
SCIENTIFIC TOOLS TO AID CANCER COMBINATION THERAPY DEVELOPMENT
NCI recently developed scientific tools that can aid cancer combination therapy development, including proof-of-concept assays for experimental agents, microarrays for testing drug combinations, an epigenetic toolbox to learn more about the biology of cancer, and an NCI drug repository that provides drugs for testing of combinations.
Mechanism-of-Action Assays
NCI is currently developing more than 50 assays for evaluating the mechanisms of action of molecularly targeted agents. Once validated, these assays will be made available to the research community at no charge. These tools include assays for many of the molecular mechanisms related to cancer, such as activation of various tyrosine kinases and oncogenes, DNA damage, and apoptosis. Some of these assays detect more than one molecular mechanism simultaneously, including an assay that has antibodies that detect all the different phosphorylation sites on MET. NCI is also funding researchers to develop multiplex assays appropriate for use in the clinic with a single biopsy. These multiplex assays could be used, for example, to assess two or more molecular pathways for drug resistance.
In addition, NCI has developed “combo plates” specially formatted for researchers to use in testing new compounds in combination with
commercially available anticancer drugs (Mayfield, 2011). A plated set of about 100 FDA-approved oncology drugs is now available without charge to the research community. Information about this resource and how to obtain it can be found on the Division of Cancer Treatment and Diagnosis Developmental Therapeutics Program website.1
Preclinical Models for Combinations
To assess the effects of combining anticancer agents on tumor growth inhibition, NCI’s toxicogenomics program is testing 5,000 unique combinations for the 100 commercially available anticancer drugs across many clinically relevant concentrations on the NCI-60 panel, which includes 60 human tumor cell lines.
For about 10 percent of the combinations, some synergistic effects appear to be greater than the additive effects alone. The researchers are trying to confirm synergistic activity in a variety of different xenograft animal models. Some of the antagonistic or additive effects that have been observed were unpredicted, Dr. Doroshow noted. For example, some cell lines that are insensitive to the individual agents used in the combination, such as dasatinib and 6-MP, are sensitive when the agents are used in combination. “This shows us that there are many things about the drugs, old and new, that we think we know and, in fact, we don’t,” Dr. Doroshow said. “Such systematic screening will provide, we hope, novel information for the investigative community to help us understand how to put some of these agents together.”
Dr. Doroshow estimated that all 5,000 combinations will be evaluated by the end of 2011, at which time the data will be made publicly available on the NCI website. NCI scientists are also modeling the effectiveness of combining investigational agents with approved agents. Furthermore, they are exploring combinations of 300 investigational agents in a variety of concentrations (Mayfield, 2011). Dr. Blackman suggested researchers correlate the findings from NCI’s combination screening program to clinical data on such combinations to assess how predictive the assays are. Dr. Doroshow responded that it certainly can be done across the NCI database of Phase II trials. Dr. Amy Abernethy, director of the Duke Cancer Care Research Program, suggested testing combinations of oncology and non-oncology commercially available drugs for their antitumor effects, and Dr. Doroshow responded, “Repurposing non-oncologic drugs is not something we are doing, but is something of significant interest to Dr.
![]()
1 See http://dtp.nci.nih.gov/branches/dscb/oncology_drugset_explanation.html (accessed December 14, 2011).
[Francis] Collins and to the NIH, and activities are under way to do this.” Dr. Bachman said that GSK is also exploring such drug combinations.
Investigational Agents Available for Preclinical Testing
Until recently, Dr. Doroshow said, the Department of Health and Human Services’ Office of General Counsel prohibited NCI from purchasing or synthesizing patented agents for research purposes. But that policy changed in 2009, and NCI has acquired more than 300 investigational agents anticancer potential for in vitro testing, including multiple representatives of each class. A program is being established to allow investigators to submit requests for in vitro studies of the effects of specific combinations of these investigational agents (Mayfield, 2011). Dr. Doroshow said NCI can supply these compounds to NCI intramural scientists and to its contractors. But at the present time, the Office of General Counsel at the NIH prohibits them from sending these agents to extramural investigators. He noted, however, that NCI does have the repository space and other resources, expertise, and willingness to provide these compounds to extramural investigators if the legal issues prohibiting this can be overcome.
I-SPY 2 TRIAL2
The I-SPY 2 TRIAL is a Phase II multisite clinical trial testing multiple experimental drugs while simultaneously assessing the effectiveness of various biomarkers to predict response to the investigational agents. The trial was launched on March 17, 2010. The I-SPY 2 TRIAL builds on I-SPY 1 TRIAL,3 which was designed to evaluate neoadjuvant chemotherapy in patients with locally advanced breast cancer, and brought together data from multiple molecular biomarker studies and biomedical imaging (Barker et al., 2009).
In the I-SPY 2 TRIAL, 800 patients with locally advanced breast cancer will have their tumor biopsies characterized by a panel of biomarkers, some of which are established and approved and some of which are exploratory or need to be qualified. The results from these biopsies will be used to divide the patients into different groups that will receive 1 or combinations of 12 experimental drugs and/or standard drug therapy
![]()
2 Information on the I-SPY 2 TRIAL is from Extending the Spectrum of Precompetitive Collaboration in Oncology Research: Workshop Summary (IOM, 2010a) and Dr. Wholley’s presentation on June 14, 2011.
3 The I-SPY 1 TRIAL was a collaboration involving NCI’s Specialized Programs of Research Excellence, the American College of Radiology Imaging Network, Cancer and Leukemia Group B, and NCI’s Center for Biomedical Informatics and Information Technology.
prior to surgery. Using biomedical imaging, the effect on the tumor will be measured at four points during the 6 months that patients receive treatment, and when the tumor is removed. The patients will then be followed for 5 years.
This innovative study uses an adaptive trial design to enable researchers to use early data from one set of patients to guide decisions about which treatments might be more useful for patients later in the trial. The study design also enables drugs to be dropped quickly from the trial if they are ineffective or harmful (FNIH, 2010). Tumor response is also assessed by biomarker category. If the data indicate drugs are not improving the tumor response in patients with particular biomarkers, patients with those biomarkers will be assigned other drugs.
In addition, the study design allows drugs to be graduated to Phase III trials sooner if they are shown to be beneficial. Once drugs graduate to Phase III testing or are dropped, new drugs will seamlessly be entered into the trial to take their place.
Promising data on biomarkers in I-SPY 2 TRIAL can be used to support an application for Premarket Approval at FDA or to request to use a biomarker to stratify patients in a Phase III validation study.
The trial is testing the most promising drugs by class across many companies, each of which is contributing the experimental agents. The unique structure of the trial and the multiple companies involved in it, however, create numerous challenges, especially in the regulatory arena. Usually multiple drugs and biomarkers require multiple trials, each with its own IND. Even when a drug is successful in the first phase of testing, the trial has to be stopped and a new one created to continue testing in the next phase. This is extremely time consuming and inefficient. To speed up the process, the Biomarkers Consortium, trial organizers, and FDA worked together to develop a plan in which the master IND being used by the trial is held by FNIH, who manages the Biomarkers Consortium along with several other large biomedical partnerships. FNIH was chosen because it was seen as a trusted, neutral third party that can sponsor and manage the trial fairly and effectively.
In addition, the initial five experimental agents that will be used in the trial were approved for testing purposes by FDA and the relevant IRBs before the trial started. Other agents that will be evaluated in the I-SPY 2 TRIAL (there will be as many as 12, contributed by more than 6 different companies) will be submitted to FDA and IRBs for approval for testing purposes as the trial progresses so that by the time investigators are ready to add new agents to the trial, they will be ready to enter. Each time a new agent is added to the trial, an appendix is added rather than changing the protocol. An effort was made to involve all the stakeholders from all the sites as early as possible. For example, in preparation for IRB
approval, 45 key stakeholders were brought together for education and feedback. This changed a traditionally long linear process, with consecutive approvals by various participants and inefficient reapproval loops, to a more streamlined team effort.
No single company stands to be the sole beneficiary of the I-SPY 2 TRIAL. The intellectual property resulting from the trial will be handled according to the Biomarkers Consortium policies:
• Preexisting IP related to agents contributed by companies will remain with the company owning that IP;
• Preexisting IP related to biomarkers and platforms will remain with the inventing companies, and be licensed for use in the project. In some cases the tests have been published and are available commercially;
• New IP will be managed by FNIH, acting as a trusted third party to hold and license the new inventions. FNIH will return a fair share of royalties (less expenses) to inventing organizations;
• FNIH prosecutes and manages resulting patents; and
• Data are expected to be broadly applicable and available as quickly as possible.
Institutions participating in the I-SPY 2 TRIAL use common data elements and a shared information technology infrastructure, which employs tools provided by caBIG.4 Within the caGRID, the underlying architecture of caBIG, the I-SPY 2 TRIAL is leveraging several bioinformatics platforms, including caTISSUE, caARRAY, and caIntegrator. Access to the data is democratized and credit is shared.
The I-SPY 2 TRIAL is expected to cost approximately $26 million over 5 years (FNIH, 2010). Some funding secured for the trial includes contributions from Eli Lilly, Genentech, Johnson & Johnson, and Safeway, Inc. FNIH is working to raise the remaining funding from pharmaceutical and other companies, nonprofit cancer organizations, and philanthropic foundations and individuals. Only some pharmaceutical companies that have funded the I-SPY 2 TRIAL are participating in the trial. As Mr. Wholley noted, “there is no pay for play around the selection of the agents. Fund-raising is separated from the contribution of the agents.” An independent agent selection committee consisting of oncologists without conflict of interests chooses which agents are tested in the trial, based on rigorous scientific criteria.
![]()
4 caBIG stands for the cancer Biomedical Informatics Grid, an information network that enables members of the cancer community to share data and knowledge. See http://cabig.nci.nih.gov (accessed December 14, 2011).
BATTLE TRIAL5
The objective of the BATTLE trial is to use biopsy tissue from lung cancer patients in real time to suggest the best treatments they should receive for their tumors. Similar to the I-SPY 2 TRIAL, BATTLE aims to treat patients more effectively with a personalized medicine approach while simultaneously discovering and validating biomarkers. As Dr. Herbst explained, BATTLE, which is funded by the Department of Defense, is a platform for translational research for testing three hypotheses:
• Real-time biopsies can more accurately reflect the aberrant signaling pathways of lung cancer;
• Matching targeted agents with abnormal pathways will improve disease control in lung cancer patients; and
• Eight-week disease control is an acceptable surrogate for efficacy in patients with pretreated lung cancer.
BATTLE 1 began in 2007 at MD Anderson Cancer Center. It consisted of four adaptive trial designs, and was available to all lung cancer patients; the only prerequisite was a core biopsy of their tumor. Following biomarker analysis of their tumor sample under an umbrella protocol, patients were adaptively randomized to one of four treatments with targeted cancer therapies, including one treatment which was a combination of two agents, and one treatment that was a multitargeted inhibitor. Because of the involvement of different research groups and pharmaceutical companies, each treatment had its own separate Phase II clinical trial. Initial published results of BATTLE 1 (Kim et al., 2010, 2011) suggest that lung cancer patients “are going to do differently, not only based on having a mutation, but the specific type of mutation and the correlate for that is it’s probably affecting different signaling pathways,” Dr. Herbst said. He and his colleagues continue to mine the frozen tumor tissue they collected to discover new biomarkers.
BATTLE 2, which became active in 2011, uses a similar protocol to BATTLE 1, but has some improvements, including the use of fine needle aspirations prior to core biopsies to ensure adequate tumor cells are accessed (see Figure A-1). This trial is being conducted at MD Anderson and Yale Cancer Centers in collaboration with Merck, AstraZeneca, and Bayer/Onyx, who are providing the five agents that will be tested in four treatments, two of which are two-agent combinations.
Dr. Herbst estimated that BATTLE 2 costs $20,000 per patient, not
![]()
5 Information on the BATTLE trial is from Dr. Herbst’s presentation (June 14, 2011) and Dr. Papadimitrakopoulou’s presentation (June 14, 2011).
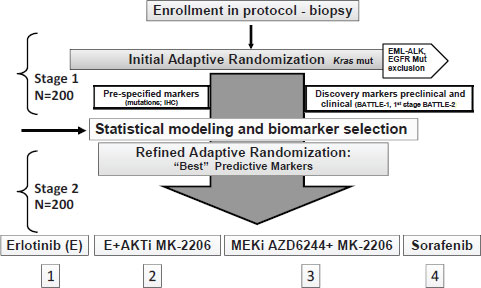
NOTE: EGFR = epidermal growth factor receptor, EML-ALK = echinoderm microtubule-associated protein-like/anaplastic lymphoma kinase fusion gene, IHC = immunohistochemistry, mut = mutation.
SOURCE: Papadimitrakopoulou presentation (June 14, 2011).
including the $7–$8 million infrastructure costs that support it. Dr. Papadimitrakopoulou stressed the complexity of BATTLE 2 and the numerous steps it took to develop the study, which can be seen in Figure A-2.
For this trial, researchers had to forge three three-way IP agreements and four contracts between MD Anderson Cancer Center and pharmaceutical sponsors, as well as submit four NCI grant applications (initial PO1 and resubmission, initial RO1 and resubmission). Protocol development and the first IRB approval were achieved in July 2009, followed by 10 protocol revisions and approved amendments, based on recent clinical trial results and the evolution of scientific knowledge. The trial was activated in June 2011.
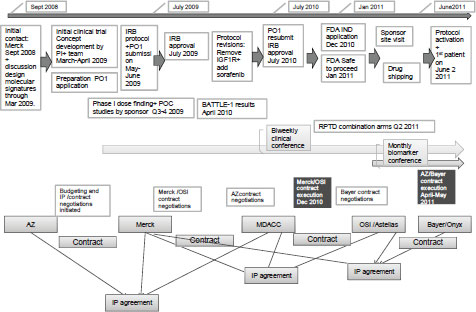
NOTE: AZ = AstraZeneca, FDA = Food and Drug Administration, IGFR1 = insulin-like growth factor receptor 1, IND = investigational new drug, IRB = institutional review board, IP = intellectual property, MDACC = MD Anderson Cancer Center, OSI = OSI Pharmaceuticals, PI = principal investigator, POC = proof of concept, RPTD = recommended phase treatment dose.
SOURCE: Papadimitrakopoulou presentation (June 14, 2011).
STAND UP TO CANCER PI3K TEAM6
Stand Up To Cancer funds an innovative platform of preclinical and clinical development of agents that target PI3K in women’s cancers. The team responsible for this platform come from eight academic institutions and cancer centers and includes pathologists, biomarker experts, clinicians, mathematical modelers, biostatisticians, and cell-based assay experts, as well as patient advocates. The treatments they are developing
![]()
6 Information on the Stand Up To Cancer PI3K Team is from Dr. Cantley’s presentation (June 14, 2011).
all target PI3K because it is mutated frequently in breast, endometrial, and ovarian cancers, on which they are focused.
The PI3K research team is exploring combinations of targeted agents in animal models and using those results combined with biomarker analyses of tumor biopsies to guide selection of patients into clinical trials that are supported by the same Stand Up To Cancer program. For example, Dr. Cantley pointed out that single-agent therapy with a PI3K inhibitor or another type of drug he called “Z” has not been effective in his animal models of breast cancer, but the combination has produced cures in a mouse model. “When the two are used together you get a dramatic effect—we can actually take the drug away and these tumors don’t come back—but if you looked at either one of these [alone], you wouldn’t get excited at all.” He added that the combination is well tolerated and he hasn’t had to reduce the dose of either drug to have the combination work.
“Our animal models are really what are driving our hypotheses,” said Dr. Cantley. These animal models involve mice genetically engineered to have mutations that are seen frequently in human cancers in combinations that mimic what occurs in the clinic. Although all the mice are genetically engineered to have an initial mutation or set of mutations, researchers assess subsequent secondary mutations that develop and these are quite heterogeneous, Dr. Cantley noted. “We’re seeing the same kind of heterogeneity that we probably see in human disease,” he said.
The genetically engineered mice are treated with the same agents being tested in the trials the investigators are designing for human patients. Such testing and genetic analysis of the animal tumors identifies resistance mechanisms, leading to hypotheses for innate or acquired resistance to PI3K inhibitors in the human trials, and suggests combination therapies to test in patients.
Patients enrolled in PI3K clinical trials are asked to provide a tumor biopsy sample at entry as well as a subsequent biopsy if the cancer progresses. These biopsies are analyzed for the same resistance mutations seen in the mouse models, and are used to guide which experimental therapy patients receive. The trials incorporate novel imaging approaches, such as functional quantitative imaging before and a few weeks after initiation of the drugs, to more quickly ascertain likely responders.
SAFE HARBORS FOR COLLABORATION
Several safe harbors have been established with the aim of fostering collaborations in the development of cancer biomarkers or drugs, including combination therapies. Organizations discussed at the workshop include the CEO Roundtable on Cancer’s Life Sciences Consortium,
the Cancer Immunotherapy Trials Network, FNIH and its Biomarkers Consortium, and the Reagan–Udall Foundation.
Life Sciences Consortium of the CEO Roundtable on Cancer7
The CEO Roundtable on Cancer was established in 2001 and consists of 17 representatives from 11 pharmaceutical companies and 26 representatives from NCI-Designated Comprehensive Cancer Centers. The Life Sciences Consortium is a task force of the Roundtable and brings together Roundtable members to further its goals, which are to:
• Develop standards across the life sciences industry to expedite the R&D process;
• Develop a pool of precompetitive intellectual property for biomarkers; and
• Diminish the regulatory burden of new cancer drug approval.
To help achieve its first goal of expediting the R&D process, the Life Sciences Consortium acted on findings that the most rate-limiting steps in the development of clinical trials were contracting and budgeting (Dilts and Sandler, 2006). To expedite the contract and budget negotiations required between industry and publicly funded investigators before the launch of a collaborative trial, the Consortium and NCI reviewed copies of 78 redacted clinical trial agreements and identified 45 key concepts related to intellectual property, study data, subject injury, indemnification, confidentiality, and publication rights. From these agreements, they then gleaned the exact language that embodied the key concepts and used it to create standardized and harmonized clauses for clinical trial agreements that are designed to serve as a starting point for contract negotiations (CEO Life Sciences Consortium and NCI, 2008). The analysis found that several key concepts showed greater than 67 percent similarity across the agreements, suggesting that negotiations frequently reach common results for these concepts. The U.S. Department of Justice gave the proposed clauses a favorable review and indicated that it had no intention to challenge the initiative (DOJ, 2008).
Nine out of eleven of the Life Sciences Consortium companies have adopted the START (Standard Terms of Agreement for Research Trial) clauses for their oncology programs, with one making it a standard operating procedure, and another using the clauses for all therapeutic areas. The Consortium plans to use the same process to develop standardized
![]()
7 Information on The Life Sciences Consortium is from IOM (2010a) and comments from Dr. Martin Murphy.
material transfer agreements for academic collaborations in the laboratory to expedite the process of preclinical development.
The Life Sciences Consortium recently began addressing its second goal of developing a pool of precompetitive intellectual property for biomarkers. It plans to work with NCI as a safe harbor for this effort because NCI currently has a robust biomarker program, according to Dr. Gregory Curt, chair of the Life Sciences Consortium and the U.S. medical science lead of emerging products at AstraZeneca-Oncology. Consortium companies will present their biomarker programs under confidentiality to NCI, which will select the most promising markers for coinvestment and collaboration. This will reduce the duplicative and expensive research that individual companies and NCI are spending on biomarker development and should, along with the START clauses, significantly reduce the amount of time needed to validate biomarkers (IOM, 2010b).
“At the CEO Roundtable, we’ve tried to create an independent safe harbor where companies can do together what otherwise it’s impossible to do, so real progress can be made,” said Dr. Martin Murphy. He added that a new initiative of the Roundtable is to give an award to the pharmaceutical company that lowers barriers to collaboration better than any other pharmaceutical company on the Roundtable. He added that often resistance to changing the culture of companies comes not from the upper echelon of the company, but rather from middle levels. “The intent is simple: To try to [emphasize] throughout the entire organization, if not now, when, and if not us, who?” Dr. Murphy said.
The Foundation for the National Institutes of Health8
FNIH was created by Congress in 1990 and incorporated in 1996 to support NIH in its mission to improve health by forming and facilitating public–private partnerships for biomedical research and training. According to the Foundation’s website, FNIH “unites experts, funding, patients and resources around common biomedical research goals identified by NIH—all in an effort to respond to the most urgent priorities in human health, both domestically and around the world. Unique in its mandate, the Foundation builds partnerships that enable ambitious, multipronged, sweeping attacks on problems that would be impossible to mount otherwise. Individuals and interests large and small can all make important contributions toward solving even the most complex health challenges” (FNIH, 2011a).
The Foundation is a non-profit, 501(c)(3) charitable organization that
![]()
8 Information about the FNIH and Biomarkers Consortium is from IOM (2010a) and Mr. Wholley’s presentation on June 14, 2011.
has raised more than $560 million in private-sector funds for more than 100 projects, including partnerships between the private sector and federal agencies such as the Biomarkers Consortium and the I-SPY 2 TRIAL. “We’re really skilled at pulling together the types of governance mechanisms that make these things work,” Mr. Wholley said.
The Biomarkers Consortium
Mr. Wholley elaborated on the Biomarkers Consortium, a project of FNIH. This consortium’s founding partners included FDA, NIH, the Centers for Medicare & Medicaid Services (CMS), the Biotechnology Industry Organization, and the Pharmaceutical Research and Manufacturers of America. The Biomarkers Consortium was prompted by the growing awareness of the importance of validated biomarkers in the success of targeted therapies. But biomarker development and validation lag far behind the development and clinical testing of the innovative treatments that depend on the biomarkers for their success. Such validation requires multiple studies with large amounts of data to ensure the integrity and reproducibility of results, and is quite expensive and time consuming. This validation is difficult to accomplish in a single institutional setting, Dr. Wholley pointed out, and thus requires partnerships and a strategic approach. In addition, there is clear direction from FDA, according to Dr. Wholley, to develop consensus across sectors with regard to validated biomarkers, and recent draft guidance from FDA (2010) cites the value of consortia in developing and validating biomarkers.
The Biomarkers Consortium was launched in 2007 to facilitate the development and validation of biomarkers using new and existing technologies in a precompetitive context. The Consortium aims to qualify biomarkers and validate the underlying analytical technologies for specific applications in diagnosing disease, predicting therapeutic response, or improving clinical practice. In the spirit of precompetitiveness, however, the Consortium will not qualify or validate biomarkers in areas that directly intersect with certain compounds being developed by a specific company.
The Consortium is expected to generate information that can inform regulatory decision making, and its results are broadly available to the entire scientific community, not just its participants. “The whole goal of the Consortium is to drive significant public health benefit,” said Mr. Wholley.
The Consortium has nearly 50 contributing members, including pharmaceutical companies, academic researchers, and numerous nonprofit organizations. The Executive Committee of the Consortium has senior
representatives from NIH, FDA, the pharmaceutical industry, FNIH, CMS, and patient advocacy groups. Steering committees for four major disease areas (cancer, immunity and inflammation, metabolic disorders, neuroscience), composed of 20 to 30 individuals each, also have equal representation from NIH, FDA, industry, and academia. These committees, along with the Executive Committee, decide what biomarker projects to pursue, and direct smaller project teams of 8 to 10 people, who also have balanced representation across all the sectors and carry out the project. Projects are approved based on their scientific merit, precompetitive quality, and feasibility.
The project plan, which is developed by both the steering committee and project team, includes governing policies for intellectual property and data sharing, confidentiality, conflict of interest, selection and award of grants and contracts, and antitrust issues, which are posted on the Internet (FNIH, 2011b). The Biomarkers Consortium has launched 12 projects aimed at validating biomarkers for cancer and metabolic disorders, as well as neuroscience biomarkers, and those linked to inflammation or immunity. “All of our projects and our governing structure is fully representative of all the stakeholders from top to bottom, including FDA, industry, NIH, academia and it’s been a pretty successful mechanism for generating these projects,” Dr. Wholley said.
Cancer Immunotherapy Trials Network (CITN)
Started in 2010, CITN is funded by NCI and the Fred Hutchinson Cancer Research Center and employs the collective expertise of academic immunologists to conduct multicenter research on agents that boost patients’ own immune systems to fight their cancer (FHCRC, 2011). By collaborating with member institutions, industry sponsors, and philanthropic foundations, CITN aims to spearhead regulatory approval of promising agents and advance the knowledge of antitumor immunity and its application in immunotherapy.
The mission of the CITN is to select, design, and conduct early-phase trials that would not otherwise be possible, using novel regimens and providing high-quality immunogenicity and biomarker data that elucidate mechanisms of immune responses to inform subsequent development pathways. CITN supplies both clinical trial facilities and increased access to agents that are on prioritized lists for testing. There are 27 member sites involved in CITN, including all the large cancer centers, and the first clinical trials are expected to be launched by the end of 2011, according to Dr. June.
Reagan–Udall Foundation
The Reagan–Udall Foundation is a potential source of support for collaborations in development and regulatory science. Created in 2007 by the FDA Amendments Act of 2007, the Foundation was designed and given statutory authority to collaborate closely with FDA on scientific priorities to advance the agency’s mission to modernize medical, veterinary, food, and cosmetic product development, thereby accelerating innovation and enhancing the safety of medical products. The Foundation collaborates or contracts with stakeholders, such as FDA, university consortia, public– private partnerships, academia, nonprofits, and industry, to efficiently and effectively advance its goals and priorities. The Foundation is currently working on regulatory issues related to developing multiple drug regimens for tuberculosis as well as identifying common mechanisms of cardiotoxicity for oncology drugs.9
![]()
9 See http://www.focr.org/component/option,com_eventlist/Itemid,41/id,27/view,details/ (accessed December 14, 2001) and http://www.gatesfoundation.org/Grants-2011/Pages/Reagan-Udall-Foundation-OPP1027026.aspx (accessed December 14, 2001).














