and fatal in one. The drug sponsor, with FDA’s support, suspended the marketing of natalizumab, and FDA placed ongoing clinical trials on hold and issued a public health advisory informing patients and health-care providers to suspend its use (FDA, 2010b). At the time of the initial warnings about natalizumab, it was estimated that approximately 8,000 MS patients had taken natalizumab, 3,000 of whom were clinical-trial participants (FDA, 2005b).The drug sponsor further examined clinical-trial participants and convened a panel of medical and scientific experts to guide its activities. A year later, after extensive re-examination of clinical-trial participants, no additional cases of PML had been found.
In February 2006, FDA allowed the resumption of the clinical trial of natalizumab contingent on continued study of the risks associated with the drug. In March 2006, FDA held a meeting of its Peripheral and Central Nervous Systems Drug Advisory Committee regarding natalizumab (FDA, 2010b). MS patients testified at the advisory committee meeting as to how natalizumab greatly improved their quality of life. The committee “recommended a risk-minimization program” that included “mandatory patient registration and periodic follow-up to identify as early as possible any cases of PML that may occur, and to try to determine the reason the infection occurs” (FDA, 2010b). In response, the drug sponsor submitted a risk-management plan, called TOUCH, to ensure the safe use of natali-
zumab. In June 2006, FDA approved resumed marketing of natalizumab with the risk-management plan.
As of January 2010, 35 confirmed cases of PML had been reported to FDA (FDA, 2010b). Natalizumab remains on the market with a medication guide and with a new drug label that includes a table summarizing the rate of PML by number of infusions, PML risk, and information on the occurrence of immune reconstitution inflammatory syndrome, a condition that has been associated with natalizumab use in multiple sclerosis patients. Despite a well-defined risk of serious adverse effects, natalizumab remains on the market because preferences of some patients support the decision that the clinical benefits of natalizumab continue to outweigh its potential risks.
In February, 2000, FDA approved alosetron (Lotronex®) for “the treatment of irritable bowel syndrome [IBS] in women whose predominant bowel symptom is diarrhea” (FDA, 2000a). Approval was made on the basis of two clinical trials (a total of 1,273 women) in which “Lotronex was significantly more effective than placebo in providing relief from IBS pain and discomfort and in reducing the percentage of days with urgency” (FDA, 2000b). As of November 10, 2000, however, FDA had “reviewed a total of 70 cases of serious post-marketing adverse events, including 49 cases of ischemic colitis and 21 cases of severe constipation. Of these 70 cases, 34 resulted in hospitalizations without surgery, 10 resulted in surgical procedures and three resulted in death” and following discussion between FDA and the drug sponsor, the sponsor withdrew Lotronex® from the market in November, 2000 (FDA, 2000b).
After the withdrawal of Lotronex® from the market, FDA and the drug sponsor “received numerous emails, letters and telephone calls from patients who related how their IBS symptoms were not responsive to any therapy other than Lotronex®, and how their quality of life was adversely affected by its withdrawal” (FDA, 2002). FDA approved “a supplemental New Drug Application (sNDA) that allows restricted marketing of Lotronex® (alosetron hydrochloride), to treat only women with severe diarrhea-predominant irritable bowel syndrome (IBS). The approved sNDA for Lotronex® includes a risk management program to ensure patients and physicians are fully informed of risks and possible benefits of Lotronex®” (FDA, 2009a).
![]()
aThe committee uses the regulatory history of these drugs to demonstrate the importance of considering the perspectives of the patients when FDA makes decisions. The committee is not commenting on, or drawing any conclusions about, the timing or nature of the regulatory decisions.
Considering stakeholder values in regulatory decisions requires a process for identifying key stakeholder groups and eliciting their views (see Appendix D for discussion of decision conferencing). Particularly for complex and controversial regulatory decisions, it may also require guidance from ethicists and others skilled at identifying sets of values from stakeholder and public comments and in methods for blending scientific and nonscientific considerations in decision-Careful analyses of the different reasons that underlie different stakeholders’ and experts’ views about which regulatory actions best advance the public health or the interests of particular patients can be instructive (NRC, 1996). To the extent that differences in the reasons reflect different technical interpretations of the relevant scientific findings, the results of a properly conducted Stage II should help reduce disagreements about appropriate regulatory policy. However, when the reasons differ because various stakeholders and technical experts use different values to attach meaning to the findings, such as how important it is to secure or avoid a particular benefit or risk, disagreements may be more difficult to bridge.
Practical Considerations
FDA should take into account a number of practical considerations when making its regulatory decisions. For example, it should be feasible to implement a restriction imposed by a REMS, any required medical testing should be affordable and accessible. As an illustration, the REMS for thalidomide contains specific certifications for health care providers prescribing and pharmacists dispensing thalidomide, and requirements for pregnancy testing (Celgene Corporation 2001), and is considered a successful REMS (IAF, 2010). In other instances, there may be substantial practical obstacles to the conduct of certain kinds of studies, limiting the extent to which they can be considered a viable regulatory option (Armitage et al., 2008; Ellenberg, 2011; GAO, 2009a; Hamburg, 2011). For example, experience from previous similar research may suggest that it will be difficult to accrue sufficient numbers of desired patients within the needed timeframe.
Deciding on a Regulatory Action
Regulatory decision-making is a qualitative process and necessitates judgments by regulators concerning the acceptability of the benefit–risk profile of a drug in light of relevant legal, ethical, and practical considerations (NRC, 1994, 1996, 2009, 2011; Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). Experts give decision-makers estimates of the benefits and risks associated with a drug—including the nature, magnitude, and likelihood of the risks and benefits—and a characterization of the confidence or uncertainty in the benefits and risks. The regulators’ difficulty in reaching a decision will depend, in part, on the degree of uncertainty in the estimates of
the benefits and risks and on the severity of the public health consequences if the wrong regulatory action is taken. Decisions are easiest when there is little uncertainty, the consequences of choosing regulatory alternatives are small, and there is broad agreement among most stakeholders and technical experts about the most appropriate regulatory action. Many of FDA’s regulatory decisions are in that category. In those cases, the decision and its rationale should be apparent, and a complex and formal process for coming to that decision is probably neither necessary nor warranted.
Decisions are most difficult when there is large uncertainty in the scientific evidence, there is a potential for severe public health consequences if a wrong regulatory action is taken, and there is considerable disagreement among stakeholders and technical experts about what the right action should be. Difficult regulatory decisions should be identified and resolved in a more formal process. The key components of the decision-making stage when the situation is contentious should be
• Ensuring that the processes used to assess the data about benefits and risks, to determine data quality, and to elicit stakeholder preferences were adequate and that the regulatory decision-makers have the pertinent information from the processes.
• Determining and characterizing the social, political, ethical, and logistic factors that are affecting regulators’ decision-making judgments.
• Determining the regulatory thresholds of evidence needed to justify alternative regulatory actions.
• Understanding the effect of regulatory actions, their effectiveness in improving the health of those who use the drug, the degree to which changes in assumptions about data or values affect the benefit–risk balance, and potential unintended consequences of the regulatory actions.
• Identifying the thresholds of evidence needed to justify alternative regulatory actions.
• Evaluating whether a postmarketing study (a postmarketing requirement or postmarketing commitment) would provide evidence sufficient to support changing a regulatory action.
• Understanding whether providers and patients are able to detect serious adverse events quickly, and particularly if an adverse event is reversible when the treatment is stopped. This information may be important for decision-makers as they consider whether the regulatory action they choose mitigates the risk to the population sufficiently. Such understanding might affect the regulatory actions under consideration. For example, a REMS might be considered to mitigate the risks posed by a drug that can cause a reversible adverse event that is easily detected.
• Assessing whether the time and resources needed to increase the level of evidence to justify a change in regulatory action are appropriate.
Sources of disagreement in FDA should be identified for each of those components so that, at a minimum, internal and external stakeholders will have a better understanding of key subjects of divergence and how the divergence might have led different decision-makers to recommend different regulatory actions. (See Chapter 3 for a discussion of potential sources of disagreement in FDA.)
Use of the framework that the committee proposes cannot eliminate disagreement about what regulatory actions are most appropriate, but it should enhance the likelihood that an appropriate decision is taken. Moreover, the framework can achieve the openness and transparency that are desired, foster a better understanding among stakeholders of how regulatory decisions are made, and enhance public trust and confidence (NRC, 1996, 2009, 2011)—all of which are consistent with FDA’s goal of improved transparency (Hamburg and Sharfstein, 2009).
Communicating, Implementing, and Evaluating Regulatory Actions
Effective communication, including communication of risks, is an important aspect of regulatory decision-making and governance (Calman, 2002; Fischhoff, 2009, 2010; NRC, 1989, 1996; Pidgeon and Fischhoff, 2011). Although FDA has a long history of describing specific regulatory concerns for a wider scientific or general audience (Ellenberg et al., 1994; Siegel, 2002; Temple and Pledger, 1980), there has been a recent push for more communication about and greater transparency in FDA’s regulatory decisions. FDAAA included a number of requirements for transparency,9 and FDA has undertaken a number of initiatives in that regard (see Box 2-6). For example, FDA has improved transparency by posting review documents on Drugs@FDA and, in the case of Avandia and other drugs, posting memorandums outlining scientific disagreements among FDA scientists and the basis of the final decision. In addition, FDA scientists have recently written and published commentaries that explain their regulatory decisions and actions (Woodcock et al., 2010). Box 2-7, which discusses a published article by FDA staff explaining the rationale behind the agency’s decision on dabigatran (Beasley et al., 2011), further illustrates efforts at increased transparency. FDA also recently published a guide, edited and authored by members of its Risk Communication Advisory Committee, that discusses the importance of and best practices in the communication of benefits and risks, including in the FDA context (FDA, 2011d). The committee commends FDA for those activities, but more needs to be done. For any particular drug, it remains difficult to find a clear, concise document that outlines the public health questions that have arisen over the drug’s lifecycle that have prompted regulatory decisions; summarizes the benefit–risk assessment, including the evidence on which it was based; outlines the scientific, ethical, and practical considerations that influenced the decision; and describes plans to manage any potential or known risks associated with a
![]()
921 USC § 360bbb–6 (2010).
BOX 2-6
Food and Drug Transparency Initiatives
The US Food and Drug Administration (FDA) has taken other actions to increase transparency not directly related to requirements under FDAAA. In 2009, FDA launched a transparency initiative. This initiative is proceeding in three phases: the first is intended to provide the public with information on how the agency works, the second on how FDA reaches decisions, and the third on how FDA can become more transparent to industry to foster a more cost-efficient regulatory process (FDA, 2012b).
In addition, FDA has published a series of perspectives and commentaries that describe agency policy positions and explain agency decisions about particular drugs. Communication of its regulatory decisions on rosiglitazone (Avandia®) is a good example. On September 23, 2010, FDA placed severe restrictionsa on the availability of rosiglitazone and discontinued its approval of the Thiazolidine Intervention with Vitamin D Evaluation (TIDE) study that was planned to compare the benefit–risk balance of rosiglitazone and pioglitazone. European regulatory authorities that analyzed the same data reached a decision to suspend the marketing authorization of rosiglitazone. To explain the basis of its regulatory action, senior FDA officials published a commentary in the New England Journal of Medicine (Woodcock et al., 2010). Because there had been much controversy within FDA about the appropriate regulatory decision, the director of the Center for Drug Evaluation and Research took the unusual step of posting on the FDA website a memorandum, Decision on continued marketing of rosiglitazone, outlining how it weighed the available information and took the various internal recommendations into account (FDA, 2010c). That coordinated communication plan provided the public with a unique insight into the agency’s decision-making process on a controversial topic.
![]()
aFDA required the drug sponsor to issue a Risk Evaluation Mitigation Strategy (REMS) according to which “the drug will be available to patients not already taking it only if they are unable to achieve glycemic control using other medications and, in consultation with their health care professional, decide not to take pioglitazone [a diabetes medication in the same class of drugs] for medical reasons. Current users of rosiglitazone will be able to continue using the medication if they appear to be benefiting from it and they acknowledge that they understand [the risks associated with the use of rosiglitazone]. Doctors will have to attest to and document their patients’ eligibility; patients will have to review statements describing the cardiovascular safety concerns” (Woodcock et al., 2010).
BOX 2-7
Dabigatran: The Importance of Transparency in Food
and Drug Administration (FDA) Decision-Making
The reaction to the approval of a higher dose rather than a lower dose of dabigatran (Pradaxa®) illustrates the importance of transparency in FDA’s drug-approval process. The approval was seen as controversial and not understood by the public.
In October 2010, FDA approved a 150-mg twice-daily dose, but not a 110-mg twice-daily dose, of dabigatran, a direct thrombin inhibitor for stroke and embolism prevention for patients who have atrial fibrillation. Approval was based primarily on data from the Randomized Evaluation of Long-Term Anticoagulation Therapy trial, an active-control trial that compared the two doses of dabigatran to warfarin (Connolly et al., 2009). The trial followed 18,113 patients for a median of 2 years. The primary outcome measure was a composite of “time to first occurrence of stroke or systemic embolic event” and the secondary outcome measures were “time to first occurrence of stroke, [systemic embolic event] or all cause death” and “time to first occurrence of stroke, systemic embolic event, pulmonary embolism, myocardial infarction or vascular death”. The 110-mg twice-daily dose was not inferior to warfarin, and the 150-mg twice-daily dose was superior. Bleeding complications and hemorrhagic stroke were more common in patients randomized to warfarin than to either dose of dabigatran. For the approved dose of 150 mg twice daily, ischemic strokes were less common in those randomized to dabigatran than in those randomized to warfarin (RR, 0.76; 95% CI, 0.60–0.98), but the risk of myocardial infarction was higher in those randomized to dabigatran (RR, 1.38; 95% CI, 1.00–1.91 for the 150-mg dose). An excess of serious coronary events was also noted in one of the ximelagatran trials (Fiessinger et al., 2005). The drug sponsor argued that dabigatran should be approved at both doses and that the 100-mg twice-daily dose should be available to people who were at risk for bleeding.
Seven months after FDA approved dabigatran, FDA staff published an article explaining its rationale for approving the 150-mg twice-daily dose, but not the 110-mg twice-daily dose, of dabigatran (Beasley et al., 2011). The article highlighted the underuse of warfarin, which was the only blood-thinner option for atrial-fibrillation patients, because of fear of bleeding, difficulties with its use, and the increased risks of strokes and disability. In the article, Beasley et al. (2011) explained that “both regimens would have been considered safe and effective if studied alone in comparison with warfarin, although the noninferiority finding for the 110-mg dose is somewhat less compelling. But given the clear differences
Continued
between the two doses, FDA’s critical regulatory decision was whether to approve both strengths or only the higher strength”. FDA reviewers were “unable to find any population for whom the availability of a lower dose would improve dabigatran’s benefit–risk profile, and it appeared clear that most, if not all, patients should receive the higher dose.” The article clearly articulated the rationale behind FDA’s decision. In this benefit and risk analysis, FDA did not take into account the possibility that patients may vary in their risk aversion for one adverse event (bleeding) rather than another (stroke).a
Although the decision was criticized, when FDA communicated the rationale for the decision, it allowed clinicians, patients, and others to understand why FDA had made the decision. That better understanding could increase compliance with the recommended dose and improve public health.
![]()
aFDA recently issued a Drug Safety Communication on dabigatran through MedWatch related to reports of serious bleeding events. It stated that it is “working to determine whether the reports of bleeding in patients taking [dabigatran] are occurring more commonly than expected, based on observations in the large clinical trial that supported [its] approval” (FDA, 2011e).
drug or any evaluations of previous regulatory decisions. FDA review documents that are posted at Drugs@FDA contain much of that information, but critical documents are difficult to locate, and the key information is often difficult to find within documents.
In addition to improving transparency and effective communication of its decision-making processes and decisions, FDA policy makers should strive to continuously improve the efficiency and effectiveness with which they function. Evaluations should be conducted on a periodic basis to identify key facilitators of and barriers to timely regulatory action and potential improvements in the decision-making process. Such evaluations should also assess whether regulatory decisions are having the intended effects on drug use and health outcomes and whether unintended consequences are occurring. Much of the health care industry and other high-risk industries (such as aviation) have adopted such a learning approach to improve the quality of care and reduce preventable harm. FDA has an opportunity to learn from past and current decisions by fostering an organizational culture of continuous learning about decision-making processes that is consistent with the lifecycle approach. One goal of the Sentinel Initiative is to facilitate such evaluations. The public, industry, and regulators would all benefit if a learning process led to a systematic approach to regulatory decision-making that is flexible, timely, and consistent in its use.
The committee therefore recommends that the plan for implementation of the framework include a mechanism for reviewing the decision-making process through monitoring and evaluation, as outlined in Science and Decisions: Advancing Risk Assessment (NRC, 2009). Continuing evaluation of the benefit and risk assessment and management decisions is important for ensuring FDA’s continued effectiveness, and the results of the evaluations would inform future improvements in the framework and in the underlying processes and timeliness of benefit and risk assessment and management.
As has been recommended to other agencies making risk-based decisions, FDA should specify which types of decisions will be evaluated, when the evaluations will be conducted, who will conduct them, and what the criteria for evaluations will be (Presidential/Congressional Commission on Risk Assessment and Risk Management, 1997). After-action reviews are of special importance for postmarketing drug-related decisions that are particularly controversial or difficult. FDA needs good outcome measures for the effects of its regulatory actions on public health, including whether the actions have the intended effect on drug use, such as an increase or decrease in prescriptions, and how they affect the occurrence of all relevant health outcomes, including adverse drug events, disease outcomes, and death. For instance, an intervention to reduce off-label use should actually reduce off-label use and thereby improve the health of the public. The importance of including a followup evaluation in Stage III of the framework is highlighted by empirical evidence on the utility of some postmarketing regulatory actions for addressing risks. For example, studies have demonstrated that providers and patients do not consistently heed safety labels, including even the most serious boxed warnings (IOM, 2007b; Lasser et al., 2006; Yu et al., 2011); in contrast, some boxed warnings have been associated with reductions in medication use and prescription (Bhatia et al., 2008). Those reductions might be appropriate or inappropriate, depending on the circumstances. The evaluations should be used to help to determine whether regulatory decisions should be revisited. All evaluations should consider whether and when it is appropriate to solicit stakeholder input.
BENEFIT AND RISK ASSESSMENT AND MANAGEMENT PLAN DOCUMENT
The three-stage decision-making framework recommended by the committee earlier in this chapter is not intended to be used only once, but rather whenever questions about the benefits or risks associated with a drug arise. In this section, the committee proposes the development of a document, akin to a technical support document, that would serve as a public record of the experience of a drug throughout its lifecycle, to be updated at regular points during its lifecycle, including whenever the framework is used to evaluate the drug’s benefits and risks. The committee calls the document a Benefit and Risk Assessment and Management Plan, or BRAMP. Because the benefit–risk profile of a drug can
change during its lifecycle, a BRAMP document serves as a living document that is updated when there is new safety or efficacy information or other information that affects the drug’s benefit–risk profile. The committee does not anticipate that the BRAMP would document every postmarketing safety signal. Rather, the BRAMP document would be updated whenever FDA determines that new information about a drug warrants consideration of regulatory action to evaluate or manage the drug’s benefit–risk profile, including consideration of whether to require the manufacturer to conduct postmarketing research.
A BRAMP document is intended to formalize and make concrete FDA’s commitment to a lifecycle approach to drug oversight and benefit–risk management. It serves, in part, as a checklist that supports organizational adherence to the lifecycle approach. The BRAMP document will add to other FDA transparency initiatives discussed previously, increasing the transparency of FDA’s decisions, and should foster collaboration between FDA and drug sponsors in the oversight of drugs and management of their risks. In addition to information about the stages of the decision-making framework whenever it is used, the BRAMP document should include a description of any boxed warnings, REMS, or other components of regulatory decisions, any future plans for managing or identifying risks, reassessing benefits and risks, and evaluating the effects of regulatory decisions. Details of the types of information that could be included in such a document are presented in Box 2-8 and discussed below.
Although FDA is responsible for making regulatory decisions about approved drugs, responsibility for ensuring that the benefits of a drug outweigh its risks is shared by FDA and the drug sponsor. For each new drug, therefore, responsibility for the initial development of some of the content that would go into the BRAMP document should rest with the drug sponsor; much of that material is already submitted as part of the documentation for approval, such as a summary and benefit and risk assessment, and any proposed postmarketing-study designs or REMS. FDA would use the company-submitted materials as a starting point for the BRAMP document and prepare the BRAMP document for posting on the agency’s website within the same timeframe that is required for FDA to post documents related to new drug applications. FDA staff involved with the drug’s premarketing application as well as staff with expertise and knowledge in postmarketing safety assessment should be responsible for describing the public health question, the process and outputs of the agency’s benefit assessment and risk assessment, the rationale for the agency’s regulatory decision, and the scientific, legal, ethical, and practical factors that went into the final decision to approve a drug. For example, if a drug was approved on the basis of a surrogate end point, that fact should be noted in the BRAMP document, as should any resulting pharmacovigilance or postmarketing commitments or requirements. Final approval of the BRAMP document would be FDA’s responsibility.
Considerations for the oversight of drugs in the postmarketing setting are different from those in the premarketing setting. First, as discussed further in
BOX 2-8
Components of a Benefit–Risk Assessment
and Management Plan Document
1. Public health question.
2. Summary of the benefit and risk assessment, including
• Description of the process used to assess the benefits and risks, including how stakeholder input was sought and incorporated.
• Summary of the available evidence used, the quality and uncertainty of the different studies, and the judgments made about the different studies, including how the studies were factored into decisions.
• A characterization of the overall consistency and uncertainty of the body of evidence.
• Explicit statements of the assumptions used in estimating benefits and risks.
• Estimates of benefits and risks, including outputs of analyses.
• Analyses of assessments sensitivity to the assumptions used and judgments made.
3. Regulatory actions and rationale, including
• A statement of the regulatory decision, such as approval of a drug.
• The rationale for the decision, including not only the final output of the benefit–risk assessment process but a description of any other factors that affected the decision, such as ethical issues, timeframe issues, the lack of an available treatment for a disease, or the risk posed by a drug in a specific population. It should also include a description of any value-of-information analysis, deci-
Chapter 4, the types of evidence likely to be available in the postmarketing setting are broader than those typically available premarketing. For example, data from surveillance and observational studies are much more likely to be available. In the postmarketing setting, therefore, expertise is needed in surveillance, epidemiology, and the evaluation of safety data collected from different observational studies, as well as clinical trial designs, biostatistics, medicine, pharmacology, risk communication, and ethics.
Second, research has demonstrated a tendency of individuals or groups to ignore or discount evidence that does not confirm a previous decision, and give more weight to evidence that confirms a previous decision; this tendency is often termed confirmation bias (Back et al., 2011; Jonas et al., 2001). Confirmation bias has been demonstrated for political (Strickland et al., 2011), medical (Mendel et
sion conferencing, or multiple-criteria decision analysis that was conducted
• Any actions that are meant to highlight or mitigate a drug’s risks, including
a. Labels or labeling changes.
b. Boxed warnings.
c. A Risk Evaluation and Mitigation Strategy.
• A description of any postmarketing surveillance, studies, or trial requirements or commitments including
a. A description of any potential safety issues or effectiveness questions that are considered potential problems.
b. The public health question and the uncertainties that need to be decreased to enable a regulatory decision.
c. Details of the design of the study, including important humansubjects protections.
d. Any other aspects of the studies that are required to be disclosed under the Food and Drug Administration Amendments Act (FDAAA).
e. The timeframe and reporting requirements outlined in accordance with FDAAA.
f. For clinical studies, the specific ClinicalTrials.gov URL where further information and results can be found.
• Schedule of future reviews, including
a. A schedule for future benefit–risk assessments, with a given timeframe or, when there are potential safety or effectiveness issues, the events being detected.
b. A plan to evaluate the effects of the regulatory decision on the public’s health.
al., 2011), and risk evaluation decisions (Cox and Popken, 2008). Such confirmation bias could affect the interpretation of new evidence by FDA’s drug approval staff after they have approved a drug. Along similar lines, Carpenter (Carpenter, 2010b) discusses criticisms of FDA for the “slowness and timidity with which they examine and question past decisions”, the conflict between those within FDA “who approve drugs and those who monitor them after approval”, and the effects on the reputation of FDA or offices within FDA when approval decisions are questioned or drugs are withdrawn.
Those two considerations suggest that postmarketing oversight should be done by FDA staff who have not been involved in premarketing, excluding all such staff would eliminate knowledge of the history of the drug, including the premarketing studies (IOM, 2007a). Seeking to balance breadth of expertise, management of con-
firmation bias, and continuity in knowledge of the drug, the committee suggests that the FDA team responsible for maintaining the BRAMP postmarketing should: (1) be led by FDA staff who did not play primary roles in the drug’s approval process, (2) include continued input and involvement from representative staff involved in the premarketing setting, and (3) include membership with expertise in the areas listed above (surveillance, epidemiology, and the evaluation of safety data collected from different observational studies, as well as clinical trial designs, biostatistics, medicine, pharmacology, risk communication, and ethics).
Postmarketing updates or revisions to the BRAMP document would be needed throughout the lifecycle of the drug, including times when refined and validated safety signals or other issues arise that might affect the benefit–risk profile of an approved drug. Responsible staff—in consultation with others within FDA, with external experts as necessary, and with relevant stakeholders, including industry—should conduct periodic reviews of the BRAMP document, oversee audits and quality control as appropriate, review final reports of all studies included in the BRAMP document, and ensure that timely policy analysis and responses to any required studies occur and are recorded in the BRAMP document.
FDA currently posts a number of documents related to the approval of individual drugs at Drugs@FDA. The materials posted depend on the date of the approval; drugs approved more recently have a summary review document that contains many of the components of a BRAMP document. For example, the summary review for tesamorelin (Egrifta®) includes a description of the available evidence and a summary of the benefit and risk assessment (Center for Drug Evaluation and Research, 2010). The summary reviews, however, are prepared at the time of approval and apparently are not updated when new regulatory actions are considered, are not consistent among all drugs, and do not always include specific details about REMSs and postmarketing requirements. Having a single, living, publicly available summary document that contains the history of all regulatory actions related to a drug and a description of the rationale and support for the decisions is critical for improving FDA’s ability to manage drugs in the postmarketing setting and to increase the transparency of FDA’s regulatory decisions.
SPECIAL CONSIDERATIONS IN THE DECISION OF WHETHER TO REQUIRE A POSTMARKETING STUDY
Balancing the desire for more certainty about drug safety against the need for timely decisions is a key challenge in drug regulation. Regulators and policy makers must see themselves as managing uncertainty and delay, as well as managing risk (NRC, 2009). In this context, qualitative or quantitative assessments of the uncertainty of drug safety should be aligned with the regulatory requirements,
the time pressure for managing a drug’s benefits and risks generated by public health interests, and the ethical acceptability of conducting further research.
One of the regulatory actions available to FDA that can decrease uncertainty is a postmarketing requirement for research. Much of the committee’s charge focuses on the scientific and ethical issues associated with postmarketing requirements, so the committee discusses requiring postmarketing research in more detail in this section. This section begins with a discussion of two specific circumstances under which FDA should give serious consideration to requiring a postmarketing requirement and should provide a public rationale if it decides not to do so. It then discusses one tool, value-of-information (VOI) analysis, which can help to determine whether additional information from a postmarketing requirement would help in decision-making.
General Ethical Considerations in a Food and Drug
Administration Decision to Require a Postmarketing Study
The decision by FDA to require a postmarketing study is associated with particular ethical obligations for FDA, and requires an ethical justification. When FDA requires a study to be conducted, its decision may seem to be based on scientific or regulatory considerations alone. However, at its root, the decision to require research can also be viewed as fundamentally an ethical one. FDA must be confident that criteria for any regulatory action are satisfied not merely because the law allows or requires it but because of its mission to protect and promote public health—an ethical responsibility. The public has vested the agency with legal authority so that it can discharge that responsibility, agreeing to sacrifice some measure of liberty in exchange for the protection that FDA will provide and reposing trust in agency officials to discharge their mission responsibly.
When FDA imposes a postmarketing requirement, it is expressing not only scientific uncertainty about the benefits or harms of a drug but a judgment that the public health interests served by requiring additional research outweigh the burdens placed on pharmaceutical manufacturers and—more importantly, from an ethical standpoint—any risk of harm to or burdens on research participants. The burdens on pharmaceutical companies are the time and money involved in conducting the study, whereas the burdens on research participants may include inconvenience, loss of opportunities for more efficacious treatment, and physical or mental harm.
A related ethical consideration has to do with accountability for research harms. All research on human participants raises ethical questions about the rights and interests of participants and about accountability when participants experience research-related harms or indignities. When research is being conducted because of an FDA mandate, FDA bears a measure of ethical responsibility for any adverse outcomes that participants experience.
In Chapter 4, the committee discusses specific steps that FDA should take to increase the likelihood of adequate protection of the rights and interests of human participants who participate in the postmarketing research that it requires. In addition to its ethical responsibilities to human participants, there are pragmatic and political reasons for FDA to provide strong guidance and oversight of postmarketing studies that it requires, particularly clinical trials. Such studies will have the imprimatur of FDA because FDA ordered that the research be conducted, but without oversight there is no guarantee that it will satisfy minimum scientific standards or even that it will serve the policy purposes that led to the study mandate. FDA incurs a serious risk to its reputation if studies that it requires are judged to be flawed by the research community or an institutional review board (IRB). Alternatively, people may wrongly perceive that a poorly designed clinical trial is well designed and safe to participate in because FDA’s “stamp” is on it. There is also a danger that investigators could use the fact that a study has been required by FDA to resist an IRB’s proposed changes in study design. An IRB may assume that FDA has conducted a thorough review of the design of a required study whereas in reality it did not provide investigators with much guidance on design.
Each of those possibilities involves a breakdown in the quality-assurance system on which public trust in the clinical research enterprise depends. Furthermore, there is a danger that if FDA does not provide strong guidance on the design of research that it requires, it will not receive the kind of information that it needs to make a responsible policy decision. When FDA requires research to be conducted, it must not defer the question of study design to pharmaceutical companies or their academic collaborators but rather should participate actively in study design and monitoring to ensure that the research will serve the purposes for which it was ordered. When the tensions between FDA’s mission to protect the public’s health and its obligation to protect potential research participants appear particularly problematic, FDA may wish to seek the counsel of an independent advisory group before making final determinations about whether or what kinds of postmarketing research to require.
When FDA requires a postmarketing study, at a minimum, it should specify to drug sponsors and the public what information is needed to help reach an appropriate answer to the public health question that prompted the research. The type of study design needed to answer the question also should be specified, as should study endpoints and inclusion and exclusion criteria (see Chapter 4). That FDA should specify key aspects of study design is consistent with FDA’s interpretation of FDAAA in its April, 2011 guidance document on postmarketing requirements: “the authority to require a responsible person to conduct a postapproval study or studies or clinical trial(s) of the drug includes the authority for FDA to describe the study or trial to be conducted, including how the study or trial is to be done and the population and indication. In other words, we can
require a study or clinical trial that is well-designed and adequate to address the serious safety concern” (FDA, 2011f).
In addition to its obligation to ensure that a study will provide the requisite information, FDA has an obligation to ensure timely use of study findings in its regulatory decisions. Finally, in the case of clinical trials, FDA should articulate safety-monitoring schemes and any other design features that it views as necessary for the ethical justification of a trial, as the committee discusses in Chapter 4.
Specific Circumstances for Considering a
Postmarketing Research Requirement
There is no absolute rule or algorithm as to when a postmarketing requirement should be required beyond the requirements in accelerated approvals and in the Pediatric Research Equity Act (PREA; see Chapter 1). In the information available during the approval process, there can be different indicators of the need for postmarketing research. After a drug is approved, there are many circumstances in which information could emerge that would suggest that, compared with the benefits and risks expected at the time of approval, either the benefits of a drug are smaller than expected or the risks posed by a drug are greater than expected.
FDA should prospectively determine and publicly identify the risk factors or conditions, including clinical drug characteristics, that are associated with greater uncertainty about the benefit–risk profile in both the premarketing and postmarketing settings. Given the conditions identified, FDA should require postmarketing research in a timely fashion unless there is a compelling reason not to and should make public the rationale for requiring or not requiring postmarketing research in each case. Value-of-information analyses (see Box 2-9), or at a minimum the conceptual framework that underlies them, can be a useful tool to help determine whether more research would improve the decision-making process.
The committee believes that the premarketing and postmarketing considerations should include the following:
• when information about several surrogate endpoints are available and they provide conflicting evidence about the likely health outcomes associated with a drug;
• when first-in-class drugs are evaluated on the basis of surrogate endpoints that are typically used to evaluate drugs in another class;
• when safety signals identified from premarketing data or postmarketing surveillance involve a substantial public health concern or a severe adverse event;
• when there is a strong biologic rationale for a particular adverse effect;
BOX 2-9
Value-of-Information Analysis
Value-of-information (VOI) analysis is a potentially useful tool for deciding whether further research is needed (Claxton et al., 2001; NRC, 2009). VOI helps determine whether or not it is worthwhile to collect additional information or conduct additional research, prior to making a decision (Ginnelly et al., 2005). The expected VOI is calculated by weighing the change in the potential net benefits to the population from the decisions from obtaining that information. If the information would not alter a regulatory decision, the VOI is zero (Ginnelly et al., 2005; Raiffa, 1968). Even if not formally applied, it provides a very helpful conceptual framework for deciding when and what types of research are needed by linking this determination to subsequent decision-making.
VOI analysis can be used to identify when and how a decision-maker’s preferred option might be changed if the decision-maker were able to incorporate additional information into the decision (NRC, 2009). In other words, VOI analysis describes the relationship between the knowledge that might come from the considered source of information and its potential for improving decision outcomes, and it can help to establish a threshold for specific regulatory decisions. The analysis enables regulators, given the current state of knowledge, to evaluate the likely health benefits of various courses of action, including a decision to gather more information.
VOI analysis allows decision-makers to weigh the value of additional evidence against the potential risks posed by delaying a regulatory decision until the information is available (NRC, 2009). That assessment is an important part of the benefit–risk management decision-making process, it helps to determine whether to seek a postmarketing commitment (PMC) or postmarketing requirement (PMR).
VOI analysis has important limitations. It does not measure the scientific merit and broader utility of a study and therefore is not a substitute for the analysis of these issues. For the same reason, traditional VOI is helpful for determining whether to seek a PMC or require a PMR and less well suited to determine which types of studies are most appropriate for producing the evidence sought.
• when a drug is expected to have a different benefit–risk profile under real-world conditions or in specific patient groups;
• when a drug belongs to a class in which a substantial safety signal has previously been identified; and
• •when evidence emerges in the postmarketing setting that suggests a lack of benefit.
The committee elaborates below on indications for requiring postmarketing research when drugs are approved on the basis of surrogate endpoints and when safety signals of concern are present in premarketing data.
Drugs Approved on the Basis of Surrogate Endpoints
Many drugs are approved on the basis of what are called surrogate endpoints, which FDA has defined as “a biomarker intended to substitute for a clinical endpoint”, one that is “expected to predict clinical benefit (or harm, or lack of benefit) based on epidemiologic, therapeutic, pathophysiological or other scientific evidence” (Atkinson et al., 2001).10 Examples include not only the measures of blood pressure, cholesterol, glucose or glycated hemoglobin (HbA1C), and serum chemistry but tumor shrinkage, electrocardiographic findings, pulmonary-function tests, and imaging studies, such as carotid ultrasonography to assess the arterial intimal–medial wall thickness as a measure of subclinical atherosclerosis.
Surrogate endpoints are commonly used in the approval of new drugs. Drugs that qualify for the accelerated approval mechanism require confirmatory postmarketing studies as a condition of approval, but about one-third of drugs that go through the traditional approval process are approved solely on the basis of evidence on surrogate endpoints (GAO, 2009b), which have been discussed extensively by scientists in and outside FDA (Fleming and DeMets, 1996; Prentice, 1989; Psaty et al., 1999; Temple, 1999). The primary advantage of surrogate-endpoint trials is the ability to evaluate drugs more quickly and in smaller studies than would be required for the demonstration of a reduction in the risk of major clinical events. In part because of the low sample size, information from trials that use surrogate endpoints remains incomplete with respect not only to uncommon risks but also to actual health benefits associated with the use of the drugs. Changes in a surrogate endpoint are sometimes poor predictors of changes in health outcomes (CAST, 1989). Surrogates for efficacy, moreover, are unlikely to capture information about off-target effects that may lead to adverse events. The 2009 Government Accountability Office (GAO) report New Drug Approval: FDA Needs to Enhance Its Oversight of Drugs Approved on the Basis of Surrogate Endpoints (GAO, 2009b) summarizes the matter well:
While the use of surrogate endpoints can expedite the approval of drugs, reliance on these endpoints also introduces uncertainty regarding the risks and benefits of a drug because the clinical effectiveness is not directly measured. Thus their use can lead to the adoption of useless or even harmful therapies if the effect on a surrogate endpoint does not accurately predict whether treatments provide benefits to patients, or if the drug has a smaller than expected benefit and a larger than expected adverse effect.
![]()
10For a discussion of how to evaluate or validate surrogate endpoints see Evaluation of Biomarkers and Surrogate Endpoints in Chronic Disease (IOM, 2010).
FDA should consider requiring postmarketing requirements when
• Information about several surrogate endpoints is available and provides conflicting evidence about the likely health outcomes associated with a drug.
• First-in-class drugs are evaluated on the basis of surrogate endpoints that are typically used to evaluate drugs in another class.
Drugs approved on the basis of surrogate endpoints have on occasion been the subjects of postmarketing drug-safety problems. For instance, the premarketing studies of rosiglitazone demonstrated a reduction in fasting glucose and glycated hemoglobin (see Table 2-1). Using only glycated hemoglobin as the surrogate endpoint, one would have predicted a clinical benefit, such as a reduction in the risk of coronary disease (Selvin et al., 2004). However, rosiglitazone increased body weight and low-density lipoprotein (LDL) cholesterol (Law et al., 2003), a major risk factor for coronary disease. Indeed, LDL cholesterol is a traditional surrogate endpoint used in the approval of lipid-lowering drugs. On the basis of its effect on LDL cholesterol, the predicted effect of rosiglitazone on heart disease is not only larger than the effect predicted on the basis of glycated hemoglobin but in the opposite direction (Law et al., 2003). In an evaluation limited to surrogate endpoints, the adverse effects on lipids present a safety signal that requires additional evaluation.
The premarketing studies of the weight loss drug sibutramine also had mixed results (see Table 2-2). Compared with placebo, the use of the drug was associated with a weight loss of about 10 lb. Traditionally, weight loss is associated with a reduction in blood pressure (Wilson, 2008), but sibutramine, despite weight loss, increased blood pressure, which is an accepted surrogate endpoint in the evaluation of antihypertensive drugs (Temple, 1999). On the basis of its effect on blood pressure, the predicted effect of sibutramine on heart disease was not only larger than the effect size predicted on the basis of weight loss but in the opposite
TABLE 2-1 Rosiglitazone in Phase 3 Trials
| Outcome | Placebo | 4-mg rosiglitazone |
8-mg rosiglitazone |
| Fasting glucose, mg/dLa | 233 | 204 | 186 |
| Glycated hemoglobin, %a | 9.7 | 8.9 | 8.6 |
| LDL cholesterol, mg/dLb | 130 | 145 | 148 |
| Weight change, kg median (25th, 75th percentile) | –0.9 (–2.8, 0.9) | 1.0 (–0.9, 3.6) | 3.1 (1.1, 5.8) |
aAdministered as once daily dose.
bOnce daily and twice daily dosing groups combined.
Abbreviation: LDL, low-density lipoprotein.
SOURCE: Data from (FDA, 2007b).
TABLE 2-2 Sibutramine Trials
| Sibutramine | ||||
| Placebo | 10 mg | 15 mg | 20 mg | |
| Weight loss, lb | ||||
| Study 1 | 2.0 | 9.7 | 12.1 | 13.6 |
| Study 2 | 3.5 | 9.8 | 14.0 | |
| Study 3 | 15.2 | 28.4 | ||
| Change in systolic blood pressure, mm HG | ||||
| Mean | -0.1 | 4.0 | 4.7 | |
| Early morning | -0.9 | 9.4 | 5.3 | |
| Change in diastolic blood pressure, mm Hg | ||||
| Mean | 0.1 | 5.0 | 5.6 | |
| Early morning | -3.0 | 6.7 | 5.8 | |
SOURCE: Data from FDA (2008e).
direction. Given evidence on surrogate endpoints pointing in opposite directions (Law et al., 2009), it is not possible to predict the drug’s effect on actual health outcomes reliably. The Sibutramine Cardiovascular Outcomes Trial (SCOUT), which demonstrated an increased risk of major cardiovascular events (James et al., 2010), led to the withdrawal of sibutramine on October 8, 2010.
Torcetrapib, a cholesterol ester transfer protein (CETP) inhibitor, presents a case in which two surrogate endpoints that were available during the premarketing evaluation pointed in opposite directions and there was no validated surrogate endpoint for a first-in-class drug (see Box 2-10 for details of studies). In Phase 2 trials, torcetrapib raised high-density lipoprotein (HDL) cholesterol, possibly reducing cardiovascular risk, but increased blood pressure, possibly increasing cardiovascular risk. As a condition of approval, FDA required a large Phase 3 outcome trial for approval; this trial was stopped early because torcetrapib increased the risk of cardiovascular events and death. An increase in HDL cholesterol remains a surrogate endpoint that lacks support for use in future trials. These examples suggest that FDA should track the experience with a variety of surrogates.
A serious study-design bias may arise from the use of a surrogate endpoint during drug approval to infer or predict its effects on actual health outcomes. This bias, when present, is detected almost exclusively in the postmarketing setting, sometimes by observational studies but more frequently by randomized trials. The findings from these studies provide critical information about the reliability and the validity of surrogate endpoints that are likely to be used for the approval of future medications. As an essential element of the lifecycle approach to improving regulatory science at FDA, the ability to refine, enhance and improve the surrogate endpoints used for future drug approvals represents an effort to protect the health of the public and prevent the adverse events occasioned by faulty or flawed inferences on the basis of trial results from surrogate endpoints that turn
BOX 2-10
Torcetrapib
Low concentrations of HDL cholesterol, the “good cholesterol”, are associated with an increased risk of cardiovascular events. Torcetrapib, an inhibitor of CETP, raised HDL cholesterol in a 4-week crossover study that included 19 participants (Brousseau et al., 2004). In another study of 1,188 participants who had coronary disease and who were taking atorvastatin (Lipitor®), torcetrapib was compared with placebo for the primary outcome of coronary atherosclerosis or atheroma volume as assessed by coronary intravascular ultrasonography (Nissen and Wolski, 2007). The increase in HDL cholesterol associated with torcetrapib was pronounced (see table below), and LDL was significantly decreased. There was no significant change in the primary outcome of atheroma volume, and torcetrapib was associated with a small increase in the risk of cardiovascular events (relative risk, 1.07; 95% confidence interval, 0.85–1.34). Both systolic and diastolic blood pressures were increased significantly by torcetrapib. FDA required the conduct of a large outcome trial for drug approval (Barter et al., 2007), but it was stopped early because torcetrapib increased the risk of cardiovascular events and death.
Torcetrapib Trial
Abbreviations: CETP, cholesterylester transfer protein; FDA, Food and Drug Administration; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
SOURCE: Data from Nissen et al. (2007).
out to be “biased,” poor predictors of actual health outcomes that are important to patients. Since FDAAA specifies “failure of expected pharmacological action” as a safety issue, improving methods to predict drug effectiveness in the postmarketing context can be regarded as part of FDA’s drug safety responsibility.
The opportunity for surrogate-endpoint bias occurs often. According to a GAO report (GAO, 2009b) between January 1, 1998, and June 30, 2008, about one-third (69 of 204) new molecular entities approved under the traditional approval process were approved on the basis of surrogate endpoints. The GAO report notes that “according to FDA officials, they do not have specific criteria for determining when they will accept a surrogate endpoint as a valid substitute for a clinical endpoint, and such decisions are made on a case-by-case basis” (GAO, 2009b). During the GAO review, the “FDA planned to develop a comprehensive inventory of all surrogate endpoints used to approve new drugs, including those under the traditional process. FDA officials told us [the GAO] that they were able to compile a partial list of such endpoints, but due to other competing priorities, this inventory was never completed” (GAO, 2009b).
Based in part on the experience with rosiglitazone, FDA revised its guidance for the approval of anti-diabetic medications (FDA, 2008f). Phase 2 and 3 trials are now required to include an evaluation of cardiovascular events by an independent cardiovascular endpoints committee, and the sponsor is expected to perform a meta-analysis of the data from these studies. The upper limit of the two-sided 95 percent confidence interval for the association with cardiovascular events now influences not only the decision about drug approval but also the requirements for postmarketing trials. This guidance represents an excellent example of the lifecycle approach to methods. Postmarketing experience and research helped to shape and improve the methods by which future medications in the same class will be evaluated.
Like the guidance on anti-diabetic therapies, the guidance on drug-induced liver injury incorporates experience from postmarketing studies to improve the use of surrogate endpoints for safety in future drug evaluations (FDA, 2009c). These models can and should be replicated and documented. For instance, the postmarketing study findings for sibutramine may suggest ways of improving the surrogate marker methods for evaluating weight loss drugs during the premarketing setting (James et al., 2010); and the postmarketing study findings for ezetimibe (Kastelein et al., 2008; Rossebo et al., 2008) and fenofibrate (Tonkin and Chen, 2010) may suggest ways of improving these methods for evaluating lipid-lowering drugs.
Drugs About Which Premarketing Data Yield Safety Signals
Although not all safety signals that are seen in premarketing trials will require postmarketing studies, there are some circumstances in which they should be considered. One is when there is a substantial public health concern, as occurred in the case of the H1N1 vaccine (DeStefano and Tokars, 2010; SteelFisher et al., 2010). Another is when a severe adverse event is seen, and a third is when there is a strong biologic rationale for a particular adverse effect. Rofecoxib (Vioxx®) is a nonsteroidal anti-inflammatory drug (NSAID) that primarily
inhibits the cyclo-oxygenase-2 (COX-2) enzyme. Rofecoxib was evaluated in 58 premarketing studies that included 5,771 patients, 3,629 of whom received rofecoxib for 1 day or more. Writing in May 1999, the medical officer reviewing rofecoxib noted “there is a theoretical concern that patients chronically treated with a COX-2 selective inhibitor may be at higher risk for thromboembolic cardiovascular adverse experiences than patients treated with COX-1/COX-2 inhibitors (conventional NSAIDs), due to the lack of effect of COX-1 inhibition on platelet function”.11 The same reviewer noted that “there was a … higher incidence of ischemic/thromboembolic events (angina, myocardial infarction, CVA, TIA) in patients taking rofecoxib when compared with patients taking placebo. … In 6 weeks [sic] studies there was one event in the placebo group (0.2%) and a total of 12 events (approximately 1%) in the rofecoxib group.”12 The summary goes on to say that “with the available data, it is impossible to answer with complete certainty whether the risk of cardiovascular and thromboembolic events is increased in patients on rofecoxib. A larger database will be needed to answer this and other safety questions.”13
After approval for marketing, the manufacturer conducted the Vioxx GI Outcomes Research (VIGOR) trial, seeking to demonstrate that rofecoxib would be associated with fewer serious gastrointestinal complications, such as perforations and major bleeds, than naproxen (Bombardier et al., 2000). Cardiovascular events were “not specified in the study design” although data on them apparently were collected and “assessed for a future meta-analysis” (Bombardier et al., 2000). Later, a colonic polyp prevention trial that was also designed to look at thrombolytic events (the Adenomatous Polyp Prevention on Vioxx [APPROVe] trial) demonstrated an increased risk of such events with rofecoxib; the study was stopped, and the manufacturer voluntarily withdrew rofecoxib from the market. Had the VIGOR trial been designed to evaluate both the gastrointestinal benefits and the thromboembolic adverse events because of the safety signal in premarketing studies and the biologic rationale for such effects, more complete information about the benefit–risk profile of rofecoxib might have been available much sooner. (Interestingly, VIGOR was conducted at twice the daily recommended chronic dose, which underscores the importance of being attentive to dose levels both for benefits and risk contrasts [Bombardier et al., 2000].) Postmarketing observational studies of rofecoxib also provided safety information (see discussion in Box 4-1).
![]()
11Villalba ML. FDA Medical Officer Review of VIOXX (rofecoxib), Part 7, NDA 21-042 (capsules) and NDA 21-052 (oral solution) available through Drugs@FDA (accessed February 19, 2011), p. 104.
12Villalba ML. FDA Medical Officer Review of VIOXX (rofecoxib), Part 7, NDA 21-042 (capsules) and NDA 21-052 (oral solution) available through Drugs@FDA (accessed February 19, 2011), p.104.
13Villalba ML. FDA Medical Officer Review of VIOXX (rofecoxib), Part 7, NDA 21-042 (capsules) and NDA 21-052 (oral solution) available through Drugs@FDA (accessed February 19, 2011), p.105.
At the time a drug is approved, uncertainties about its benefits and risks necessarily remain. Adequate protection of the public’s health requires a lifecycle approach to the management of the benefit–risk profile of drugs, including an increasing role for postmarketing surveillance and required postmarketing research. The three-stage decision-making framework and BRAMP document presented in this chapter provide guidance to FDA on how to respond to new information about a drug’s benefits and risks as experience with the drug grows. The framework and BRAMP document, with its division of oversight responsibility between FDA offices, are mechanisms for incorporating sound science, ethical considerations, high-quality benefit and risk assessments, and the principles and practices of regulatory science, including public accountability and transparency, into FDA’s decision-making processes and oversight practices.
Finding 2.1
FDA’s current approach to drug oversight in the postmarketing setting is not sufficiently systematic and does not ensure consistent assessment of benefits and risks associated with a drug over its lifecycle. Use of a standardized regulatory decision-making framework that is flexible enough to adapt to decisions of different complexity could make FDA’s decision-making process more predictable, transparent, and active, allowing FDA to better anticipate postmarketing research needs and to plan for such research early when more design options with fewer ethical tensions might be possible.
Recommendation 2.1
FDA should adopt a consistent decision-making framework for regulatory actions across the lifecycle of all drugs that includes opportunities for input from patients and other stakeholders. This framework should be employed in making the initial drug approval decision and, in the postmarketing context, whenever new information that could affect the drug’s benefit–risk profile emerges. The framework should include three stages:
Stage I: Define the public health question that requires a regulatory decision or agency response.
Stage II: Assess the drug’s confirmed or potential benefits and risks by using a systematic process to evaluate and characterize existing evidence and any sources of disagreement about that evidence.
Stage III: Determine the appropriate regulatory response to the public health question specified in Stage I, including whether further research should be
required, by integrating the evaluation of the evidence of benefits and risks from Stage II with legal and ethical considerations and input from stakeholders; communicate to the public the reasoning behind the decision; implement the regulatory response; and, particularly for difficult or controversial decisions (see Recommendation 2.5), evaluate the impact of the regulatory response.
Finding 2.2
No single, clear, comprehensive, and public document currently captures FDA’s assessments of a drug’s benefits and risks over the course of its lifecycle, nor does any documentation help to standardize FDA’s decision-making processes or describe FDA’s rationale for its regulatory actions. Capturing such information in a living document would formalize the lifecycle approach to drug regulation, improve regulatory oversight, and improve the transparency of FDA’s decisions.
Recommendation 2.2
FDA should require and maintain, for each new drug and for already approved drugs for which questions about the benefit–risk profile are raised, a publicly available and understandable Benefit and Risk Assessment and Management Plan (BRAMP). For new drugs, the BRAMP document should be initiated during the drug-approval phase and updated over the lifecycle of the drug at pre-specified times in the postmarketing setting and whenever questions about the drug’s benefit–risk profile arise. The document should include a description of: any public health questions raised during the drug’s lifecycle; the benefit and risk assessment specific to each public health question; key stakeholder input specific to each question; any regulatory decisions or actions and the rationale for each decision, including requirements for postmarketing research or a risk evaluation and mitigation strategy (REMS); a schedule for future assessments of benefits and risks; and plans for and results of evaluating the effectiveness of any regulatory decisions or actions.
• In the premarketing phase, the drug sponsor should provide a summary of the drug’s benefits and risks, any uncertainties in the evidence, and plans for decreasing those uncertainties. FDA should use that information as a starting point to develop the BRAMP document. FDA staff involved with the drug’s premarketing application and staff with expertise and knowledge in postmarketing safety assessment should finalize the initial entry to the BRAMP document.
• In composing teams to monitor the safety of a drug and maintain its BRAMP in the postmarketing phase of the drug’s lifecycle, FDA should
consider the real or perceived confirmation bias of staff that played a significant role in approving the drug. This should be managed by ensuring that the leader of the postmarketing safety monitoring team is without the potential for such bias. The monitoring team should have expertise in surveillance, epidemiology, and the evaluation of safety data collected from different observational and clinical trial designs. The team should review and modify the BRAMP document at specified intervals throughout the lifecycle of the drug, including when new information warrants re-evaluation of the drug’s benefit–risk profile.
Finding 2.3
In the premarketing setting, evidence is derived primarily from randomized controlled trials. In the postmarketing setting, however, evidence may be derived from surveillance, observational studies, patient registries, published and unpublished clinical trials, meta-analyses, and relevant case reports or series. Data sources, study designs, and analytic approaches for the postmarketing context are evolving rapidly. Given those differences, the expertise needed to evaluate and characterize the quality of evidence in the postmarketing setting is different from and broader than that needed in the premarketing setting.
Recommendation 2.3
In making determinations about appropriate regulatory decisions to be implemented in the postmarketing context, FDA should ensure that the full range of methodologic expertise is used to evaluate the strength of evidence of a drug’s benefits and risks from a wide range of designs. For complex regulatory decisions, including decisions about requiring additional postmarketing research, such expertise should include, but not be limited to
• Clinical medicine and clinical practice, such as pharmacy.
• Biostatistics: Bayesian, frequentist, and causal inference methods.
• Epidemiology and pharmacoepidemiology.
• Clinical trials.
• Benefit–risk analysis.
• Research and public health ethics.
• Risk communication.
Finding 2.4
Section 901 of FDAAA14 stipulates the purposes for which FDA has the authority to require postmarketing observational studies and RCTs, and 2011 FDA guid-
![]()
14USC § 355(o) (2010).
ance for industry provides information on FDA’s implementation of that section of FDAAA. Although FDA’s decisions to require postmarketing research need to be made case by case, there are some identifiable conditions that are concordant with but more specific and detailed than those outlined in FDAAA and FDA guidance, which make information from additional postmarketing research important.
Recommendation 2.4
FDA should prospectively determine and publicly identify specific conditions, including drug characteristics and other features, that are associated with greater uncertainty about a drug’s benefit–risk profile in the postmarketing setting. Under those identified conditions, FDA should require postmarketing research in a timely fashion unless there is a compelling reason not to, and should make public the rationale for requiring or not requiring postmarketing research in each case. Those premarketing and postmarketing conditions should include the following
• A drug is approved when several surrogate endpoints provide conflicting evidence about the likely health outcomes associated with the drug.
• A first-in-class drug is approved on the basis of surrogate endpoints used in drugs of a different class.
• A drug is associated with safety signals from premarketing data or postmarketing surveillance when
o there is a substantial public health concern,
o a severe adverse event is seen, or
o there is a strong biologic rationale for a particular adverse effect.
• A drug is expected to have a different benefit–risk profile in a subgroup or under real-world conditions.
• A drug is in a class for which a substantial safety signal has previously been identified.
• Evidence of a lack of benefit of a drug in the whole population or in identifiable subgroups emerges in the postmarketing setting.
Finding 2.5
Some FDA decisions in response to postmarketing public health questions are controversial or difficult. Complex instances tend to occur when FDA must make a decision despite scientific disagreement about the relevant evidence or when the likely effects of a given regulatory action are uncertain. These cases serve as important opportunities for FDA, external scientists, and the public to learn about the complexities of the decision-making process and the consequences of a regulatory decision, and for FDA to improve its processes and practices.
Recommendation 2.5
FDA should conduct after-action reviews of postmarketing drug-related decisions that are particularly controversial or difficult or when a major regulatory decision is made after marketing. Such a review should include an assessment of the decision-making process itself and the effects of the final decision on the public’s health.
Finding 2.6
Surrogate endpoints are often relied on in the drug-approval process, and their use has been related to a number of high-profile drug-safety problems. The findings of postmarketing studies can be used to revise the approval process and improve the endpoints and methods used in it.
Recommendation 2.6
As part of a continuing effort to improve regulatory science, FDA should maintain and annually update a list of surrogate endpoints allowed for use in the approval of drugs, the rationale for their use, the postmarketing experience regarding their correlation with health outcomes of interest, and any revisions of approval requirements that may have been suggested by the results of the postmarketing studies. The list should accumulate the postmarketing experience of the successes and failures of various surrogates so that for each major drug class, the regulatory science related to approval methods can be modified and improved. FDA should also revise or develop guidance documents for the use of selected surrogate endpoints that, on the basis of postmarketing studies, appear to be inconsistently predictive of clinical outcomes.
Abraham, J. 2010. Pharmaceuticalization of Society in context: Theoretical, empirical and health dimensions. Sociology 44(4):603-622.
AHRQ (Agency for Healthcare Research and Quality). 2002. Systems to rate the strength of scientific evidence. Washington, DC: Department of Health and Human Services.
Armitage, J., R. Souhami, L. Friedman, L. Hilbrich, J. Holland, L. H. Muhlbaier, J. Shannon, and A. Van Nie. 2008. The impact of privacy and confidentiality laws on the conduct of clinical trials. Clinical Trials 5(1):70-74.
Asamoah, A. M., and J. M. Sharfstein. 2010. Transparency at the Food and Drug Administration. New England Journal of Medicine 362(25):2341-2343.
Atkinson, A. J., W. A. Colburn, V. G. DeGruttola, D. L. DeMets, G. J. Downing, D. F. Hoth, J. A. Oates, C. C. Peck, R. T. Schooley, B. A. Spilker, J. Woodcock, and S. L. Zeger. 2001. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clinical Pharmacology & Therapeutics 69(3):89-95.
Back, E. A., P. Esaiasson, M. Gilljam, O. Svenson, and T. Lindholm. 2011. Post-decision consolidation in large group decision-making. Scandinavian Journal of Psychology 52(4):320-328.
Barter, P. J., M. Caulfield, M. Eriksson, S. M. Grundy, J. J. Kastelein, M. Komajda, J. Lopez-Sendon, L. Mosca, J. C. Tardif, D. D. Waters, C. L. Shear, J. H. Revkin, K. A. Buhr, M. R. Fisher, A. R. Tall, and B. Brewer. 2007. Effects of torcetrapib in patients at high risk for coronary events. New England Journal of Medicine 357(21):2109-2122.
Beasley, B. N., E. F. Unger, and R. Temple. 2011. Anticoagulant options—why the FDA approved a higher but not a lower dose of dabigatran. New England Journal of Medicine 364(19):1788-1790.
Bhatia, S. K., A. J. Rezac, B. Vitiello, M. A. Sitorius, B. A. Buehler, and C. J. Kratochvil. 2008. Antidepressant prescribing practices for the treatment of children and adolescents. Journal of Child and Adolescent Psychopharmacology 18(1):70-80.
Bombardier, C., L. Laine, A. Reicin, D. Shapiro, R. Burgos-Vargas, B. Davis, R. Day, M. B. Ferraz, C. J. Hawkey, M. C. Hochberg, T. K. Kvien, and T. J. Schnitzer. 2000. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR study group. New England Journal of Medicine 343(21):1520-1528.
Borer, J. S., H. Pouleur, E. Abadie, F. Follath, J. Wittes, M. A. Pfeffer, B. Pitt, and F. Zannad. 2007. Cardiovascular safety of drugs not intended for cardiovascular use: Need for a new conceptual basis for assessment and approval. European Heart Journal 28(15):1904-1909.
Brian Haynes, R. 2006. Forming research questions. Journal of Clinical Epidemiology 59(9):881-886. Brousseau, M. E., E. J. Schaefer, M. L. Wolfe, L. T. Bloedon, A. G. Digenio, R. W. Clark, J. P. Mancuso, and D. J. Rader. 2004. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. New England Journal of Medicine 350(15):1505-1515.
Calman, K. C. 2002. Communication of risk: Choice, consent, and trust. Lancet 360(9327):166-168.
Carpenter, D. 2010a. Reputation and power institutionalized: Scientific networks, congressional hearings, and judicial affirmation, 1963-1986. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Cambridge, NY: Princeton University Press.
Carpenter, D. 2010b. The other side of the gate: Reputation, power, and post-market regulation. In Reputation and power: Organizational image and pharmaceutical regulation at the FDA. Cambridge, NY: Princeton University Press.
CAST (The Cardiac Arrhythmia Suppression Trial Investigators). 1989. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. New England Journal of Medicine 321(6):406-412.
Celgene Corporation 2001. Thalomid REMS. http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM222649.pdf],%20and%20is%20considered%20a%20successful%20REMS (accessed December 12, 2011).
Center for Drug Evaluation and Research. 2010. Division director review: Summary review. Application number 22-505. Washington, DC: Food and Drug Administration.
CHMP (Committee for Medical Products for Human Use). 2008. Reflection paper on benefit-risk assessment methods in the context of the evaluation of marketing authorization applications of medicinal products for human use. London, UK: European Medicines Agency.
Claxton, K., P. J. Neumann, S. Araki, and M. C. Weinstein. 2001. Bayesian value-of-information analysis. International Journal of Technology Assessment in Health Care 17(10):38-55.
Connolly, S. J., M. D. Ezekowitz, S. Yusuf, J. Eikelboom, J. Oldgren, A. Parekh, J. Pogue, P. A. Reilly, E. Themeles, J. Varrone, S. Wang, M. Alings, D. Xavier, J. Zhu, R. Diaz, B. S. Lewis, H. Darius, H. C. Diener, C. D. Joyner, and L. Wallentin. 2009. Dabigatran versus warfarin in patients with atrial fibrillation. New England Journal of Medicine 361(12):1139-1151.
Coplan, P. M., R. A. Noel, B. S. Levitan, J. Ferguson, and F. Mussen. 2011. Development of a framework for enhancing the transparency, reproducibility and communication of the benefit-risk balance of medicines. Clinical Pharmacology & Therapeutics 89(2):312-315.
Cox, L. A., Jr., and D. A. Popken. 2008. Overcoming confirmation bias in causal attribution: A case study of antibiotic resistance risks. Risk Analysis 28(5):1155-1172.
DeStefano, F., and J. Tokars. 2010. H1N1 vaccine safety monitoring: Beyond background rates. Lancet 375(9721):1146-1147.
Ellenberg, S. S. 2011. Accelerated approval of oncology drugs: Can we do better? Journal of the National Cancer Institute 103(8):616-617.
Ellenberg, S. S., J. S. Epstein, J. C. Fratantoni, D. Scott, and K. C. Zoon. 1994. A trial of RSV immune globulin in infants and young children: The FDA’s view. New England Journal of Medicine 331(3):203-205.
EPA (US Environmental Protection Agency). 2005. Human health risk assessment protocol for hazardous waste combustion facilities, support materials. http://www.epa.gov/osw/hazard/tsd/td/combust/riskvol.htm (accessed December 12, 2010).
EPA. 2009. General guidance on risk management programs for chemical accident prevention (40 CFR Part 68). Washington, DC: Environmental Protection Agency.
Evans, B. 2010. Seven pillars of a new evidentiary paradigm: The Food, Drug, and Cosmetic Act enters the genomic era. Notre Dame Law Review 85(2):419-524.
FDA (US Food and Drug Administration). 1999. Food and Drug Administration 09 June 1999 Trovan (trovafloxacin/alatrofloxacin mesylate). http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/drugsafetyinformationforheathcareprofessionals/publichealthadvisories/ucm053103 (accessed June 6, 2011).
FDA. 2000a. Drugs@FDA: Lotronex (alosteron hydrochloride). 02/09/2000 Approval Letter. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails (accessed March 7, 2012).
FDA. 2000b. Questions and answers about Lotronex. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110887.htm (accessed March 7, 2012).
FDA. 2002. Questions and answers about Lotronex. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110859.htm (accessed March 7, 2012).
FDA. 2004. Pharmaceutical cGMPs for the 21st century—a risk-based approach (final report). Washington, DC: Department of Health and Human Services.
FDA. 2005a. Joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee. Paper read at Joint Meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee, Hilton Gaithersburg.
FDA. 2005b. FDA news release: FDA issues public health advisory on Tysabri, a new drug for MS. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2005/ucm108413.htm (accessed July 7, 2011).
FDA. 2007a. Transcript of FDA press conference on Trasylol. November 5, 2007. http://www.fda.gov/NewsEvents/Newsroom/MediaTranscripts/ucm122244.htm (accessed August 23, 2011).
FDA. 2007b. Avandia (rosiglitazone maleate) label revision. Supplement number 028, 08/14/2007. http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021071s028lbl.pdf (accessed March 17, 2012).
FDA. 2008a. Manufacturer removes remaining stocks of Trasylol: Access limited to investigational use. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116895.htm (accessed March 20, 2012).
FDA. 2008b. FDA alerts health care providers to risk of suicidal thoughts and behavior with antiepileptic medications. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100200.htm (accessed June 15, 2011).
FDA. 2008c. Statistical review and evaluation: Antiepileptic drugs and suicidality. Washington, DC: Department of Health and Human Services.
FDA. 2008d. Transcript. Joint meeting of the Peripheral and Central Nervous System Drugs Advisory Committee (PCNS) and the Psychopharmacologic Drugs Advisory Committee (PDAC). Beltsville, MD: Department of Health and Human Services.
FDA. 2008e. Meridia (sibutramine hydrochloride monohydrate) label revision. Supplement number 030, 05/29/2008. http://www.fda.gov/downloads/Drugs/DrugSafety/PublicHealthAdvisories/UCM130745.pdf (accessed March 7, 2012).
FDA. 2008f. Guidance for industry. Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Washington, DC: Department of Health and Human Services.
FDA. 2009a. Suicidal behavior and ideation and antiepileptic drugs. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm074939.htm (accessed January 24, 2012).
FDA. 2009b. Lotronex (alosetron hydrochloride) information. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110450.htm (accessed March 2012).
FDA. 2009c. Guidance for industry: Drug-induced liver injury. Premarketing clinical evaluation. Washington, DC: Department of Health and Human Services.
FDA. 2010a. Natalizumab (marketed as Tysabri) questions and answers. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm107202.htm (accessed March 20, 2012).
FDA. 2010b. FDA drug safety communication: Risk of progressive multifocal leukoencephalopathy (PML) with the use of Tysabri (natalizumab). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm199872.htm#_Ref252346632 (accessed March 20, 2012).
FDA. 2010c. Decision on continued marketing of rosiglitazone (Avandia, Avandamet, Avandaryl). http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM226959.pdf (accessed March 20, 2012).
FDA. 2011a. Office of Special Health Issues. http://www.fda.gov/AboutFDA/CentersOffices/OC/OfficeofExternalAffairs/OfficeofSpecialHealthIssues/default.htm (accessed February 18, 2012). FDA. 2011b. Guidance for industry warnings and precautions, contradictions, and boxed warning sections of labeling for human prescription drug and biological products-content and format. Washington, DC: US Food and Drug Administration.
FDA. 2011c. Driving biomedical innovation: Initiatives for improving products for patients. Washington, DC: US Food and Drug Administration.
FDA. 2011d. Communicating risks and benefits: An evidence-based user’s guide. Edited by B. Fischhoff, N. Brewer and J. S. Downs. Washington, DC: Department of Health and Human Services.
FDA. 2011e. FDA drug safety communication: Safety review of post-market reports of serious bleeding events with the anticoagulant Pradaxa (dabigatran etexilate mesylate). http://www.fda.gov/Drugs/DrugSafety/ucm282724.htm#sa (accessed March 7, 2012).
FDA. 2011f. Guidance for industry postmarketing studies and clinical trials—implementation of section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act. Washington, DC: Department of Health and Human Services.
FDA. 2012a. PDUFA reauthorization performance goals and procedures fiscal years 2013 through 2017. Washington, DC: US Food and Drug Administration.
FDA. 2012b. FDA transparency initiative. http://www.fda.gov/AboutFDA/Transparency/TransparencyInitiative/default.htm (accessed March 20, 2012).
Fergusson, D. A., P. C. Hebert, C. D. Mazer, S. Fremes, C. MacAdams, J. M. Murkin, K. Teoh, P. C. Duke, R. Arellano, M. A. Blajchman, J. S. Bussieres, D. Cote, J. Karski, R. Martineau, J. A. Robblee, M. Rodger, G. Wells, J. Clinch, and R. Pretorius. 2008. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. New England Journal of Medicine 358(22):2319-2331.
Fiessinger, J. N., M. V. Huisman, B. L. Davidson, H. Bounameaux, C. W. Francis, H. Eriksson, T. Lundstrom, S. D. Berkowitz, P. Nystrom, M. Thorsen, and J. S. Ginsberg. 2005. Ximelagatran vs low-molecular-weight heparin and warfarin for the treatment of deep vein thrombosis: A randomized trial. JAMA 293(6):681-689.
Fischhoff, B. 2009. Risk perception and communication. In Oxford textbook of public health. Fifth ed., edited by R. Detels, R. Beaglehole, M. A. Lansang and M. Gulliford. Oxford: Oxford University Press. Pp. 940-952.
Fischhoff, B. 2010. Judgment and decision making. Wiley Interdisciplinary Reviews: Cognitive Science 1(5):724-735.
Fleming, T. R., and D. L. DeMets. 1996. Surrogate end points in clinical trials: Are we being misled? Annals of Internal Medicine 125(7):605-613.
Fung, M. 2001. Evaluation of the characteristics of safety withdrawal of prescription drugs from worldwide pharmaceutical markets—1960 to 1999. Drug Information Journal 35(1):293-317.
GAO (Government Accountability Office). 2009a. Drug safety: FDA has begun efforts to enhance postmarket safety, but additional actions are needed. Washington, DC: Government Accountability Office.
GAO. 2009b. New drug approval: FDA needs to enhance its oversight of drugs approved on the basis of surrogate endpoints. Washington, DC: Government Accountability Office.
Ginnelly, L., K. Claxton, M. J. Sculpher, and S. Golder. 2005. Using value of information analysis to inform publicly funded research priorities. Applied Health Economics and Health Policy 4(1):37-46.
Greenberg, H. B. 2011. Rotavirus vaccination and intussusception—act two. New England Journal of Medicine 364(24):2354-2355.
Hamburg, M. A. 2011. Commentary: The growing role of epidemiology in drug safety regulation. Epidemiology 22(5):622-624.
Hamburg, M. A., and J. M. Sharfstein. 2009. The FDA as a public health agency. New England Journal of Medicine 360(24):2493-2495.
Health Canada. 2000. Health Canada decision-making framework for identifying, assessing, and managing health risks. Ottawa: Health Canada.
Hemminki, E., H. K. Toiviainen, and L. Vuorenkoski. 2010. Co-operation between patient organizations and the drug industry in Finland. Social Science & Medicine 70(8):1171-1175.
Henry, D. A., A. J. Moxey, P. A. Carless, D. O’Connell, B. McClelland, K. M. Henderson, K. Sly, A. Laupacis, and D. Fergusson. 2001. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database System Review(1): CD001886.
Hiatt, W. R. 2006. Observational studies of drug safety—aprotinin and the absence of transparency. New England Journal of Medicine 355(21):2171-2173.
IAF (Institute for Alternative Futures). 2010. Optimal futures for Risk Evlauation and Mitigation Strategies (REMS)—workshop report. Alexandria, VA: Institute for Alternative Futures and Society for Women’s Health Research.
IOM (Institute of Medicine). 2007a. The future of drug safety: Promoting and protecting the health of the public. Washington, DC: The National Academies Press.
IOM. 2007b. Understanding the benefits and risks of pharmaceuticals: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2010. Evaluation of biomarkers and surrogate endpoints in chronic disease. Washington, DC: The National Academies Press.
IOM. 2011a. Finding what works in health care: Standards for systematic reviews. Washington, DC: The National Academies Press.
IOM. 2011b. Advancing regulatory science for medical countermeasure development: Workshop summary. Washington, DC: The National Academies Press.
James, W. P., I. D. Caterson, W. Coutinho, N. Finer, L. F. Van Gaal, A. P. Maggioni, C. Torp-Pedersen, A. M. Sharma, G. M. Shepherd, R. A. Rode, and C. L. Renz. 2010. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. New England Journal of Medicine 363(10):905-917.
Jonas, E., S. Schulz-Hardt, D. Frey, and N. Thelen. 2001. Confirmation bias in sequential information search after preliminary decisions: An expansion of dissonance theoretical research on selective exposure to information. Journal of Personality and Social Psychology 80(4):557-571.
Jones, K. 2008. In whose interest? Relationships between health consumer groups and the pharmaceutical industry in the UK. Sociology of Health & Illness 30(6):929-943.
Karkouti, K., W. S. Beattie, K. M. Dattilo, S. A. McCluskey, M. Ghannam, A. Hamdy, D. N. Wijeysundera, L. Fedorko, and T. M. Yau. 2006. A propensity score case-control comparison of aprotinin and tranexamic acid in high-transfusion-risk cardiac surgery. Transfusion 46(3):327-338.
Kastelein, J. J. P., F. Akdim, E. S. G. Stroes, A. H. Zwinderman, M. L. Bots, A. F. H. Stalenhoef, F. L. J. Visseren, E. J. G. Sijbrands, M. D. Trip, E. A. Stein, D. Gaudet, R. Duivenvoorden, E. P. Veltri, A. D. Marais, and E. de Groot. 2008. Simvastatin with or without ezetimibe in familial hypercholesterolemia. New England Journal of Medicine 358(14):1431-1443.
Kessler, D. A., and D. C. Vladeck. 2008. A critical examination of the FDA’s efforts to preempt failure-to-warn claims. Georgetown Law Journal 96:1-47.
Lasser, K. E., D. L. Seger, D. T. Yu, A. S. Karson, J. M. Fiskio, A. C. Seger, N. R. Shah, T. K. Gandhi, J. M. Rothschild, and D. W. Bates. 2006. Adherence to black box warnings for prescription medications in outpatients. Archives of Internal Medicine 166(3):338-344.
Law, M. R., N. J. Wald, and A. R. Rudnicka. 2003. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: Systematic review and meta-analysis. BMJ 326(7404):1423.
Law, M. R., J. K. Morris, and N. J. Wald. 2009. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: Meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ 338:1245-1261.
Levi, M., M. E. Cromheecke, E. de Jonge, M. H. Prins, B. J. de Mol, E. Briet, and H. R. Buller. 1999. Pharmacological strategies to decrease excessive blood loss in cardiac surgery: A meta-analysis of clinically relevant endpoints. Lancet 354(9194):1940-1947.
Lofgren, H. 2004. Pharmaceuticals and the consumer movement: the ambivalences of ‘patient power’. Australian Health Review 28(2):228-237.
Mangano, D. T., I. C. Tudor, and C. Dietzel. 2006. The risk associated with aprotinin in cardiac surgery. New England Journal of Medicine 354(4):353-365.
McClellan, M. 2007. Drug safety reform at the FDA—Pendulum swing or systematic improvement? New England Journal of Medicine 356:1700-1702.
Mendel, R., E. Traut-Mattausch, E. Jonas, S. Leucht, J. M. Kane, K. Maino, W. Kissling, and J. Hamann. 2011. Confirmation bias: Why psychiatrists stick to wrong preliminary diagnoses. Psychological Medicine 41:2651-2659.
Miksad, R. A., M. Gonen, T. J. Lynch, and T. G. Roberts, Jr. 2009. Interpreting trial results in light of conflicting evidence: A Bayesian analysis of adjuvant chemotherapy for non-small-cell lung cancer. Journal of Clinical Oncology 27(13):2245-2252.
Nissen, S. E., and K. Wolski. 2007. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356(24):2457-2471.
Nissen, S. E., J. C. Tardif, S. J. Nicholls, J. H. Revkin, C. L. Shear, W. T. Duggan, W. Ruzyllo, W. B. Bachinsky, G. P. Lasala, and E. M. Tuzcu. 2007. Effect of torcetrapib on the progression of coronary atherosclerosis. New England Journal of Medicine 356(13):1304-1316.
NRC (National Research Council). 1983. Risk assessment in the federal government: Managing the process. Washington, DC: National Academy Press.
NRC. 1989. Improving risk communication. Washington, DC: National Academy Press.
NRC. 1994. Science and judgment in risk assessment. Washington, DC: National Academy Press.
NRC. 1996. Understanding risk: Informing decisions in a democratic society. Washington, DC: National Academy Press.
NRC. 2009. Science and decisions: Advancing risk assessment. Washington, DC: The National Academies Press.
NRC. 2011. A risk-characterization framework for decision-making at the Food and Drug Administration. Washington, DC: The National Academies Press.
O’Reilly, J. T. 2008. Losing deference in the FDA’s second century: Judicial review, politics, and a diminished legacy of expertise. Cornell Law Rev 93(5):939-980.
Patel, M. M., V. R. López-Collada, M. M. Bulhões, L. H. De Oliveira, A. B. Márquez, B. Flannery, M. Esparza-Aguilar, E. I. Montenegro Renoiner, M. E. Luna-Cruz, H. K. Sato, L. d. C. Hernández-Hernández, G. Toledo-Cortina, M. Cerón-Rodríguez, N. Osnaya-Romero, M. Martínez-Alcazar, R. G. Aguinaga-Villasenor, A. Plascencia-Hernández, F. Fojaco-González, G. Hernández-Peredo Rezk, S. F. Gutierrez-Ramírez, R. Dorame-Castillo, R. Tinajero-Pizano, B. Mercado-Villegas, M. R. Barbosa, E. M. C. Maluf, L. B. Ferreira, F. M. de Carvalho, A. R. dos Santos, E. D. Cesar, M. E. P. de Oliveira, C. L. Osterno Silva, M. de los Angeles Cortes, C. Ruiz Matus, J. Tate, P. Gargiullo, and U. D. Parashar. 2011. Intussusception risk and health benefits of rotavirus vaccination in Mexico and Brazil. New England Journal of Medicine 364(24):2283-2292.
PhRMA (Pharmaceutical Research and Manufacturers of America). 2012. PhRMA transitions management of benefit-risk action team framework to CIRS. http://www.phrma.org/media/releases/phrma-transitions-management-benefit-risk-action-team-framework-cirs (accessed February 18, 2012).
Pidgeon, N., and B. Fischhoff. 2011. The role of social and decision sciences in communicating uncertain climate risks. Nature Climate Change 1:35-41.
Prentice, R. L. 1989. Surrogate endpoints in clinical trials: Definition and operational criteria. Statistics in Medicine 8(4):431-440.
Presidential/Congressional Commission on Risk Assessment and Risk Management. 1997. Risk assessment and risk management in regulatory decision-making. Final report. Volume 2. http://www.riskworld.com/Nreports/1997/risk-rpt/volume2/pdf/v2epa.PDF (accessed September 15, 2008).
Psaty, B. M., N. S. Weiss, C. D. Furberg, T. D. Koepsell, D. S. Sisčovick, F. R. Rosendaal, N. L. Smith, S. R. Heckbert, R. C. Kaplan, D. Lin, T. R. Fleming, and E. H. Wagner. 1999. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA 282(8):786-790.
Raiffa, H. 1968. Decision analysis. In Tools for making acute risk decisions: With chemical process safety applications. Hoboken, NJ: John Wiley & Sons, Inc.
Ray, W. A., and C. M. Stein. 2006. Reform of drug regulation—beyond an independent drug-safety board. New England Journal of Medicine 354(2):194-201.
Richardson, W. S., M. C. Wilson, J. Nishikawa, and R. S. Hayward. 1995. The well-built clinical question: A key to evidence-based decisions. ACP Journal Club 123(3):A12-13.
Rossebo, A. B., T. R. Pedersen, K. Boman, P. Brudi, J. B. Chambers, K. Egstrup, E. Gerdts, C. Gohlke-Bärwolf, I. Holme, Y. A. Kesäniemi, W. Malbecq, C. A. Nienaber, S. Ray, T. Skjærpe, K. Wachtell, and R. Willenheimer. 2008. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. New England Journal of Medicine 359(13):1343-1356.
Rothman, S. M., V. H. Raveis, A. Friedman, and D. J. Rothman. 2011. Health advocacy organizations and the pharmaceutical industry: an analysis of disclosure practices. American Journal of Public Health 101(4):602-609.
Schneeweiss, S., J. D. Seeger, J. Landon, and A. M. Walker. 2008. Aprotinin during coronary-artery bypass grafting and risk of death. New England Journal of Medicine 358(8):771-783.
Sedrakyan, A., T. Treasure, and J. A. Elefteriades. 2004. Effect of aprotinin on clinical outcomes in coronary artery bypass graft surgery: A systematic review and meta-analysis of randomized clinical trials. The Journal of Thoracic and Cardiovascular Surgery 128(3):442-448.
Selvin, E., S. Marinopoulos, G. Berkenblit, T. Rami, F. L. Brancati, N. R. Powe, and S. H. Golden. 2004. Meta-analysis: Glycosylated hemoglobin and cardiovascular disease in diabetes mellitus. Annals of Internal Medicine 141(6):421-431.
Siegel, J. P. 2002. Assessing the use of activated protein C in the treatment of severe sepsis. New England Journal of Medicine 347(13):1030-1034.
Steel-Fisher, G. K., R. J. Blendon, M. M. Bekheit, and K. Lubell. 2010. The public’s response to the 2009 H1N1 influenza pandemic. New England Journal of Medicine 362(22):e65(1)-e65(6).
Straus, S. E., P. Paul Glasziou, W. S. W. Scott Richardson, and R. B. R. Brian Haynes. 2010. Evidence-based medicine: How to practice and teach it, 4th edition. Edinburgh: Churchill Livingstone.
Strickland, A. A., C. S. Taber, and M. Lodge. 2011. Motivated reasoning and public opinion. Journal of Health Politics, Policy and Law 36(6):935-944.
Temple, R. 1999. Are surrogate markers adequate to assess cardiovascular disease drugs? JAMA 282(8):790-795.
Temple, R., and G. W. Pledger. 1980. The FDA’s critique of the anturane reinfarction trial. New England Journal of Medicine 303(25):1488-1492.
Tonkin, A. M., and L. Chen. 2010. Effects of combination lipid therapy in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 122(8):850-852.
Transparency Task Force, HHS, and FDA. 2010. FDA transparency initiative: Draft proposals for public comments regarding disclosure policies of the U.S. Food and Drug Administration. Washington, DC: Department of Health and Human Services.
Wilson, P. W. 2008. Cardiovascular risk assessment—where are we going? Preventive Cardiology 11(3):183-184.
Woodcock, J., J. M. Sharfstein, and M. Hamburg. 2010. Regulatory action on rosiglitazone by the U.S. Food and Drug Administration. New England Journal of Medicine 363(16):1489-1491.
Yu, D. T., D. L. Seger, K. E. Lasser, A. S. Karson, J. M. Fiskio, A. C. Seger, and D. W. Bates. 2011. Impact of implementing alerts about medication black-box warnings in electronic health records. Pharmacoepidemiology and Drug Safety 20(2):192-202.




























































