







































Below is the uncorrected machine-read text of this chapter, intended to provide our own search engines and external engines with highly rich, chapter-representative searchable text of each book. Because it is UNCORRECTED material, please consider the following text as a useful but insufficient proxy for the authoritative book pages.
7 Tropospheric Chemical Cycles TROPOSPHERIC CHEMISTRY AND BIOGEOCHEMICAL CYCLES BY C. C. DEEWICHE The composition of the troposphere is determined to a large extent by reactions in the biosphere that main- tain a quasi-stable tropospheric composition that would not persist in the absence of biological activity. The exist- ence of molecular oxygen and nitrogen in the atmo- sphere is the most obvious consequence of this activity. These substances are present as a result of slow reaction kinetics and are not at thermodynamic equilibrium. The concept of geochemical "cycles" of elements is not new, but only recently has the significance of biolog- ical activity in these cycling processes been appreciated. Elemental cycles developed by the atmospheric chemist, the biologist, the geochemist, and others all have differ- ent features of importance, depending on the interests of the reporting scientist. In the sections that follow we will present only a brief overview of some of these cycles to place them in perspective from the standpoint of the atmospheric chemist. Most of the elements considered here have at least one volatile component of biological origin. Most of the cy- cles reflect the alternate oxidation and reduction of com- pounds in the energy metabolism of one or another life form. The process most commonly recognized is that of the photosynthetic-respiration sequence involving car- bon and oxygen. Other reactions, such as those of nitro- gen fixation and denitrification and the reactions of sul- fur oxidation and reduction, are ancillary expressions of the primary processes involving carbon. They are gen 101 erally dependent upon the carbon cycle for their opera- tion, although the compounds of sulfur are themselves grist for a photosynthetic energy input. Most of these major cycles have been altered in some of their features on the global scale by a factor of 2 or more as the result of human activity. Fossil fuel burning, although only 10 percent of respiration as a source of . · _ . atmospheric (~)2, gives an annual increment of about 0.3 percent. Industrial nitrogen fixation and the use of legumes have about equaled "natural" nitrogen f~xa- tion, and sulfur from fossil fuel combustion and mining activities has about equaled the natural sources of atmo- spheric sulfur. Other processes, such as erosion of soil and the injection of some heavy metals like lead into the atmosphere, probably have altered natural cycles even more. The closeness with which these cycles are coupled frequently is not appreciated in attempting to predict the consequence of their perturbation by human or other influences. Fundamentally, this coupling has its source in the energy demands of living organisms. The total system is drained of all the energy extractable from any reaction that can yield energy in significant amounts, and so tends to move toward a median energy level, expressed otherwise by Lovelock and Margulis in their treatment of the Gaia hypothesis. Many of the compartments involved in biogeochemi- cal cycles are shown in Figure 7.1. For our purposes, we
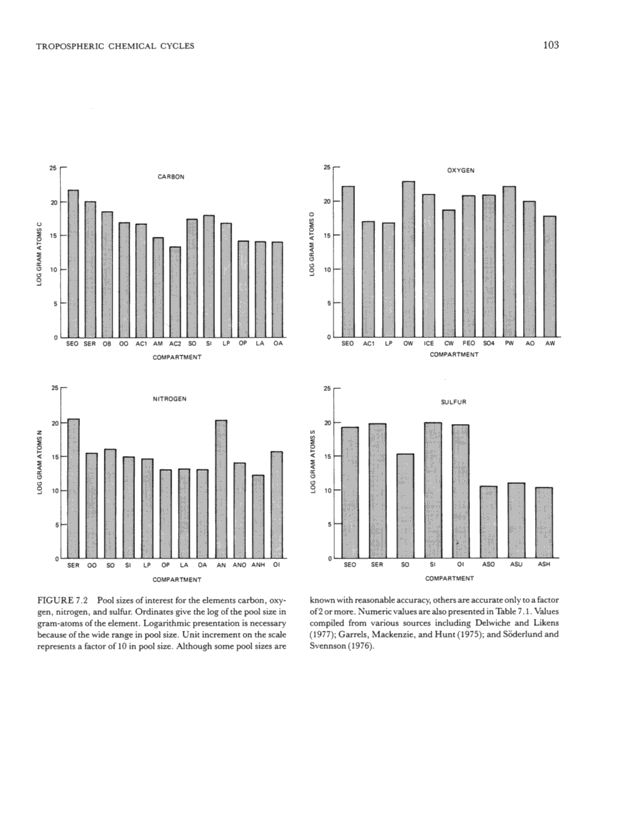
102 FIGURE 7.1 Diagrammatic representa- tior~ of major compartments of biogeo- chem~cal cycles as discussed in the text. Although movement of oceanic plates, vol- canic activity, and many other processes involved in these cycles are discontinuous, they are treated as steady-state processes for the purpose of developing mean estimates of their significance. Continental Plate will not consider long-term (hundreds of thousands of years) cycle features, such as the sedimentary cycle or processes of subduction and subsequent volatilization through volcanism, except as the results of these proc- esses contribute to the annual flux of a given element. For comparison, Figure 7.2 and Tables 7.1 and 7.2 give the distribution of four of the primary elements of inter- est (carbon, nitrogen, oxygen, and sulfur) between vari- ous "compartments" or "pools" in the environment and the estimated rates of transfer between them. It is important to remember that models of this type are intended as thinking tools, giving only the best estimates of the magnitude of the fluxes and burdens. Uncertain- ties of a factor of 2 or more are not unusual, and only in a secondary way is this uncertainty important to the anal- ysis of problems or to planning. A number of features have been omitted from Figure 7.2 for the sake of clarity. For example, the large pool of volatiles in magma is not considered except for an indi- cation of volcanic sources where appropriate. Most of these volcanic sources are assumed to be the return of volatiles subducted with sediments, but some probably are truly "juvenile, " representing an out-gassing of the magma that has been taking place (at a diminishing rate) since the earth was formed. The magnitude of this juvenile source relative to the recycling of subducted materials is controversial and not pertinent to the argu- ments we explore here. Several points are evident from an examination of this table: ~ Land Plants I | Sediments | ~ _ . . ~ . I Land A - als | 1 1 - Subduction Wedge _: - / - PART II ASSESSMENTS OF CURRENT UNDERSTANDING | Atmosphere | Ocean Plate 1. The major pools of the various elements are a function of their chemistry. Most of the nitrogen is in a partially "reduced" form in the atmosphere; most ofthe carbon is in carbonate rocks, bicarbonate ion in the ocean, or more reduced materials in soil sand sedi- ments, with only a small (but important) fraction in the atmosphere. Sulfur is divided between the sulfate of the oceans and evaporites or in sediments, the atmosphere containing only a small amount in transit between these pools. 2. Biological processes are major factors in the move- ment of elements between the various pools, but, in general, the biosphere constitutes only a small fraction of the total. 3. Oxidation-reduction reactions in biological sys- tems are responsible for most of the transfer taking place. The separation of charge of biological processes orobablv has created a broader range of oxidation potentials than existed before life developed on the planet. Thus there probably are both more oxidizing and more reducing conditions than existed before the appearance of life. The former is part of scientific lore, but the latter frequently is overlooked. 4. The concentration of oxygen in the atmosphere is determined not by the rate of photosynthesis, but by the degree to which reduced compounds (particularly those of carbon, nitrogen, sulfur, and iron) can be kept buried. 5. The partition of compounds between various compartments is a function of the energy balances in- volved. Thus, for example, the concentration of CH4 in
TROPOSPHERIC CHEMICAL CYCLES 20 en ~ 15 o a: ~ 10 o o o 6 15 a: a: ° 10 5 o 103 SE R 00 LP CARBON SEO SE R OB 00 AC1 AM AC2 SO COM PA RTM E N1 N ITROGEN Sl LP OP OP LA OA AN ANO ANH COMPARTM ENT OA FIGURE 7.2 Pool sizes of interest for the elements carbon, oxy- gen, nitrogen, and sulfur. Ordinates give the log of the pool size in gram-atoms of the element. Logarithmic presentation is necessary because of the wide range in pool size. Unit increment on the scale represents a factor of 10 in pool size. Although some pool sizes are OXYGEN SEO AC1 SEO SER SO OW ICE CW FEO COM PA RTM E NT SU LFU R S04 PW AO AW S I 01 ASO ASU ASH COMPARTMENT known with reasonable accuracy, others are accurate only to a factor of 2 or more. Numeric values are also presented in Table 7. 1. Values compiled from various sources including Delwiche and Likens (1977); Garrels, Mackenzie, and Hunt (1975); and Soderlund and Svennson (1976).
104 PART II ASSESSMENTS OF CURRENT UNDERSTANDING TABLE 7.1 Pool Sizes of Interest for Carbon, Oxygen, Nitrogen, and Sulfur Element Compartment Carbon Oxygen Nitrogen Sulfur SEO Oxidized sediments 50.8 152. 0.21 SER Reduced sediments 1.04 0.1 0.60 OB Ocean bicarbonate 0.032 OO Ocean organic 0.00083 6.2 E-5 AC 1 Atmospheric CO2 0.00054 0.00 ~ 1 AM Atmospheric CH4 5.2 E-6 AC2 Atmospheric CO 2.0 E-7 SO Soil organic 0.0025 0.00021 2.2 E-5 SI Soil inorganic 0.010 1.1 E-5 0.81 LP Land plants 0.00069 0.00065 5.7 E-5 OF Ocean plants 1.5 E-6 1.2 E-7 LA Land animals 1.2 E-6 1.4 E-7 OA Ocean animals l.lE-6 1.2E-7 OW Ocean water 761 ICE Ice 9.16 CW Continental water 0.055 FEO In iron oxice 6. SO4 In sulfates 8.5 PW Sediment pore water 177 AO Atmospheric O2 0.76 AW Atmospheric water 0.0058 AN Atmospheric N2 2.8 ANO Atmospheric N2O 1.3 E-6 ANH Atmospheric NH3 2.0 E-8 OIOceaninorganic 7.1 E-5 0.41 ASO Atmospheric SO2 3.4 E- 10 ASU Atmospheric sulfate ion 1.0 E-9 ASH Atmospheric reduced S 1.9 E- 10 NOTES: Values are in units of 102° gram-atoms ofthe element. Elements of igneous rock and magma are not included in this compilation. Where no values are given, the pool is not applicable, insignificantly small, or unknown. The code letters used correspond with those of Figure 7. 2. the atmosphere probably is a direct reflection of the energy relatior~ships of microbial processes. 6. The consequences of human alteration of these cycles are best interpreted in terms of rates. Although the total system probably could accommodate large per- turbations if sufF~cient time were allowed, the rate con- stants for many of the processes considered here are of the order of tens of thousands of years or more, and human activities on time scales of decades or centuries are not accommodated. This short overview of biogeochemical cycles is in- tended to serve as a backdrop against which to examine atmospheric cycles of more immediate concern to this report. Details of these chemical cycles are available elsewhere (see bibliography at the end of each cycle section). BIBLIOGRAPHY Ahrens, L. H. (1979~. Origin and Distribution of the Elements. Perga mon, New York,537 pp. Bremner, J. M., andA. M. Blackmer(1978~. Nitrous oxide: emis- sion from soils during nitrification of fertilizer nitrogen. Science 199:295-296. Broda, E. (1975~. The history of inorganic nitrogen in the bio- sphere. J. Mol. Evol. 7:87-100. Broda, E. (1975~. The Evolution oftheBioenergetic Process. Pergamon, Oxford, 211 pp. Broecker, W. S., T. Takahashi, H. M. Simpson, and T.-H. Peng (1979~. Fate of fossil fuel carbon dioxide and the global carbon budget. Science206:409-418. Delwiche, C. C. (1970~. The nitrogen cycle. Sci. Amer. 223:137- 146. Delwiche, C. C., and B. A. Bryan (1976~. Denitrification. Ann. Rev. Microbiol. 30: 241 -262. Delwiche, C. C., and G. E. Likens (1977) Biological Response to Fossil Fuel Combustion Products, in Global Chemical Cycles and Their Alterations by Man, Werner Stumm, ed. Dahlen Konferen- zen, Berlin, pp. 73-88. Garrels, R. M., F. T. Mackenzie, and C. Hunt (1975~. Chemical Cycles in the Global Environment. William Kaufmann, Los Altos, California. Garrels, R. M., A. Lerman, and F. T. Mackenzie (1976~. Controls of atmospheric O2 and CO2: past, present and future. Amer. Sci. 64:306-315. Holland, H. D. (1978~. The Chemistry of the Atmosphere and Oceans. Wiley, New York.
TROPOSPHERIC CHEMICAL CYCLES TABLE 7.2 Selected Transfer Rates Between Compartments I II III IV V Process From, To Quantity Source Ratio Sink Ratio Carbon Photosynthesis land ACT, LP 4036 0.0747 0.0585 ocean ACT, OF 2080 6.4 E-4 13.7 Fossil fuel combustion SER, AC1 388 3.7 E-6 7.2 E-3 Biological CH4 production SO(?), AM 26 1.1 E-4 0.69 Atmosphere-ocean (CO2) exchange ACT, OB 8190 0.074 2.6 E-3 Wildfire LP, AC1 126 1.8 E-3 2.3 E-3 Oxygen Photosynthesis land CW, AO 8072 1.5 E-3 1.1 E-4 Fossil fuel AO, OW 1160 1.5 E-5 1.5 E-8 combustion Nitrogen N fixation land AN, LP 6.9 2.5 E-8 0.12 ocean AN, OF 0.724 2.6 E-9 0.060 industrial AN, SI 2.83 1.0 E-8 2.6 E-4 Denitrif~cation land SI,AN 8.5 7.7 E-3 3.0 E-8 ocean OI, AN 2.86 4.0 E-4 1.1 E-8 Sulfur Fossil fuel combustion SER, ASO 2.0 3.3 E-8 58. Wildfire LP, ASO 0.82 1.0 E-3 24.1 Biological reduction land SI, ASH 0.12 1.5 E-9 6.3 ocean OI, ASH 0.085 2.1 E-9 4.47 Volcanic return SER, ASO 0.12 2.0 E-9 3.5 NOTES: The symbols used in Column II correspond with those of Table 7. 1. Rates are in units of teragram ( 1 E- 12 grams) atoms per year. Column IV gives the ratio of the quantity transferred to the source quantity; Column V gives the ratio of the quantity transferred to the sink quantity. Holser, W. T. (1977~. Catastrophic chemical events in the history of the ocean. Nature 267:403-408. Junge, C. E. (1972~. The cycles ofatmospheric gases naturaland man-made. Quart. I. Roy. Meteorol. Soc. 98:711-729. Kellogg, W. W., R. D. Cadle, E. R. Allen, A. L. Lazrus, and E. A. Martell (1972~. The sulfur cycle: man's contributions are com- pared to natural sources of sulfur compounds in the atmosphere and oceans. Science 175:587-596. Kvenvolden, K. A., ed. (1974~. Geochemistry and the Origin of Life. Dowden, Hutchinson and Ross, 422 pp. Li, Yuan-Hui (1972~. Geochemical mass balance among litho- sphere, hydrosphere, and atmosphere: the Gaia hypothesis. Tel- 1us26:1-10. 105 Margulis, L., and J. E. Lovelock (1978~. The biota as ancient and modern modulator of the earth's atmosphere. Pure Appl. Geophys. 116:239-243. Ponnamperuma, C. (1977~. Chemical Evolution of the Early Precam- brian. Academic, New York, 221 pp. Soderlund, R., and B.;H. Svensson (1976~. The global nitrogen cycle, in Nitrogen, Phosphorus and Sulphur Global Cycles, B. H. Svensson and R. Soderlund, eds. SCOPE Report 7. Ecol. Bull. Stockholm 22:23-72. Sokolova, G. A., and G. I. Karavaiko (1964~. Physiology and Geo- chemical Activity of Thio~oacilli. Translated from Russian, 1968. 283 pp.
106 WATER (HYDROLOGICAL CYCLE) BY R. DICKINSON Water is such an important component of the envi- ronment that it is not surprising to realize that it is also one of the more important atmospheric species from the viewpoint of chemistry. Furthermore, the general framework that is used in this section to consider the cycles of other atmospheric trace constituents is also appropriate for water. Its distribution in the atmosphere is determined by the balances between sources, sinks, and transport, as illustrated in Figure 7.3. Water occurs in the atmosphere in three phases vapor, liquid, and solid and all three phases interact strongly with the other chemical cycles. The transformations between phases need special emphasis in viewing water as an atmospheric chemical. SOURCES With the exception of a small source by CH4 oxida- tion in the stratosphere, and minuscule amounts yielded by some tropospheric reactions, the sources for water are entirely at the earth's surface. Water is removed from the earth's surface because of higher vapor pres- sures maintained at surface interfaces than within the atmosphere. On the global average, 1 .0 m of water per year moves from the surface to the atmosphere and falls again as precipitation. The water vapor at wet interfaces is maintained at the saturation vapor pressure by equi- librium between the wet surface and its immediately adjacent molecular boundary layers. However, trans- port and removal processes in the atmosphere act to me) ! :i Su bl l motion //// °~snOl - - Bare Sea Ice ~ - _ a ce reduce water vapor pressure over much of the atmo- sphere to values below saturation. Furthermore, surface materials are often warmer than the overlying atmo- sphere because they absorb solar radiation. Some com- bination of lower temperature and lower relative hu- midity for the overlying air makes its water vapor partial pressure and mixing ratio lower than that of the surface. The consequent gradient in free energy drives water from the surface. Meteorologists often approximate the upward flux of water from the surface, Fw, by an expres- sion of the form Fw=CwpaV~qs-qa)' where Cw is a bulk transfer coefficient (under some con- ditions deductible from micrometeorological theory); Pa = density of the air; qS = water vapor mixing ratio at the surface (e.g., the saturation mixing ratio evaluated at the temperature of the surface); qa = water vapor mixing ratio in the air, evaluated at some reference level, usually 2 m above the ground or 10 m above the ocean; and V = magnitude of the wind at the reference level. Over oceans, Cw = 1.4 x 10-3 with some dependence on wind speed and wave height. About 70 percent of the earth is covered by water and about 75 percent of the water entering the atmosphere comes from the ocean surface. The remaining 25 per- cent undergoes the interesting and complex physics of hydrological processes on land. At the simplest level, we can distinguish between evaporation from nonphoto- synthesizing surfaces and transpiration. Evaporation Atmospheric - Transport Cloud Condensation ~_ U' ~ ' 1 ~ I l 1 1 1 '1 / Snow Covered Planetary / /~: ~ Vegetation Boundary Layer / /~ ~ /1/ / ~ Low Shadow Shadow /~ - p;~/ Transpiration Leaf Temperature Leaf Evaporation Am/ ~ Dew Formation b. I Surface Wind q ~ in Canopy ~POLE O~FAN EQUATOR FIGURE 7.3 Features of the hydrological cycle in the atmosphere.
TROPOSPHERIC CHEMICAL CYCLES occurs as described above, with two additional compli- cations: (1) the remaining water on relatively dry sur- faces will be bound by surface tension and other stronger forces that will lower the water vapor pressure, and (2) the rate of water removal may be limited by the maxi- mum rate at which water can diffuse from the interior of the soil or other object to the surface. Water transpiring from plants passes through the stomata of leaves, which generate enough diffusional resistance to lower signifi- cantly the water vapor on the outside of a leaf from its saturation value. The stomata! resistance changes with various environmental factors, including inability of roots to supply enough water because of soil dryness. Water loss by vegetation through stomata is believed to be primarily an accidental consequence of the need for plants to move CO2 into their leaves to supply their photosynthetic cycles. Most other gas exchange be- tween higher plants and the atmosphere also occurs through the stomata. Interception is another process involving vegetation that is of concern to hydrologists. The leaves and other plant parts become coated with water that can reevaporate, without the water progress- ing farther into the ground. The saturation vapor pressure of water varies strongly with temperature according to the Clausius- Clapyron relationship. Thus saturation water vapor pressures near the surface and hence evaporation are much larger in the tropics than in high latitudes. TRANSPORT AND DISTRIBUTION Water vapor moves from the surface, through the planetary boundary layer, and then into the free atmo- sphere, where it is redistributed horizontally and verti- cally by atmospheric wind systems until it undergoes gas-to-droplet conversion. On a global average, a column of air holds about 27 kg/m2 of water. Water in vapor form has an average lifetime of about 10 days and can move large distances (thousands of kilometers or more) before conversion to droplets. Liquid and ice par- ticles generally have lifetimes of several hours or less and so are carried distances of 100 hen or less before recon- version to the gas phase or removal by precipitation. Because precipitation rates do not have as strong a latitudinal dependence as evaporation rates, large-scale atmospheric transport moves a significant fraction of the water evaporated in tropical latitudes into middle and high latitudes. This transport is one of the major proc- esses for maintaining temperatures at high latitudes warmer than implied by radiative-convective equilib- rium within a vertical column. Motion processes on various scales are intimately connected to the gas-to-droplet conversions and droplet 107 removal processes of precipitation systems described be- low. The mixing ratio of water vapor in the troposphere varies over 4 orders of magnitude, from a few parts per hundred in the tropics near the surface to less than one part per thousand over the poles at the surface and to a few parts per million near the tropopause. This variabil- ity is explained to zeroth order by assuming a fixed relative humidity and noting that the mixing ratio varies with its saturated value. The reason relative humidity is not too variable, with sufficient averaging, is under- stood in terms of the role of atmospheric motion sys- tems. By continuity, at any one time about half of the atmosphere is moving upward and is constrained to relative humidity near 100 percent by precipitation processes. The rest of the atmosphere is moving down- ward and drying the air to values much lower than saturation (e.g., near 10 percent). Combining the up- ward and downward streams gives an average relative humidity near 50 percent. As suggested by this discus- sion, instantaneous water concentrations at a given at- mospheric level in the free atmosphere and given loca- tion have about a factor of 10 variability depending on the instantaneous vertical motion patterns. TRANSFORMATION AND SINKS In terms of chemical reactions of the water molecule itself, the most important role of water in the atmo- sphere is as a source for OH through the reaction, H2O + O(iD) ~ 20H. The production of the OH radical is fundamental to all the elemental cycles and is discussed in more detail in each of the other cycles sections. In the form of drop- lets, water provides the medium for numerous heteroge- neous and homogeneous aqueous-phase reactions that are also fundamental to all element cycles in the troposphere. On the microscopic scale, atmospheric water vapor is converted to droplets or snowflakes by migration to condensation centers, initially submicrometer cloud condensation nuclei. Growth of the droplets or flakes continues by further water vapor diffusion. When sizes of several micrometers or so are reached, further droplet growth occurs by collisional coalescence until the drops reach sufficiently large size (~100 Em) that their fall velocity exceeds the velocity of upward air motion. Their fall velocity is determined by the balance between downward gravitational acceleration and viscous (Stokes) drag, and so increases with increasing radius. From a macroscopic viewpoint, water vapor con- denses because atmospheric motions have produced wa- ter mixing ratios near their saturation values. The satu
108 ration mixing ratio decreases strongly with altitude because of its temperature dependence. Thus water con- densation is driven primarily by upward transport via atmospheric motions. Conversely, sinking air tends to be cloudfree and of low relative humidity. The latent heat released by condensation processes can be of major importance in maintaining or enhancing atmospheric vertical motions. Two kinds of precipitation systems are distinguished, according to whether the latent heat is their primary drive or merely a positive feedback. Con- vective precipitation systems are driven by the latent heat they release. These generally occur on a horizontal scale with a fine structure of the order of 1 km and a PART II ASSESSMENTS OF CURRENT UNDERSTANDING large-scale organization of the order of 10 to 100 km. Layered precipitation systems are driven by upward motions in large-scale atmospheric wind systems forced by other modes of atmospheric instability. Convective precipitation can occur within layered systems. BIBLIOGR APHY Baumgartner, A., and E. Reichel (1975~. The World Water Balance. Elsevier, Amsterdam. L'vovich, M. I. (1979~. World Water Resources and Their Future. American Geophysical Union, Washington, D.C., 415 pp.
109 OZONE BY H. LEVY II Ozone (03) is both an important oxidant in its own right and a prerequisite for the production of hydro- peroxyl and hydroxyl radicals. These radicals play key roles in most of the elemental cycles and control the atmospheric lifetimes of many of the short- and me- dium-lived trace gases. Besides its chemical role, O3 is a significant absorber of long-wave radiation. Changes in the concentration of tropospheric O3 will not only change the chemical lifetimes of many trace gases, but may also affect the climate. SOURCES The major sources of tropospheric O3 are strato- spheric injection and in situ photochemical production. There is also a small indirect contribution from the com- bustion source of NO2 The stratospheric injection of O3 has been observed directly in the region of "tropopause folds," inferred from radioactivity measurements, and calculated from general circulation/transport models. These different approaches all arrive at a cross-tropopause flux in the range 3-12 x 10~° molecules per square centimeter per second. In situ photochemical production occurs both in the polluted boundary layer and in the free troposphere as a whole. Significant production of oxidant, in particular 03, has been clearly demonstrated in polluted urban environments. Not only has the production been simu- lated in smog chambers, but highly elevated O3 concen- trations are frequently observed in areas with high con- centrations of hydrocarbons and NOX. What is not known at this time is the importance of this smog source to the global troposphere. Summertime measurements of O3 at 500 mb would suggest that this production extends up into the middle troposphere over regions of surface pollution. Positive correlations between fluctua- tions in simultaneous CO and O3 vertical profiles have also been observed in the free troposphere, particularly over land at midlatitude in the northern hemisphere. This has been interpreted as demonstrating that O3 has the same source region, the polluted boundary layer, as CO. A realistic estimate of the contribution from the polluted boundary layer is not yet available. These same smog reactions, HO2 + NO ~ NO2 + OH, and followed by NO2 + hi' ~ NO + 03, should occur throughout the troposphere. Numerous theoretical calculations have predicted column produc- tion rates in the background troposphere of the range 1- 10 x 10~' molecules per square centimeter per second or more. These calculations are, however, completely de- pendent on theoretical predictions of the peroxy radical concentrations and on predicted or measured concen- trations of NO. At this time, there are many uncertain- ties in both the calculations and the measurements. Therefore, while the calculated production rates are much higher than the stratospheric injection rates, they are also less certain. On the other hand, they are so much larger that they suggest an important role for photochemical production in the troposphere. SINKS The two demonstrated removal paths for O3 are de- struction at the earth's surface and in situ photochemical destruction. A third, the fast reaction of O3 with biologi- cally emitted organics in the surface layer, is very diff~- cult to separate from surface deposition and may end up being included in many measurements of surface de- struction rates. The surface destruction rate, frequently expressed as a surface deposition velocity, is highly variable depend- ingnot only on the nature of the surface, but, in the case of vegetation, on the type of vegetation, time of year, and even time of day. Various methods have been used to measure deposition velocity over a number of surfaces. These methods include: a direct measure of loss inside a box that covers a particular surface; indirect measure- ments based on inferring a flux from a measured vertical gradient; and an indirect measurement using the eddy correlation technique that calculates an eddy flux. Sur- face deposition velocities, while highly variable, do ap- pear to separate into two main categories: (1) Land either bare or covered with vegetation has values of deposition velocity (W0) that range from 2.0 cm/s for daytime forests and cultivated crops to 0.2 cm/s over nighttime grassland. Bare land falls in the low end ofthis range. (2) Water, snow, and ice surfaces have values in the range 0. 1 to 0.02 cm/s. Estimates of average global fluxes to the surface have been made with different val- ues for deposition velocity as a function of surface type and different values for O3 concentration in the surface RO2 + NO ~ NO2 + OH,
110 layer. There is considerable uncertainty in the distribu- tion of surface types over the globe, deposition velocities for particular surfaces, and the global distribution of O3 in the surface layer. Nonetheless, these calculations pre- dict deposition fluxes in the same range as predictions of . . . . stratosp :lerlc 1nJectlon. The other halfof O3 photochemistry is photochemical destruction. At this time, the major removal path is -- thought to be: O3 + he ~ O(~D) + 02, followed by O(iD) + H2O ~ 2 OH. A number of other mechanisms have been suggested: the destruction of O3 by HOX radicals; the oxidation of NOX to nitrate and HNO3 and their resulting deposi- tion; the reactions involving the oxidation of halogens, particularly I, in the maritime boundary layer. Recent analyses of some regional boundary layer data in the equatorial Pacific strongly supports the existence of a photochemical removal process with an effective column removal rate of the order of 1-2 x 10~i mole- cules per square centimeter per second. This removal rate is much larger than the estimated surface deposition flux and is needed to explain the extremely low O3 mix- ing ratios (5 to 10 ppb) that were observed at that time. Again, as for photochemical production, a realistic esti- mate of the global importance of this process requires accurate calculations of radical concentrations and de- tailed knowledge of other trace gas concentrations. DISTRIBUTION/CLIMATOLOGY A global data- base, sufficient to produce a coarse resolution O3 climatology, is urgently needed for the field of tropospheric chemistry. Not only is it needed to produce a global distribution of OH, the principal oxi- dizing species in the various elemental cycles, but it is needed to provide a framework for tropospheric photo- chemistry as a whole. Due to its high variability in the troposphere, relative standard deviations in the range of 25 to 100 percent, a realistic global data base will require relatively high spatial and temporal resolution. Both the Dobson network and satellite observations provide a global field oftotal O3. Unfortunately, approx- imately 90 percent of the total O3 resides in the strato- sphere, and existing techniques are not able to accu- rately extract the small fraction of the signal that represents the troposphere. Therefore these global fields are, at best, of qualitative use. The best existing tropospheric data set is provided by individual ozonesonde stations that are now measuring or have in the past measured vertical profiles of O3 from PART II ASSESSMENTS OF CURRENT UNDERSTANDING the ground to the middle stratosphere on a more or less regular basis. Data from stations still operating are be- ing archived by the Canadian Department of the Envi- ronment. Unfortunately, there are a number of very serious problems with this data set: 1. A number of different types of sensors have been used, many of which were never accurately intercali- brated. Serious doubts have recently been raised about the absolute accuracy of the ozonesonde measurements in the troposphere, particularly for the older types of sondes that are no longer in operation or available for intercomparisons with current devices. Previous inter- comparisons of operational devices alone have raised serious doubts about combining measurements from different research groups with different devices into a single data set. 2. Even if all the available data were useful, the global coverage is completely inadequate. Almost all the stations are in the northern hemisphere, and most of them are at midlatitude. There are a few in the high latitudes ofthe northern hemisphere, one in the tropics, and one operating (we hope) in the southern hemisphere at Aspendale, Australia. There are no stations in opera- tion in any of the oceans, even at midlatitudes in the northern hemisphere. If all the sites that are no longer operating are included, there is minimal improvement in the global coverage. While a global data set does not exist, careful analysis of either individual station data or individual networks using a common sensor and measurement protocol has produced many useful insights: 1. In all cases the mean proB0es of O3 increase with height. 2. Where it has been analyzed, the variance profile has a maximum in the upper troposphere and in the boundary layer with a minimum in the middle tropo- sphere. A more detailed analysis of variance in the As- pendale, Australia, data finds in the troposphere that it is dominated by synoptic and shorter time scales. 3. The profiles show a spring maximum with, in the case ofthe midlatitude northern hemisphere continental stations, a continuation of this maximum into the sum- mer. When analyzed, the variance also appears to be higher in the spring. 4. Both mean values and variability are greater at midlatitudes in both hemispheres than in the tropics, but the tropical data base is very limited. When data from a common instrument are considered, there is still some evidence for a midlatitude maximum, particularly in the lower troposphere. 5. An analysis of the North American network finds a significant east-west asymmetry on even the regional
TROPOSPHERIC CHEMICAL CYCLES scale However, there is not enough longitudinal resolu- ISSUES ton In the data to determine the extent of zonal asym metry in the global O3 field. A number of north-south transects through the mid dle and upper troposphere are available. The one data set that provides more than a single snapshot has severe troubles with absolute calibration of the sensor and was from the upper troposphere with the likelihood of air craft incursions into the lower stratosphere at midlati tude and high latitude. A few single snapshot transects with relatively accurate O3 sensors in the middle tropo sphere are also available. For this very sparse data set, O3 iS a minimum in low latitudes, the values either level offor decrease from midlatitude to highlatitude, and the maxima occur at midlatitude in the hemispheric spring. Although these few data sets are not time mean latitudi nal fields, they may have captured certain latitudinal features of O3. Given the 25 percent relative standard deviations observed in profile data in the middle tropo sphere, it is also possible that these few profiles are atypi cal. A repeated series of flights over the same path with an accurate and validated sensor is certainly needed. By far, the best time series data are available from continuous measurements of surface O3. Unfortu nately, the boundary layer is frequently very unrepre sentative of the troposphere as a whole. The mean val ues may be strongly affected by local meteorology and surface removal, as well as local photochemical produc tion and destruction. It is not clear what, if anything, can be inferred about the global troposphere from these excellent time series. In remote clean regions, the data appear to have many of the features observed in O3 profiles with the addition of significant and currently unexplained diurnal fluctuations. They do appear to have significantly lower variability than is observed in the boundary layer of the profile data. In regions with pollution sources of NOX, they show concentration max . . ~ . . ma extent sing trom spring into summer. The final sources of distribution data are field gener ated by general circulation/transport models. A recent calculation of tropospheric O3 with only stratospheric injection and surface removal, photochemical produc tion and destruction having been excluded, has pro duced a tropospheric climatology of O3 representative of the model meteorology. To the extent that the model me teorology is representative of atmospheric meteorology, the model field should represent the transport contribu tion to the real O3 field. Outside of the boundary layer in general and continental regions with significant anthro pogenic pollution in particular, the real O3 field may be dominated by real atmospheric transport. This should be particularly true of the variance in the O3 field. Therefore, the model variance fields may be quite useful in designing observational networks for O3. 111 . . The three key issues involving tropospheric O3 are as follows: 1. Its climatology (i.e., tropospheric distribution of mean values and higher moments); 2 . The process or combination of processes that exert dominant control on its climatology; 3. The possible existence of long-term changes in the mean concentration and the causes of such trends if they do exist. It is obvious that these three issues are intertwined. Fur- thermore, it is obvious that the first requirement is the development of a reliable data set. A few stations mak- ing very accurate long-term measurements for the de- tection of trends are needed, along with a significantly larger number making accurate measurements for a few years to establish at least a coarse global climatology. Coupled with this is the continued development and refinement of both the theory and numerical modeling of tropospheric transport and fast photochemistry. The earliest view of tropospheric O3 had it being transported down from the stratosphere and being de- stroyed at the earth's surface. Other than boundary layer variability, which would result from the large inho- mogeneity of the surface destruction process, the distri- bution and variability would be controlled by meteoro- logical processes on all scales. This view is still supported by much of the observational data. In the early 1970s, an active photochemistry was pro- posed for the clean troposphere, which led to the predic- tion of large photochemical production and destruction rates. These predictions depended on many reactions that have not been quantitatively confirmed in the real troposphere and have as inputs species concentrations that were not well known. Nonetheless, the calculated photochemical production and destruction rates were much larger than measurements and estimates of strato- spheric injection and surface removal. Furthermore, there were observations, particularly at midlatitude, that supported a strong role for photochemistry in the summer. Recently, a unification of the transport and photo- chemical theory was proposed in which photochemical production occurred primarily in the upper troposphere with the precursor NOX being injected from the strato- sphere. Ozone destruction would then dominate in the lower troposphere where NOX was very low. This O3 destruction has been observed in one set of data taken in the tropical Pacific boundary layer. This theory depends critically on a tropospheric NOX distribution, which increases strongly with height. A recent general circulation/transport model study
112 reexamines the classical transport theory. It finds con siderable agreement between observations and model results. At this time, there are not enough data to estab lish any theory, and the gathering of such data has the highest priority in O3 research. BIBLIOGRAPHY Chameides, W. L., and I. C. G. Walker (1973~. A photochemical theory of tropospheric ozone. I. Geophys. Res. 78:8751 -8760. Chatf~eld, R., and H. Harrison (1977~. Tropospheric ozone, 2: variations along a meridional band. I. Geophys. Res. 82:5969 5976. Crutzen, P. I. (1974~. Photochemical reactions initiated by and influencing ozone in unpolluted tropospheric air. Tellus 26:47 57. Fabian, P., and P. G. Pruchniewicz (1977~. Meridional distribution of ozone in the troposphere and its seasonal variation. 1. Geophvs. Res. 82:2063-2073. Fishman, l., end W. Seiler(1983~. Correlative nature ofozone and 103:575-584. PART II ASSESSMENTS OF CURRENT UNDERSTANDING carbon monoxide in the troposphere: implications for the tropo- spheric ozone budgeted. Geophys. Res. 88:3662-3670. Galbally, I. R., and C. R. Roy (1980~. Destruction of ozone at the earth's surface. Quart.~. Roy. Meteorol. Soc. 106:599-620. Junge, C. E. (1962~. Global ozone budget and exchange between stratosphere and troposphere. Tellus 14:364-377. Lenschow, D. H., R. Pearson, Jr., and B. B. Stankov (1981~. Estimating the ozone budget in the boundary layer by use of aircraft measurements of ozone eddy flux and mean concentra- tion. I. Geophys. Res. 86:7291-7298. Liu, S. C., D. Kley, M. McFarland, I. D. Mahlman, and H. Levy, II (1980~. On the origin of tropospheric ozone.~. Geophys. Res. 85: 7546-7552. Mahlman, J. D., H. Levy, II, and W. i. Maxim (19803. Three- dimensional tracer structure and behavior as simulated in two ozone percursor experiments.~J. Atmos. Sci. 37:655-685. Oltmans, S. I. (1981~. Surface ozone measurements in clean air. J. Geophys. Res. 86: 1174- 1180. Pittock, A. B. (1977~. Climatology of the vertical distribution of ozone over Aspendale (38S, 145E). Quartz. i. Roy. Meteorol. Soc.
113 FIXED NITROGEN CYCLE BY S. LIU AND R. CICERONE CURRENT ISSUES The global nitrogen cycle is similar to the cycle of carbon and sulfur in that it has important atmospheric, marine, and soil components. In the atmosphere, as in the biosphere, fixed (or odd) nitrogen compounds are especially important. Further, fixed nitrogen is a limit- ing nutrient in many soils and water bodies. Its scarcity has caused man to intervene by producing artificially fixed nitrogen. This industrial production, when added to the nitrogen oxides fixed inadvertently in high-tem- perature combustion processes, has brought about the present situation in which it can be demonstrated that man is strongly perturbing the nitrogen cycle regionally and globally. We will focus the discussion of the nitrogen cycle on individual fixed nitrogen compounds of atmospheric importance. Species of interest include odd-nitrogen compounds (NO + NO2 + NO3 + N205 + HNO2 + HNO3 + HNO4 + peroxyacetyl nitrate (PAN) + other organic nitrates), NH3, HCN, and N2O, and the aque- ous-phase species NO3-, NO2, and NH4 . Nitrous oxide is included because of its significance as the principal source of stratospheric odd nitrogen, which flows down- ward and can affect the upper troposphere. Odd-nitrogen species play a central role in tropo- spheric O3 and HxO' chemistry. It is well known that odd-nitrogen species together with nonmethane hydro- carbons are the major precursors of high urban O3. There is also evidence that odd-nitrogen species emitted by anthropogenic activities may contribute to the for- mation of high rural O3 that is frequently observed in the summer over rural North America and Europe. Ozone, NO2, PAN, and HNO3 are major air pollutants that may be health hazards and/or damage plants under some conditions. There are also suggestions that the O3 throughout the northern hemisphere might have been increased by pollution-derived nitrogen oxides and CO. This is of interest because infrared absorption by tropo- spheric O3 is a significant factor in the energy balance of the atmosphere. Nitrate is a major anion in regions of high acid deposi- tion in Europe and North America. There are indica- tions that long-range transport deposition of NO3 along with SO4- takes place over distances of hundreds to thou- sands of kilometers. In addition, through the effect of odd-r~itrogen species on HxOy chemistry, odd-nitrogen species may also influence the rate of oxidation of SO2 to SO4-. It is also possible that odd-nitrogen species play key roles in the oxidation of hydrocarbons, CO, and other reduced gases in the troposphere. Ammonia is the primary basic gas in the atmosphere, and it partially neutralizes sulfuric and nitric acid in precipitation. On the other hand, NH3 can probably be oxidized to odd-nitrogen species in the troposphere, and the products contribute to the acidity of precipitation. The photochemistry of atmospheric NH3 is not well understood, and a quantitative evaluation of the pro- duction rate of odd nitrogen from NH3 is not yet avail- able. Ammonium is an important component of conti- nental aerosol particles and may help stabilize other compounds in the aerosol particles. Deposition of atmo- spheric NH3, NH4, and nitrate represents an important nutrient input to the biosphere in some regions. The importance of N2O lies mainly in its control of stratospheric O3. Changes in stratospheric O3 may have significant effects on tropospheric chemistry and on bio- spheric processes. Furthermore, NOx produced from N2O in the stratosphere is transported down to the troposphere and may affect tropospheric NO distribu- tions, especially in unpolluted areas. The observed global increases of N2O indicate that the global nitrogen cycle is not in a perfectly balanced steady state. SOURCES Emissions from fossil fuel burning (primarily high- temperature combustion), stratospheric intrusion, sub- sonic aircraft, biomass burning, lightning, soil exhala- tion, oxidation of atmospheric NH3, and photolysis of marine NO2 are the likely major sources of odd-nitro- gen species. Annual fluxes from the first three sources are probably known within a factor of 2. Uncertainties for fluxes from the other sources are much greater. The estimated global annual source strength for each of these sources is presented in Table 7.3. Despite the large uncertainties in these estimates, it can be seen that anthropogenic odd-nitrogen emissions (i.e., fossil fuel burning and most of the biomass burning) are signifi- cant if not dominant sources of odd-nitrogen species. In view of the importance of atmospheric odd-nitrogen species, there Is an urgent need to evaluate the effect of these anthropogenic emissions on the distribution of tropospheric 03, HxOy' CO, hydrocarbons, and so on. Because the removal of odd-nitrogen species is primarily through wet and dry deposition, which is more efficient in the lower troposphere, upper tropospheric sources such as lightning, stratospheric intrusion, and aircraft may exert important control on the global odd-nitrogen distribution. Significant sources of NH3 may be animal waste, ammonification of humus followed by emission from
114 TABLE 7.3 Global Odd-Nitrogen Sources . . ~ Sources Source Strength (Tg N/yr) 1 5-25 0. 15-0.3 0.5-1 .5 1-10 2-20 1-10 Fossil fuel burning Subsonic aircraft Stratospheric intrusion Biomass burning Lightning Soil exhalation Oxidation of NH ~ Phdtolysis of marine NO2 soils, losses of NH3-based fertilizers from soils, and industrial emissions. Estimates for natural NH3 emis- sions are available, but are not reliable. An upper limit of about 50 Tg N/yr has been estimated for global emis- sions of NH3 from undisturbed land. The other NH3 sources (primarily from animal waste) are due to anthropogenic activities. Yearly anthropogenic emis- sions from the United States are estimated to be about 3 Tg N/yr. Apparently, there is not enough NH3 emitted to neutralize the production of H2SO4 over North America. Because NH3 is readily absorbed by surfaces such as water, plants, soil, and atmospheric aerosol particles, its residence time should be very short in the lower tropo- sphere. In fact, NH3 emitted from vegetation-covered soil may have little chance of escaping to the atmo- sphere. Thus considerable caution must be used in esti- mating the emission rates of NH3. Nitrification and denitrification of natural soluble fixed nitrogen, industrial fertilizers, and fixed marine nitrogen, and power plant emissions are the primary sources of N2O. All these sources are poorly quantified. Hydrogen cyanide is emitted from cyanogenic plants, steel industry processes, and smoldering combustion of vegetation. No quantitative estimates are available. DISTRIBUTIONS Modeling the distribution of odd-nitrogen species has been severely hampered by the poor understanding of heterogeneous removal processes and the present inabil- ity to model realistically hydrological processes in the atmosphere. Current knowledge of the distribution of odd-nitrogen species is primarily from some infrequent and sparsely distributed measurements of uneven . qua lty. The distributions of NO, NO2, HNO3, and particu- late NO3 are relatively better known, at least in conti- nental regions, because there are established measure- mer~t techniques available. There have been a few long path optical absorption measurements of NO3 in the surface air. PAN, HNO2, and other organic nitrates PART II ASSESSMENTS OF CURRENT UNDERSTANDING have been observed but not adequately quantified in the troposphere. The distribution of odd-nitrogen species in the upper troposphere is probably more uniform than in the lower troposphere since this region is relatively far from large surface sources and sinks, as well as surfaces on which heterogeneous reactions can take place. Observed upper tropospheric NOX (NO + NO2) mix- ing ratios range from 0.1 to 0.4 ppbv. For HNO3 the range is about 0.3 to 1.5 ppbv. The major sources of upper tropospheric odd-nitrogen species are probably stratospheric intrusion and lightning. It has been sug- gested that O3 produced in the upper troposphere may be the major natural source for tropospheric O3. According to model calculations, there are probably sig- nificant amounts of HNO4 and PAN in the upper tropo- sphere. Measurements of these species are needed. In the lower troposphere, observed mixing ratios of NOX range from 0.01 ppbv in clean oceanic air to 500 ppb for highly polluted urban air. Similar mixing ratio ranges can be found for HNO3 and NO3-. There has been no attempt to model the atmospheric distribution of NH3 or particulate NH4. As in the case of odd-nitrogen species, observed data are too few. Most of the measurements to date are for particulate NH4; gas- phase measurements of NH3 are practically nonexist- ent. Sensitive and specific analytical techniques are needed for NH3 and should be feasible with some effort. The distribution of N2O in the troposphere is well known. There is slightly more in northern latitudes, and the global concentration is rising slowly. About 160 pptv of HCN has been observed at midlatitudes, and little, if any, vertical or latitudinal variation is expected. TRANSFORMATIONS Gas-phase reactions of odd-nitrogen species have been studied extensively. Additional laboratory kinetic studies of the reactions of NO3, N2O5, HNO3, PAN, and other organic nitrates are still needed, especially under tropospheric conditions. Reactions of odd-nitro- gen species with nonmethane hydrocarbons and their products also need to be quantified. Field studies of NO3, the ratio of NO to NO2, and their relation to O3, HNO3, and NO3 have revealed many interesting, per- plexing results that may have important implications to tropospheric photochemistry. Heterogeneous processes, including atmospheric het- erogeneous transformations and surface deposition, play crucial roles as sinks of odd-nitrogen species and in interconversions among odd-nitrogen species. Lack of reliable laboratory determinations of the key reaction paths and rates is probably the most serious difh~culty in attempting to understand heterogeneous processes involving odd-nitrogen species. In particular, sticking
TROPOSPHERIC CHEMICAL CYCLES coefficients, aqueous reaction rates, and equilibrium constants need more attention. Most of the natural and anthropogenic odd-nitrogen species emissions are in the form of NO. Usually NO reaches photochemical equilibrium quickly with NO2 through reactions NO + O3 - ' NO2 + O2 (7.1) NO + HO2 (RO2) ~ NO2 + OH(RO) (7. 2) NO2+hv~NO+O(3P), (7.3) where R denotes a hydrocarbon radical. Reactions (7. I) and (7.3) are fast but do not result in net gain or loss of O3 because O quickly associates with O2 to form O3. Under most circumstances (when NO > 10 pptv) in the troposphere, reactions (7.2) and (7.3) result in net pro- duction of O3 over urban as well as rural areas. These reactions may also play an important role in the budget of O3 in the entire troposphere. Oxidation of NO2 by OH will form HNO3, which is readily removed from the troposphere by heterogeneous processes such as precipitation scavenging and surface deposition. This is one of the major sinks of odd-nitro- gen species and a major source of acidity in precipita- tion. PAN and HNO2 formed through reactions (7.4) and (7.5) and nighttime reactions play important roles as temporary reservoirs for NOX and hydrogen radicals in the photochemistry of the polluted atmosphere. CH3COO2 + NO2 ~ CH3COO2NO2 (PAN) (7.4) OH + NO + M ~ HNO2. (7 5) Unlike HNO3, PAN is apparently not scavenged by precipitation. The major sink for PAN is thermodisso- ciation followed by reaction of CH3COO2 with NO. Because of the low temperature in the upper tropo- sphere, PAN may have a tropospheric lifetime of a few months or longer. Therefore, if PAN is transported to or manufactured in the upper troposphere, it may serve as an important odd-nitrogen reservoir and thus a carrier for long-range transport of odd-nitrogen species. Other organic nitrates and HNO3 may play the same role as PAN in the upper troposphere. Reaction of HNO4 with OH may be an important sink for OH in the upper troposphere. This reaction is probably not important in the lower troposphere because of the fast thermal disso- ciation rate for HNO4. Formation of NO3 and N2O5 by reactions NO2 + O3 ~ NO3 + O2 (7.6) NO3 + NO2 + M ~ N205 + M (7.7) are important nighttime reactions for odd nitrogen. If N2O5 or NO3 is scavenged by aerosol particles as indi- cated by recent field measurements of NO3, they would 115 constitute a significant sink for odd nitrogen and a major source for HNO3 or particulate NO3 . The photochemical transformations among odd- nitrogen species discussed above are summarized in Figure 7.4. The more speculative transformations are indicated by dashed arrows. Minor reactions have been omitted. The most important gas-phase reaction for NH3 is probably oxidation by OH. Little is known about subse- quent reactions that involve NH2 and its products. Reaction of NH3 with HNO3 to form NH4NO3 may be a significant sink for both NH3 and HNO3. However, the reaction rate constant has not been measured. Most of atmospheric NH3 is believed to be removed by wet and dry deposition. Like odd-nitrogen species, quanti- tative information on heterogeneous processes is not available. Quantitative aspects of cycling N2O through the atmosphere are known within a factor of 2. Nitrous oxide exhibits no known tendency to react in the tropo- sphere. Accordingly, its chemical lifetime is long (about 150 years), and its accumulation in the atmosphere can lead to signif~cant infrared greenhouse effects. Discov- ered only recently, HCN could be the dominant odd- nitrogen compound in the background troposphere. Currently at about 160 ppt, its oxidation comprises an in situ source of NOX. Its apparent slow reactions probably imply a small source (0.5 Tg N/yr) of tropospheric NOX. REMOVAL Odd-nitrogen species, NH3, and their counterparts in the particulate form are removed by cloud and precip- itation scavenging, as well as dry deposition. These processes are poorly understood. Important constraints on removal rates and/or sources can be obtained through measurements of deposition rates of NH4 and NO3 . Because of recent emphasis on acid rain research, wet deposition rates for NH4 and NO3 are routinely measured over Europe and North America. Dry deposi- tion may account for more than half of the total deposi- tion of these species, but a routine measurement pro- gram does not exist. Deposition data are extremely sparse over the oceans. Atmospheric NH4 and NO3 may be important sources of f~xed nitrogen to the oceans in some regions, along with NO3 runoff in rivers. Photolysis and oxidation by O(~D) in the stratosphere are the only known sinks for N2O. This removal rate (about 10 Tg N/yr) is known within a factor of 2; accord- ingly, one can calculate from the background concentra- tion of N2O of 300 ppb an atmospheric chemical lifetime (about 150 years) to within a factor of 2. Oxidation by atmospheric OH radicals is probably the major sink for HCN. Mechanistic understanding of the reactions involved is not yet complete.
~6 BIBLIOGRAPHY PART II ASSESSMENTS OF CURRENT UNDERSTANDING M HO2 1 O3, HO2, RO2 1 _ HO2NO2 1 - OH OH // GOOD Go/ = Do Heterogeneous ~ loss ~N Heterogeneous // loss ~K . _ ~ AE R OSO L \ '~v ~ . . he OH I he H NO2 | Bauer, E. (1982~. Natural andAnthropogenic Sources of Oxides of Nitrogen (NO0for the Troposphere. P-1619. Institute for Defense Analysis, Arlington, Va. Calvert, I. G., and W. R. Stockwell (1982~. The mechanism and rates of the gas phase oxidation of sulfur dioxide and the nitrogen oxides in the atmosphere, in Acid Precipitation: SO2, NO, NO2 Oxidation Mechanisms: Atmospheric Considerations, Ann Arbor Sci- ence, Ann Arbor, Mich. Crutzen, P. I. (1979~. The role of NO and NO2 in the chemistry of the troposphere and stratosphere. Menu. Rev. Earth Planet. Sci. 7:443-472. Dawson, G. A. (1977~. Atmospheric ammonia from undisturbed land. I. Geophys. Res. 82:3125-3133. Demerjian, K. L., A. I. Kerr, and I. G. Calvert (1974~. The mechanism of photochemical smog formation. Adv. Environ. Sci. Techrzol. 4:1-262. Ehhalt, D. H., and I. W. Drummond (1982~. The tropospheric cycle of NOX, in The Proceedings of NATOAdvanced Study Institute on he I ~ I EVAPORATION ~1 NO3 / N2O5 / .e ' Heterogeneous ,~/ loss FIGURE 7.4 Photochemical transformations among odd-nitrogen species. Chemistry of the Unpolluted and Polluted Troposphere. Reidel, Hingham, Mass. Galbally, }. E., and C. R. Roy (1978~. Loss of fixed nitrogen from soils by nitric oxide exhalation. Nature 275: 734-735. Huebert, B. J., and A. L. Lazrus (1980~. Tropospheric gas-phase and particulate nitrate measurements. I. Geophys. Res. 85:7322- 7328. Kley, D., J. W. Drummond, M. McFarland, and S. C. Liu (1981~. Tropospheric profiles of NOx ~J Geophys. Res. 86:3153-3161. Logan, J. (1983~. Nitrogen oxides in the troposphere: global and regional budgets. J. Geophys. Res. 88: 10785- 10808. Platt, U., D. Perner, J. Schroder, C. Kessler, and A. Toennissen (1981~. The diurnal variations of NO3. J. Geophys. Res. 86: 11965-11980. Singh, H. B., and L. J. Salas (1983~. Peroxyacetyl nitrate in the free troposphere. Nature 302:326-328. Soderlund, R., and B. H. Svensson (1976~. The global nitrogen cycle, in Nitrogen, Phosphorus, arid Sulfur: Global Cycles. SCOPE Report 7. Ecol. Bull. (Stockholm) 22:23-74. J
117 SULFUR CYCLE BY R. HARRISS AND H. NIKI CURRENT ISSUES Human activities strongly influence the tropospheric sulfur cycle in certain regions of the world, particularly in and downwind of populated areas. The literature on the reaction and transformation of SO2 and its distribu- tion and transport in eastern North America and west- ern Europe is voluminous. A reading of even selected portions of the literature on sulfur cycling illustrates that measurements and models of sulfur on the regional scale are providing a relatively consistent understanding of the sources, transport, and fate of anthropogenic emis- sions of SO2. However, with the possible exception of reasonably well-understood processes related to local and mesoscale impacts of anthropogenic SO2, global sulfur cycle studies are in the infancy stage. Among the general categories of tropospheric sulfur sources, anthropogenic sources have been quantified the most accurately, particularly for the OECD coun- tries. Research on fluxes of sulfur compounds from vol- canic sources is now in progress. However, very few generally accepted measurements are available for either concentrations or fluxes of SO2, H2S, DMS, DMDS, CS~, COS, and other sulfur species derived from natural biogenic sources. Measurement tech- niques have been inadequate until recently; serious questions still remain concerning flux determinations. Tables 7.4, 7.5, and 7.6 summarize most of the data available in the open literature. What do the existing data indicate in terms of interest- ing hypotheses and the design of future global studies? First, natural sources of reduced sulfur compounds are highly variable in both space arid time. Variables, such as soil temperature, hydrology (tidal and water table), and organic flux into the soil, all interact to determine microbial production and subsequent emissions of reduced sulfur compounds from anaerobic soils and sed- iments. For example, fluxes of H2S, COS, CS2, (CHESS, (CH3~2S2, and CH3SH can vary by several orders of magnitude on time scales of hours and space scales of meters in a coastal environment. A second interesting aspect of existing data on biogenic sources of reduced sulfur relates to the origin of relatively high SO2 values measured in the mid-troposphere over the tropics and in the southern hemisphere during GAMETAG. SOURCES AND DISTRIBUTIONS Current estimates of global sources of atmospheric sulfur are based on very few data and will not be dis- cussed in detail here. Several recent comprehensive reviews are cited at the end of this section for the reader unfamiliar with previous attempts to estimate sulfur sources. We briefly summarize available information on sources of COS, CS2, DMS, and H2S to the troposphere in the following paragraphs; these are the major bio- genic sulfur species with a clearly identified role in tro- pospheric chemistry. Carbonyl Sulfide (COS) Carbonyl sulfide is the most abundant gaseous sulfur species in the troposphere. Concentrations of COS are approximately 500 + 50 pptv, with no detectable sys- tematic variations vertically or latitudinally. This con- stant concentration with altitude and latitude suggests a relatively long atmospheric lifetime, estimated to be around 2 years. Current estimates of global sources and sinks of COS are summarized in Table 7.6. It is important to note that these source and sink estimates are derived by extrapola- tion of a very limited data base and are subject to large uncertainties. Recent data suggest that oceanic regions of high biological productivity and organic content, par- ticularly coastal waters and upwelling areas, are a major global source of COS. Experiments in coastal waters TABLE 7.4 Approximate Tropospheric Concentration Range of Selected Sulfur Compounds in Unpolluted Air Atmospheric Concentrationsa (ng/m3) Location H2S DMS CS2 COS SO2 - Ocean boundary layer Temperate continental boundary layer Tropical coastal boundary layer Free troposphere < 5-150 20-200 1 00-9000 PD < 2-200 PD PD < 2(PD) 1200-1550 1200-1550 1200-1550 1200-1550 50-70 PD PD <10 < 15-300 < 15-300 PD 30-300 aPD indicates poorly determined at this review.
118 PART II ASSESSMENTS OF CURRENT UNDERSTANDING TABLE 7.5 Biogenic Emissions of Sulfur Compounds (emission rate in g S/m21yr) - - DMS CS2 COS Location Avg. Max. Avg. Max. Avg. Max. Avg. Max. Salt marsh 0.55 41.5 3.84 0.006 0.5 100 0.66 2.5 0.2 0.03 72 381 0.093 1.13 6.36 Freshwater marsh 0.6 1.27 Inland soils (U. S. ~0.001 0.001 0.002 Swamps and tidal flats 0.044 Sediments of shallow coastal area ~ 19 ~ 2000 Soils of humid equatorial forests 0.07 2.6 Soils of temperate regions 0.044 0.24 Open ocean 0.106 indicate that COS is produced by photooxidation of dissolved organic matter independent of salinity, plant metabolism, or bacterial activity. Some authors have suggested that the oceans may be a net sink for COS from the atmosphere. An intensive research program concerned with the production, distri- bution, and emissions of COS from coastal and oceanic environments will be required to quantify the role of the marine environment as a source of this compound. Soils can also be a source of COS to the troposphere. Coastal salt marsh soils appear to be a "hot spot" for COS emissions, but the small area of these soils limits the role of marshes as a major global source. Measure- ments from a variety of soils in the United States were used to calculate the global soil source of COS shown in Table 7.6. The total absence of data on COS emissions from tropical soils introduces significant uncertainties into estimates of the global soil source. Efforts to quan- tify COS emissions from soils will probably be compli- cated by large variations in both space and time. Micro- bial processes that produce COS are influenced by soil moisture, nutrients, soil organic content, and other physiochemical variables. Combustion processes are also thought to be a signifi- cant global source of COS. These processes include bio- mass burning, fossil fiael burning, and high-tempera- ture industrial processes involving sulfur compounds. Again, it must be emphasized that these estimates are based on very limited data and may change significantly as new data become available. During periods of low volcanic activity, COS may be a major source of sulfur to the stratosphere, resulting in the formation of the stratospheric aerosol layer that influences the earth's climate. The anthropogenic sources of COS identified in Table 7.6 represent approx- imately 25 percent of the total source strength, support- ing speculations of possible effects on climate within the next century. Because of the importance of COS in the global sulfite cycle, its sources, atmospheric chemistry, . . . . ~ . . ~ . and sin as are a critical scents issue. Carbon Disulf.~de (CS2) The abundance and distribution of CS2 in the tropo- sphere are not well known. Available measurements in the literature at this date show a typical range from approximately 15 to 30 pptv in surface nonurban air to 100 to 200 pptv in surface polluted air. The concentra TABLE 7.6 Global Sources and Sinks of Carbonyl Sulfide Estimate Range Sources (Tg/yr) Oceans Soils Volcanoes Marshes Biomass burning Coal-fired power plants Automobiles, chemical industry, and sulfur recovery processes Subtotal CS2-COS: CS2-photochemistry and OH reactions Total Global burdens (Tg) Lifetime (yr) Sinks (Tg/yr) OH reaction Stratospheric photolysis O reaction Other 0.60 0.40 0.02 0.02 0.20 0.08 0.06 1.4 0.60 2 4.7 (500 pptv) 2-2.5 21 0.8 0.1 0.03 1.1 0.3-0.9 0.2-0.6 0.01-0.05 0.01-0.06 0.1-0.5 0.04-0.15 0.01-0.3 0-2 3.8-5.2 0. 1-1 .5 C0.2 c3.3 NOTES: The estimated emissions are consistent with observed distribu- tions of COS and CS2 according to a global mass balance. All combina- tions of emissions within the ranges given above may not be consistent. SOURCE: From Khalil and Rasmussen, 1984.
TROPOSPHERIC CHEMICAL CYCLES lion of CS2 appears to decrease rapidly with altitude, indicating ground sources and a relatively short atmo- spheric lifetime. The primary removal mechanism for CS2 in the troposphere is thought to be reaction with OH, producing COS and SO2. The reaction rate con- stants for oxidation of CS2 are poorly known, and the relative importance of CS2 as a precursor for atmo- spheric COS and SO2 is an unresolved issue. The primary natural sources of COS and CS2 are thought to be similar. The one available set of measure- ments of CS2 in seawater indicates that concentrations are highest in coastal waters. Dimethylsulfide (CHINS Dimethylsulfide (DMS) is the most abundant volatile sulfur compound in seawater with an average concen- tration of ~ 100 x 10-9 g/l. This compound is produced by both algae and bacteria. The evidence for a biogenic origin for DMS has come from laboratory measure- ments of emissions produced in pure, axenic cultures of marine planktonic algae and field measurements of emissions from soils, benthic macroalgae, decaying algae, and corals. Extensive oceanographic studies have shown direct correlations between DMS concentrations in seawater and indicators of phytoplankton activity. The vertical distribution, local patchiness, and distribu- tion of DMS in oceanic ecozones exhibit a pattern very similar to primary productivity. Selected groups of marine organisms such as coccolithophorids (i.e., a type of marine planktonic algae) and stressed corrals are par- ticularly prolific producers of DMS. The calculated global sea-to-air flux of sulfur as DMS is ~0.1 g S/m2/ yr, which totals to approximately 39 x 10~2 g S/yr. A more limited set of measurements has been made in coastal salt marshes with DMS emissions commonly in the range of 0.006 to 0.66 g S/m2/yr. Hydrogen Sulfide (H2S) Knowledge of natural sources of H2S to the tropo- sphere is still rudimentary. Preliminary studies have shown that anaerobic, sulfur-rich soils (e.g., coastal soils and sediments) emit H2S to the atmosphere, albeit with strong temporal and spatial variatons. Hydrogen sulfide fluxes at a single location can vary by a factor of up to 104 depending on variables such as light, temperature, Eh, pH, 02, and rate of microbial sulfate reduction in the sediment. The presence of active photosynthetic organ- isms or a layer of oxygenated water at the sediment surface can reduce or stop emissions due to rapid oxida- tion of H2S. Agricultural and forest soils can also be a source of H2S to the atmosphere. Measurements by several inves 119 tigators suggest that maximum emissions from nonma- rine soils are associated with wet tropical forest soils. It is likely that many soils that appear to be aerated contain anaerobic microhabitats suitable for microbial sulfate reduction; the magnitude of H2S emissions will depend on the net effects of many processes that influence pro- duction, transport in the soil, oxidation rates, and exchange at the soil-air interface. Photochemical sources for atmospheric H2S have been proposed to occur through a combination of the following reactions: OH+COS OH + CS SH + HO SH + CH2O SH + H2O SH + CO2, COS + SH, H2S + O2, H2S + HCO, H2S + HO2, SH + CH3OOH ~ H2S + CH3O2, and SH + SH ~ H2S + S. Removal of H2S is thought to be accomplished by OH + H2S ~ H2O + SH, resulting in a lifetime of approximately 1 day. In situ photochemical production from COS and CS2 precur- sors is the most likely source of H2S measured in remote ocean air. Atmospheric concentrations of H2S in conti- nental air are highly variable, resulting from a complex interaction of factors determining ground emissions, in situ photochemical production, and atmospheric life- t~me. TRANSFORMATIONS AND SINKS The oxidation of SO2 to H2SO4 can often have a .,- . · ~. r slgnlilcant impact on t ne aclulty o~ preclpltatlon, cur- rently an issue of national and international concern. A schematic representation of some important transfor- mations and sinks for selected sulfur species is illustrated in Figures 5. 1 1, 5.13, and 7.5. The oxidation of reduced sulfur compounds, such as H2S, CS2, (CH3~2S, and others, leads to the production of acids or acid precur- sors such as SO2, SO4-, and CH3SO3H. Subsequent oxidation steps involving a combination of homogene- ous and heterogeneous reactions lead to the production of H2SO4, which is removed from the atmosphere by wet and dry deposition processes (see Chapter 5). Car- bonyl sulfide appears to be relatively inert in the tropo- sphere and is primarily destroyed in the stratosphere. In terms of experiments to elucidate the fast photo- chemistry of this system, measurement schemes will be
120 Multistep ~ Q ~Stratosphere \\ D ~ Multistep ~Troposphere D / At\ ~S°2:o ~ It so i: a_ _ _ ~ _ l ~ i' Ra inout, Wet and Dry Deposition ,~W W~ ~ V MV~ SURFACE SOURCES FIGURE 7.5 A tentative scheme for the oxidation and removal of atmospheric sulfur species. needed to verify the chemical pathways by which reduced sulfur species are oxidized to SO2 and SO4- . It is probable that it will be useful to carry out these experi- ments in a variety of different environments, including areas of intense sulfur emissions (e.g., swamps, tidal flats, and marshes) as well as remote marine areas. Unfortunately, present understanding of the distribu- tions of atmospheric sulfur species and the elementary chemical reactions involved in the previously described oxidation chains is quite poor. In addition, the instru- mentation necessary to measure many of the key atmo- spheric constituents has yet to be developed. Once this task is completed, it will be possible to design specific fast-photochemistry experiments to selectively study various facets of the atmospheric sulfur system. In the case of H2S oxidation, for instance, it is believed that oxidation is initiated by reaction with OH, Be., H2S + OH ~ H2O + SH, and is followed by an as yet unconfirmed reaction sequence that produces SO2 as an end product. The lifetime of H2S in the atmosphere seems highly variable based on limited field measurements. In situ studies of H2S oxidation kinetics in a variety of environments (e.g., swamps, salt marshes, and mangroves) would be extremely useful to improved understanding of the sul- fur cycle. Recent studies of DMS photooxidation provide important data on reaction mechanisms and products. The major gas-phase sulfur product produced in out- door smog chamber experiments was SO2. Substantial formation of light-scattering aerosol particles was PART II ASSESSMENTS OF CURRENT UNDERSTANDING observed, with inorganic sulfate and methane sulfonic acid as major components of the aerosol. Fourier trans- form infrared methods have been used to quantify products of the reaction of HO + CH3SCH3 in the presence of C2H5ONO and NO. Methyl thionitrite (CH3SNO) was observed as an intermediate product, with SO2 and CH3SO3H as major products. These stud- ies serve as models of important photooxidation sinks for reduced sulfur species. ROLE OF CLOUDS AND AQUEOUS-PHASE CHEMISTRY As indicated in Chapter 5 of this report, aqueous- phase chemistry (i.e., in cloud and raindrops) plays a major role in the oxidation of SO2 to H2SO4. Current thinking also suggests that clouds may be the dominant transport conduit for movement of SO2 and other rela- tively short-lived reduced sulfur species to mid-tropos- pheric altitudes. Sulfur dioxide produced below cloud base may be injected directly into the free troposphere by updrafts associated with clouds or may dissolve or react with cloud droplets, depending on a variety of poorly quantified physical and chemical variables. Evaporation of cloud droplets may produce small sul- fate-rich aerosol particles that subsequently act as cloud condensation nuclei. If the transport and reaction mech- anisms mentioned in this paragraph are active over large areas of the nonurban troposphere, they contrib- ute to explanations for acid rain in remote oceanic regions and higher SO2 in the free troposphere than in underlying ocean boundary layer air. Once in the mid- dle to upper troposphere, SO2 may have a much longer lifetime with potential for long distance transport beyond the synoptic scale. Future field experiments will need to measure a vari- ety of species including SO2, (CHAPS, H2O2, and meth- ane sulfonic acid in gas, liquid, and solid phases where appropriate. Combined ground and aircraft measure- ments focused on the role of cloud and aqueous-phase processes are a high priority. BIBLIOGRAPHY Andreae, M. O. (1980). Dimethylsulfoxide in marine and fresh- waters. Limnol. Oceanogr. 25: 1054- 1063. Andreae, M. O., and H. Raemdonck (1983~. Dimethyl sulfide in the surface ocean and the marine atmosphere: a global view. Science 221: 744-747. Aneja, V. P., A. P. Aneja, and D. F. Adams (1982~. Biogenic sulfur compounds and the global sulfur cycle. J. Air Pollut. ControlAssoc. 32:803-807. Barnard, W. R., M. W. Andreae, W. E. Watkins, H. Bingemer, and H. W. Georgi (1982~. The flux of dimethyl sulfide from the oceans to the atmosphere.J. Geophys. Res. 87:8787-8793.
TROPOSPHERIC CHEMICAL CYCLES Brown, K. A. (19823. Sulfur in the environment: a review. Environ. Pollut. 3:47-80. Chatfield, R. B., end P. J. Crutzen(1984~. Sulfur dioxidein remote oceanic air: cloud transport of reactive precursors. [. Geophys. Res. (in press). Delmas, R., i. Baudet, l. Servant, and Y. Baziard (1980~. Emis- sions and concentrations of hydrogen sulfide in the air of the tropical forest of the Ivory Coast and of temperate regions in France. I. Geophys. Res. 85:4468-4474. Ferek, R. I., and M. O. Andreae (1984~. Photochemical produc- tion of carbonyl sulfide in marine surface waters. Nature307: 148- 150. Graedel, T. E. (1977~. The homogeneous chemistry of atmospheric sulfur. Rev. Geophys. Space Phys. 15:421-428. Graedel, T. E. (1979~. Reduced sulfur emission from the open oceans. Geophys. Res. Lett. 6:329-331. Herrmann, I., and W. Jaeschke (1984~. Measurements of H2S and SO2 over the Atlantic Ocean. ]. Atm. Chem. 1: 111 - 123. Husar, R. B., I. P. Lodge, and D. i. Moore (1978~. Sulfur in the Atmosphere. Pergamon, New York. Ingvorsen, K., and B. B. Jorgensen (1982~. Seasonal variation in H2S emission to the atmosphere from intertidal sediments in Denmark. Atm. Environ. 16:855-864. Jones, B. M. R., R. A. Cox, and S. A. Penkett (1984~. Atmo- spheric chemistry of carbon disulphide. [. Atm. Chem. 1:65-86. Jorgensen, B. B. (1982~. Ecology of the bacteria of the sulfur cycle with special reference to anoxic-oxide interface environments. Phil. Trans. Roy. Soc. LondonB298:543-561. Khalil, M. A. K., and R. A. Rasmussen (1984~. Global sources, lifetimes, and mass balances of carbonyl sulfide (COS) and car- bon disulf~de (CS2) in the earth's atmosphere. Atm. Environ. (in press). Kritz, M. A. (1982~. Exchange of sulfur between the free tropo- sphere, marine boundary layer, and the sea surface.. Geophys. Res. 87:8795-8803. Lawson, D. R., and I. W. Winchester (1979~. Atmospheric sulfur 121 aerosol concentrations and characteristics from the South Amer- ican continent. Science 205: 1267- 1269. Logan, l. A., M. B. McElroy, S. C. Wofsy, and M. I. Prather (1979~. Oxidation of CS2 and OCS: source for atmospheric SO2. Nature 281: 185-188. Maroulis, P. I., A. L. Torres, A. B. Goldberg, and A. R. Bandy (1980~. Atmospheric SO2 measurements on Project GAME- TAG. /. Geophys. Res. 85:7345-7349. McElroy, M. B., S. C. Wofsy, and N. Dak Sze (1980~. Photochemi- cal sources for atmospheric H2S. Atm. Environ. 14: 159- 163. Moller, D. (1984~. On the global natural sulphur emission. Atm. Environ. 18:29-39. National Research Council (1978~. Sulfur Oxides. Committee on Sulfur Oxides, Assembly of Life Sciences, National Academy of Sciences, Washington, D.C. National Research Council (1981~. Atmosphere-BiosphereInteractions: Toward a Better Understanding of the Ecological Consequences of Fossil Fuel Combustion. Commission on Natural Resources, National Academy of Sciences, Washington, D.C. Nguyen, B. C., B. Bonsang, and A. Gaudry (1983~. The role ofthe ocean in the global atmospheric sulfur cycle. [. Geophys. Res. 88:10903-10914. Niki, FI., P. D. Maker, C. M. Savage, and L. Breitenbach (1980~. Fourier transform study of the OH radical initiated oxidation of SO2 . J. Phys. Chem. 84: 14- 16. Rasmussen, R. A., M. A. K. Khalil, and S. D. Hoyt (1982~. The oceanic source of carbonyl sulfide. Atm. Environ. 16:1591-1594. Servant, i-, and M. Delapart (1982~. Daily variations of the H2S content in atmospheric air at ground level in France. Atm. Envi- ron. 16:1047-1052. Shriner, D. S., C. R. Richmond, and S. E. Lindberg (1980~. Atmospheric Sulfur Deposition. Ann Arbor Science, Ann Arbor, Mich. Slatt, B. I., D. Natusch, I. M. Prospero, and D. L. Savoie (1978~. Hydrogen sulfide in the atmosphere of the northern equatorial Atlantic Ocean and its relation to the global sulfur cycle. Atm. Environ. 12:981-991.
122 CURRENT ISSUES CARBON CYCLE BY H. NIKI, R. DUCK, AND R. DICKINSON Although CO2 is the primary carbonaceous trace gas in the biogeochemical cycle of carbon, it is relatively unreactive in the troposphere. For this reason, we will separate the discussion of the carbon cycle into two parts" reactive carbon compounds currently recog- nized to have potentially important atmospheric and biospheric components, and CO2. The two most abun- dant reactive carbon compounds in the global atmo- sphere are CH4 and CO. Both compounds play central roles in O3 and HxO' cycles through chemical interac- tion involving common reactive intermediates (free rad- icals, e.g., OH, HO2, and CH3OO). It is highly sig- nificant that the photochemical oxidation of CH4 can lead to the formation of a large variety of oxygenated products (e.g., CH2O, CH3OOH, HCOOH, and CH3OOOH). Atmospheric in situ formation rates of these individual compounds are critically controlled by HxOy and NOx chemistry. Moreover, some ofthese com- pounds can be removed from the atmosphere heteroge- neously prior to the subsequent oxidation to CO and, eventually, to CO2. Thus the atmospheric transforma- tion of CH4 involves complex chemical feedbackmecha- nisms. Anthropogenic perturbation of CO and CH4 cycles and its overall atmospheric impact are of great current interest. In addition, numerous~nonmethane hydrocarbons (NMHCs) (e.g., saturated and unsatu- rated C2-Cg compounds, isoprene, and terpenes), par- ticulate organic carbon (POC) (including Cg-C28 alkanes and C~2-C30 fatty acids), and elemental or soot carbon are present in the global troposphere. Interest in NMHCs in the global troposphere has been increasing in recent years because oftheir potential importance as a source of CO and as reactants interacting with 03, HxOy' and NOx chemistry. However, at present, the global sources, distribution, fluxes, and transformations of NMHCs represent perhaps the greatest deficiency in understanding the active carbon cycle in the tropo- sphere. Elemental carbon is primarily of concern because of its radiative properties and its surface characteristics. It is the only important aerosol component that can absorb a significant amount of visible light. Changes in the global distribution of elemental carbon may thus be related to changes in the retention of solar radiative energy in the troposphere. Elemental carbon particles also have a very active surface that effectively adsorbs many trace gases. The role of elemental carbon in the global troposphere as a sink for trace gases and in hetero geneous reactions in general has yet to be evaluated. Of particular importance may be its activity as a catalyst for . . . . . reactions 1n c ouc s anc preclpltatlon. Estimates of budgets of several active carbon species are summarized in Table 7.7. Key issues and existing uncertainties are indicated below for the global cycles of these individual or classes of compounds. THE CYCLES OF REACTIVE CARBON COMPOUNDS Sources Major direct sources of CO appear to be primarily anthropogenic, e.g., fossil fuel use and tropical biomass burning. The latter source is still poorly quantified in terms of spatial and seasonal variations. Photochemical production from CH4 is well recognized as a major indi- rect source of CO. This source is probably known within a factor of 3, reflecting uncertainties in existing esti- mates of the global yea-rly average OH concentration and ofthe transformation mechanism. Oxidation of nat- ural NMHCs, e.g., isoprene and terpenes, leading to CO production is potentially very important, but the magnitude of this source is highly uncertain, as men- tioned earlier. Other minor global sources thus far iden- tified include oxidation of anthropogenic hydrocarbons, emissions by plants, wood used as fuel, forest wildfires, and the ocean. Several sources may be contributing comparable quantities of CH4 to the global troposphere, e.g., rice paddy fields, natural wetlands (swamps and marshes), enteric fermentation (ruminants and termites), and bio- mass burning. However, available data on all these sources are severely limited, and more extensive flux measurements are needed for reasonably quantitative estimates. Clearly, many of these sources have direct or indirect anthropogenic components and are subjected to significant perturbation, a probable cause for the recent increase in the atmospheric CH4 level. The major volatile NMHCs in the global atmosphere are probably isoprene (C5H~) and various types of monoterpenes emitted from vegetation. Although there exist relevant baseline data on emission rates of vegeta- tion classes and effects of meteorological conditions (sunlight, temperature, and humidity), more extensive flux measurements, especially in tropical regions, are needed for sufficiently accurate estimates of global annual emission rates. Note that ongoing deforestation in tropical jungles may already be having a significant
TROPOSPHERIC CHEMICAL CYCLES TABLE 7.7 Budgets of Carbon Species Direct Source per Year and Gas Source Identification CO 4-16 x 10~4 g CO Biomass burning 6.4 x 10~4gCO Industry 0.2-2 x 10~4gCO Vegetation CH4 0.3-0.6 x 10~4 g CH4 Rice paddy fields 0.3-2.2 X 10~4gCH4 Natural wetlands 0.6 x 10~4 g CH4 Ruminants <1.5 x 10~4gCH4 Termites 0.3-1.1 X 10~4 g CH4 Biomass burning 0.2 x 10~4gCH4 Gas leakage C,,Hs, 8.3x10~4gC C INCH ~6 Trees Secondary Source per Year and Source Identification 3.7-9.3x10~4gCO Methane oxidation 4-13 X 10~4gCO C5He, C~(,H~6 Oxidation Atmospheric Removal by Lifetimes 30 x 10 ~ 4 g CO 2 months OH 4.5 x 10~4g CO Uptake by soils Transport Distances Ax, /\y, Az (km); Volume Mixing Ratios in the Unpolluted Troposphere 4000, 2500, 10 50-200 ppbv 4 x 1014 g CH4 7 years Global OH 1.5-2.0 ppmv 8.3 x 10~4 g C 10hours 400, 20O,1 OH 0-10 ppbv NOTES: Diffusion distances in E-W, S-N, and vertical directions (in km) over which concentrations are reduced to 30 percent by chemical reactions. Lifetimes and removal rates calculated with [OH] = 6 x 105 molecules per cubic centimeter. SOURCE: Crutzen, 1983. impact on these sources. In addition, numerous light hydrocarbons (C2-C5) are emitted in potentially signifi- cant amounts from a variety of sources including the ocean, plants, natural gas leakage, biomass burning, and fossil fuel combustion. However, for the majority of these compounds, the major contributors have not been identified. For compounds such as C2H2 and C6H6, there are no known significant biogenic sources. Exist- ing estimates of the -global emission rates of these NMHCs are largely based on their observed atmo- spheric concentrations and removal rates by OH radi- c~s. Particulate organic carbon (POC) consists of a com- plex mixture of hydrocarbons, alcohols, acids, esters, organic bases, and other polar organic compounds. Gas-to-particle conversion appears to be the main source of POC in the global atmosphere, since the total POC mass is generally in the small particle range with radii <1 ~m. Photochemically produced species, including free radicals, may participate in this process. Several potentially significant sources for the gaseous precursors of POC are land (vegetation), aquatic sys- tems (microbiota of marine and lacustrine environ- ments), petroleum seepages, and pollution (fossil fuel combustion and biomass burning). Existing specula- tions on the total source strength for POC are based on rough estimates of the tropospheric POC distribution 123 and the mean tropospheric residence time of aerosol particles (4 to 7 days). Thus the range of uncertainty is believed to be greater than one order of magnitude for the natural sources. To unravel complex interactions of numerous, as yet poorly characterized, factors control- ling POC formation, concerted efforts in several research areas are needed. In particular, many solubili- ties and rate parameters need to be determined. The transfer processes across the interface at the aerosol par- ticle surface require better quantitation. The primary source of elemental carbon is combus- tion processes. While industrial combustion processes are significant in many urban areas, biomass burning- both natural and as a result of human activities is also very important globally. Elemental carbon is proving to be an excellent tracer of the long-range transport of combustion aerosol particles. Distribution The tropospheric distribution of CO has been studied in considerable detail and is known to be rather variable. It exhibits an interhemispheric gradient of about a factor of 3, with the highest (150 to 200 ppbv) at the middle and higher latitudes of the northern hemisphere correspond- ing to the large anthropogenic fluxes there. Seasonal variations with summer maxima and winter minima
124 have been recently identified. Further extensive mea- surements of the seasonal and latitudinal distribution in both the boundary layer and the free troposphere, par- ticularly in the tropics where the major sources are sus- pected to exist, are critical for obtaining a better under- standing of its sources, transformation, and sinks. It is reasonably well established that CH4 is distrib- uted fairly uniformly throughout the global troposphere (excepting urban atmospheres) at approximately 1.6 ppmv. However, as much as 0. ~ ppmv interhemispheric gradient with higher values in the northern hemisphere has been observed. There is also recent evidence for approximately a ~ to 2 percent annual global increase in CH4 in the late 1970s and early 1980s. Thus accurate long-term measurements of this radiatively active gas are warranted. Carbon isotope ratio measurements may provide a useful clue to this increase. In addition, it is crucial to obtain information on atmospheric concen- trations of oxygenated products, e.g., CH2O and CH3OOH, derived from CH4. Very limited measure- ments are currently available on the diurnal variation of CH2O. There are no field data on CH3OOH because of the lack of adequate monitoring instrumentation. Isoprene and terpenes (e.g., o`-pinene, ,B-pinene, b-3- carene, camphene, and d-limonene) are very short- lived in the atmosphere, and their distribution is con- fined well within the boundary layer. Up to a few parts per billion by volume of these compounds have been observed in warm and humid forested areas. Thus far, there have been no field flux determinations of these compounds because of the lack of fast-response detec- tion instrumentation to utilize eddy correlation or profil- ing techniques. Concomitant product measurements are also required, but at present, little is known about the ensuing oxygenated species (e.g., carbonyls and organic peroxides3. On the other hand, some baseline data are becoming available for the vertical and latitudi- nal distribution of C2-C5 NMHCs. Since chemical life- times of these compounds are short or at least compara- ble to the time scale of tropospheric transport, their distributions are highly variable, with higher values in the middle and high latitudes of the northern hemi- sphere. Light alkanes such as ethane and propane reach concentrations as large as 2 to 3 ppbv. Relatively high concentrations of such very reactive alkenes as propene ~ ~ O. 2 ppb) are found rather uniformly distributed over the world ocean suggesting an important marine source for these hydrocarbons. More extensive, simultaneous monitoring of these NMHCs and oxygenated products (e.g., carbonyls, peroxides, and organic nitrates) is needed to determine their atmospheric roles. Carboxylic acids, particularly formic and acetic acids, are a common constituent of both aerosol particles and rain and have also been observed in the vapor phase. There is evidence that these weak organic acids PART II ASSESSMENTS OF CURRENT UNDERSTANDING can contribute a significant fraction of the free acidity of precipitation at locations far from urban areas. Consid- erable additional research is needed to evaluate accu- rately the role of organic acids in global-scale precipita . . tlon c ~emlstry. The relatively few data available for POC indicate concentrations of 0. ~ to 0.5 ~g/m3 STP in marine air and about 1 ,ug/m3 STP in nonurban continental air. The composition of POC in terms of neutral com- pounds (60 percent), acids (30 percent), and bases (10 percent) appears to remain remarkably constant in ground-level air. For n-alkanes (C~0-C2~), the relative portions in the particulate phase are often much less than expected from the saturation vapor pressures, pos- sibly due to heterogeneous reactions. Elemental carbon concentrations are often over ~ ,ug/m3 in continental regions, particularly near urban areas. There are rela- tively few data available in remote areas. Concentra- tions over the North and South Atlantic range from about 0.05 to 0.2 ,ug/m3, while concentrations during the winter at Barrow, Alaska, averaged ~ 0.3 ,ug/m3. Considerably more data are required on the spatial and temporal distribution of both individual and classes of vapor phase and particulate organic compounds in remote areas over a wide variety of potentially signifi- cant ground-level sources. Anthropogenic contribu- tions should be better characterized by comparing emis- sion estimates with measured ambient concentrations. liansformations an`1 Sinks The main removal process for CO is in situ oxidation to CO2 by OH radicals. Inherent to existing estimates of the global rate of this process are uncertainties associ- ated with OH concentration and with the rate constant. Estimates of the global annual OH production and dis- tribution are commonly assumed to be accurate within a factor of 2 to 3. The rate constant for this reaction may be uncertain by as much as 50 percent. Although it has been measured extensively with great accuracy in the presence of inert diluent gases at low pressures, it is known to exhibit peculiar dependence on O2 pressure. To date, there have been no direct measurements of this rate constant under tropospheric conditions. Further study of the combined pressure and temperature effects is needed. Soil bacterial uptake is an additional sink for CO. The removal rate by this sink appears to be rather minor on the global scale, but may be substantial in the boundary layer over land areas. The OH-radical-initiated oxidation of CH4 is pri- marily responsible for the removal and transformation of this compound, although recent studies have shown that CH4 is consumed in the soils of both temperate and tropical forests. In the presence of suff~cient NO, the
TROPOSPHERIC CHEMICAL CYCLES OH oxidation of CH4 leads to the formation of CO via photooxidation of the oxygenated intermediate CH2O. With little NO present, CH3OOH becomes a major intermediate in place of CH2O, but its subsequent fate is uncertain. The CH3OOH does not photodissociate readily, but may undergo sufficiently fast in situ removal by OH. The rate constant and mechanism for the HO- CH3OOH reaction, as well as heterogeneous removal of CH3OOH, must be established. In addition, because of its relatively long tropospheric lifetime (~ 7 years), CH4 escapes into the stratosphere in significant amounts and plays an important photochemical role in the O3 cycle. Further quantitation of the stratospheric removal rate is needed. Nonmethane hydrocarbons are generally far more reactive with OH than is CH4. In addition, some of the double-bonded NMHCs, particularly terpenes, can be removed by O3 at a rate comparable to or even greater than those of the corresponding OH reactions. For light alkanes, there is a fair body of laboratory evidence that their oxidation mechanisms are somewhat analogous to that of CH4, although they give rise to a large variety of classes of oxygenated products including carbonyls (RCHO and RCR'O; R and R' = organic group), peroxides (ROOH), peroxynitrates (ROONO2), per- oxyacids (RC(O)OOH), and acids (RC(O)OH). Again, large uncertainties exist in their oxidation mech- anisms at low NO concentrations and in the subsequent gas phase and heterogeneous removal processes for the oxygenated products. Not much is known about the mechanisms and key intermediate products for the OH- and O3-initiated oxidation of unsaturated compounds, e.g., isoprene and terpenes, either under laboratory conditions or in the atmosphere, primarily because of the lack of adequate methodologies and detection sys- tems. Cloud and precipitation processes appear to be most important for the removal of POC. Aqueous photo- chemistry involving OH and O3 may play a particularly important role in transforming POC, as indicated by the presence of a variety of oxygenated compounds in POC. Dry deposition processes involving the sea, soil, and vegetation are also potentially significant sinks. More quantitative knowledge is required on both gas- eous and particulate fluxes associated with all of these processes. Since some of the needed analytical tech- niques and methodologies are available, further field and laboratory studies in this area should be encour- aged. THE CYCLE OF CARBON DIOXIDE The species discussed up to this point represent the minute fraction ofthe total environmental carbon that is 125 . chemically active in the atmosphere. However, it should be recognized that the cycling of these active species is embedded in and ultimately controlled by the slower biological, oceanographic, and geologic reservoirs of carbon. These slower processes are briefly summarized here. Carbon dioxide is the dominant form of carbon stor- age in the atmosphere. Vegetation and soils store organic carbon, whereas the oceans store some organic carbon but mostly inorganic carbon in the form of car- bonate ions. Ocean sediments and terrestrial rocks store vast amounts of carbonate and organic carbon, mostly accumulated over millions of years from oceanic biologi- cal debris. Carbon is mostly of atomic weight 12, but about 1 percent is of weight 13 and a minute amount of weight 14. This ~4C iS unstable with an approximately 5000- year lifetime and is constantly being created in the atmo- sphere by galactic cosmic rays transforming nitrogen. It serves as a useful clock and thus a tracer of transfer from the atmosphere to other reservoirs. The ~3C sewes as a tracer for sources of carbon since its relative concentra- tion differs between organic and inorganic carbon. Over land, carbonate and silicate rocks are dissolved by terrestrial waters, which in doing so act to convert atmospheric CO2 into bicarbonate ions. This carbon is converted back to CO2, in part by the conversion of bicarbonate into the carbonate of sea creatures. Further CO2 is released by volcanoes, presumably after having been squeezed from carbonate rocks in their conversion to silicate rocks as a result of subduction of oceanic plates under continental plates. Such processes are of interest in explaining how there could have been 5 to 10 times more CO2 in the atmosphere in past geological times than there is now. Such an abundance of CO2 is cur- rently the most favored hypothesis for explaining the warm Cretaceous climates of 100 million years ago. On intermediate time scales of thousands of years, deep oceanic processes involve the concentrations of Ca++ ions and possibly were related to biological activity dom- inant in controlling atmospheric CO2. Such processes may have reduced atmospheric concentrations of CO2 near the end of the last ice age to values not much more than half of current concentrations. On the time scales of seasons to centuries, large car- bon exchanges occur between the atmosphere, oceanic near-surface waters, and living and dead vegetation, the latter mostly in soils. All these reservoirs are of roughly the same magnitude. The atmosphere loses and gains annually about 10 percent of its carbon content by exchange with the other reservoirs. It is these relatively short-term exchanges and the physical and biological processes accompanying them that are of most rele- vance to questions of global tropospheric chemistry. First, the transfers of CO2 between the atmosphere r
126 and the land biosphere or oceans occur by means of the same physical processes whereby other trace gases are transferred. To the extent that more accurate measure- ments can be made of CO2 transfer processes than of those of other gases, study of CO2 fluxes improves the understanding ofthe fluxes of other gases. For example, terrestrial vegetation removes 03, SO2, and NO2 by absorption through leaf stomata, the primary function of the stomata being to capture CO2 from the atmo- sphere as a major plant nutrient. As another example, atmospheric CH4 comes from soils, where it is produced by anaerobic bacterial decomposition of organic car- bon. However, most of the soil bacterial decomposition is aerobic, so that more than 99 percent of the soil organic carbon released to the atmosphere is in the form of CO2. The CH4 is oxidized in the atmosphere to CO and eventually CO2 as discussed earlier, thus complet- ing the cycle between biosphere and atmosphere that was initiated by plant stomata! uptake. Second, the seasonal and latitudinal variations of CO2, although relatively small, are measured with con- siderable accuracy so that CO2 would provide a useful tracer of atmospheric motions if its sources and sinks could be adequately modeled. Seasonal variations are largely driven by northern hemisphere seasonal growth of plants whose rapid removal of CO2 from the atmo- sphere from May to August or so depletes atmospheric concentrations by about ~ ppm. Decay processes, which restore CO2 to the atmosphere, vary more smoothly over the annual cycle though operating faster during the warmer seasons. Latitudinal atmospheric variations are driven in part by the excess of fossil fuel combustion in the northern hemisphere and in part by transfers from warm tropical oceans which are supersaturated with respect to CO2. The addition of CO2 to the atmosphere from fossil fuel combustion has increased atmospheric CO2 concentrations from 315 to 340 ppmv in the last 25 years since measurements began and has led to a national program to study the possible climate change due to the warming from those increases, as well as details of the exchange of CO2 among atmosphere, oceans, and biosphere. INTERACTION WITH OTHER CYCLES Carbon monoxide interacts directly with the HxO' cycle via free radical reactions, thereby profoundly affecting the cycles of virtually all elements of atmo- spheric interest. For example, OH radicals are removed primarily by CO in the global troposphere. This process serves as the principal source of HO2 radicals, which, in turn, interact with species such as NOx and O3. The OH radicals are regenerated to some extent by some of these secondary reactions (e.g., HO2 + NO ~ OH + NO2 and H2O2 + he ~ ASH). Thus, because of complex PART II ASSESSMENTS OF CURRENT UNDERSTANDING ities in the chemical feedback mechanism, the overall atmospheric impact of CO requires thorough numerical evaluation. Methane interacts with cycles of other elements pri- marily through CO formation. The major oxygenated product CH2O can provide an additional significant source of HO2 radicals. It may also be possible that the short-lived NO2-containing product CH3OONO2 serves as an intermediate for accelerating the gas-to- particle conversion of NOx. Similarly to H2O2, the peroxy product CH3OOH may play a catalytic role in the aqueous chemistry of other elements. In general, the OH-initiated oxidation of NMHCs involves oxygenated free radicals and intermediates of the form RxOy (R = organic group), which can interact directly or indirectly with NOx, HxO'' and O3. Conspic- uous formation of 03, PAN, and organic aerosols by the photochemical reactions of NMHC with NOX is a well- known phenomenon in urban atmospheres, i.e., photo- chemical smog, and is probably occurring in the tropical regions during biomass burning. These relevant reac- tions can take place to a significant extent in the global atmosphere, e.g., PAN formation. A large variety of RxO' species are also produced by the reactions of O3 with double-bonded NMHCs. Some of these RxO compounds appear to interact with SO2 and with H2O as well. However, the identity and chemical behavior of the majority of these species remain largely unknown. As in the gaseous phase, organic compounds in both particulate and aqueous phases may undergo significant photo- and dark-reactions with HxOy' 03, NOx' SOx' and trace metals. A detailed assessment of chemical ir~teractions in atmospheric aerosol particles is ham- pered by a scarcity of relevant rate data. BIBLIOGRAPHY Andreae, M. O. (1983~. Soot carbon and excess f~ne particle potassium: Long-range transport of combustion-derived aero- sols. Science 220: 1148- 1151. Atkinson, R., K. R. Darnal, A. Lloyd, A. M. Winer, and J. N. Pitts, Jr. (1979~. Kinetics and mechanism of the reaction of the hydroxyl radical with organic compounds in the gas phase. Adv. Photochem. 11: 375-488. Berner, R. A., A. S. Lasaga, and R. M. Garrels (1983~. The carbonate-silicate geochemical cycle and its effect on atmo- spheric carbon dioxide over the past 100 million years. Amer. J. Sci. 283:641-684. Brewer, P. G., G. M. Woodwell, L Machta, and R. Revelle (1983~. Past and future atmospheric concentrations of carbon dioxide, Chapter 3 in Changing Climate. National Academy Press, Washington, D.C., pp. 186-265. Broecker, W. S. (1982~. Ocean chemistry during glacial time. Geochim. Cosmochim. Acia 46:1 689- 1 705. Bufalini, J. J., and R. R. Arnts, eds. (19815. Atmospheric Biogenic Hydrocarbor~s, Vol.1, Emissions. Ann Arbor Science, Ann Arbor, Mich. Clark, W. C., ed. (1982~. Carbon Dioxide Review: 1982. Oxford University Press, New York.
TROPOSPHERIC CHEMICAL CYCLES Crutzen, P. J. (1983~. Atmospheric interactions-homogeneous gas reactions of C, N. and S containing compounds, Chapter 3 in The Major Biogeochemical Cycles and Their Interactions. SCOPE 21. B. Bolin and R. Cook, eds., Wiley, New York, pp. 67-112. Duce, R. A., V. A. Mohnen, P. R. Zimmerman, D. Grosjean, W. Cautreels, R. Chatfield, R. Jaenicke, J. A. Ogren, E. D. Pel- lizzari, and G. T. Wallace (1983~. Organic material in the global troposphere. Rev. Geophys. Space Phys. 21:921-952. Ehhalt, D. H., R. }. Zander, and R. A. Lamontagne (1983~. On the temporal increase of tropospheric CH4. ]. Geophys. Res. 88:8442-8446. Graedel, T. E. (1979~. Terpenoids in the atmosphere. Rev. Geophys. Space Phys. 17: 937-947. Keller, M., T. J. Goreau, S. C. Wofsy, W. A. Kaplan, and M. B. McElroy ~ 1983~. Production of nitrous oxide and consumption of methane by forest soils. Geophys. Res. Lett. 10:1156- 1159. Khalil, M. A. K., and R. A. Rasmussen (1983~. Sources, sinks, and seasonal cycles of atmospheric methane. [. Geophys. Res. 88:5131-5144. Logan, }. A., M. }. Prather, S. C. Wofsy, and M. B. McElroy 127 ~ 1981). Tropospheric chemistry: a global perspective. J. Geo- phys. Res. 86:7210-7254. National Research Council (1983~. Charging Climate. National Academy Press, Washington, D.C., 496 pp. Ogren, T A., and R. J. Charlson (1983~. Elemental carbon in the atmosphere: cycle and lifetime. Tellus 35B:241-254. Peterson, E., and D. T. Tingey (1980~. An estimate ofthe possible contribution of biogenic sources to airborne hydrocarbon con- centrations. A tmos. Envirorz. 14: 79-81. Rudolph, J., and D. H. Ehhalt (1981~. Measurements of C2 and C5 hydrocarbons over the northern Atlantic. i. Geophys. Res. 86: 11959- 11964. Seiler, W., and }. Fishman (1981~. The distribution of carbon monoxide and ozone in the free troposphere. [. Geophys. Res. 86:7255-7266. Wolff, G. T., and R. L. Klimiseh, eds. (1982~. Particulate Carbon. Atmospheric Life Cycle, Plenum, New York. Zimmerman, P. R., R. B. Chatfield, J. Fishman, P. J. Crutzen, and P. L. Hanst (19789. Estimates on the production of CO and H2 from the oxidation of hydrocarbon emissions from vegeta- tion. Geophys. Res. Lett. 5:679-682.
128 CURRENT ISSUES HALOGENS BY R. CICERONE The family of halogen elements (fluorine, chlorine, bromine, and iodine) display many similarities in their chemical behavior, and they occur widely in the natural geochemical environment. Although regular progres- sions and order appear in the properties of cases and solutions of one halogen versus another (i.e., ~nter-haio- gen differences), there are also many accessible forms for each halogen in the atmosphere. For example, each halogen exists in a number of volatile forms; thus there are halogen-containing gases in the earth's atmosphere. Also, for each halogen there are volatile inorganic and organic species. In the liquid phase, there are dissolved halides and halide ions, and there are halide condensates on aerosol particles. This spectrum of halogen compounds and phases and chemical reactivities gives halogens a correspondingly wide range of atmospheric behavior and of atmospheric residence times. For example, large sea-salt particles rich in halogens are airborne only for hours; some perhaloalkanes have residence times over 100 years. Both natural and man-made halogen-containing sub- stances are now involved in atmospheric chemistry. The outstanding questions and concerns (outlined below) in atmospheric halogen chemistry are thus a mixture of basic scientific issues and much more practical pollution problems. Thirty years ago, there was little basis for practical concern over halogenated compounds in the atmosphere. Today, there is a growing list of problems with halogen compounds. Although some important localized pollution problems exist, we are more con- cerned with global or semiglobal issues here. Examples include the surface sources and tropospheric sinks of CH3C1 (an important source of stratospheric chlorine, active in stratospheric O3 chemistry), and the potential climatic effects of chlorofluorocarbons and 1,1,1-trichlo- roethane, also known as methyl chloroform. Other current questions are more qualitative because too few field data are available to permit completely quantitative hypotheses. It can also be said that these questions are profound. For example, what is the source of tropospheric HC1? In the marine atmosphere, it is plausible that sea-salt aerosol chloride can be liberated as volatile HC1 as the aerosol particle becomes acidified by evaporation or by accumulation of NO2 and SO2 from dirtier air, but this is far from proven. What is the origin of gaseous CH3C1, CH3I, and CH3Br? While supersat- urated surface waters have been observed and while there are industrial sources of CH3C1 and CH3Br, what other sources are there? Is biomass burning or direct biological emission important? What is the cause of the recently observed springtime Arctic atmosphere pulse of gaseous and particulate bromine? Are tropospheric photochemicalreactionsofiodine, chlorine, or bromine potentially important to tropospheric O3 levels? What physical and chemical factors control the gas/particle partitioning of tropospheric halogens? What are the dominant gaseous inorganic species? In the remainder of this discussion, we review the available information on the distributions of atmo- spheric halogen gases and particles, their sources, atmo- spheric transformations and removal processes, and their interactions with other atmospheric cycles. Many important topics of stratospheric interest are omitted here; our focus is on the troposphere. DISTRIBUTION OF HALOGENS IN THE ATMOSPHERE In the paragraphs that follow, the discussion pro- ceeds through chlorine, fluorine, bromine, and iodine, roughly in descending order of existing knowledge. For each halogen, there is a further subgrouping: organic gases, inorganic gases, and aqueous or particulate halide. Of all the halogen-bearing gaseous species in the atmosphere, the best measured are the organic chlorine cases, C1°. These data are largely from electron-capture Detection gas chromatography. Global concentration patterns are well known for CC13F and CC12F2, at least in the boundary layer. Less detailed data are available forCH3CCl3, CC14, CH3C1, CH2C12, CHC13, andvari- ous chloroethanes and chloroethylenes. Interhemi- spheric gradients exist, especially for the man-made compounds with shorter residence times. A wide range of residence times characterizes the organic chlorine gases from approximately 100 years for CC1F3 and CC12F2 to perhaps a month for CHC13. Temporal trends (increases) have been observed and are well docu- mentedforCCl2F2, CC13F, CH3CC13, CCl4, end several other synthetic species. Numerical data on concentra- tions, gradients, and trends are available in articles listed in the bibliography. The most abundant chloro- carbon is CH3C1 (approximately 700 pptv in both hemi- spheres). In 1983 the concentrations of CC12F2 and CC13F were about 320 and 200 pptv, respectively. Other organic chlorine species are present in lesser concentra- tions. Although very few quantitative data are available, certain relatively stable aromatic compounds like chlo- robenzenes are nearly ubiquitous in the global tropo- sphere.
TROPOSPHERIC CHEMICAL CYCLES Inorganic chlorine gases are known to exist in the troposphere, apparently in higher quantities in marine air and in polluted regions. Early evidence came from trapping air in chemically basic traps and analyzing the elemental contents ofthe trapped samples. Acids such as HC1 are efficiently trapped this way, but other unidenti- fied inorganic gases can easily yield signals that appear as HC1. Chemically specific measurement methods have been employed rarely, if at all. Spectroscopic meth- ods, quite specific to HC1, have been employed to detect and measure tropospheric HC1, but only two brief reports of such work have ever been published. Indeed, HC1 is the only inorganic chlorine species ever to have been detected with chemically or spectroscopically spe- cific instrumentation in the troposphere. ScantY evi- dence suggests that there is more HC1 in the marine atmosphere than in clean continental air. Chloride in precipitation and aerosol particles were studied extensively in the 1960s, but not much since then. In marine aerosols, C1- is a major constituent by mass (1 to 10 ,ug/m3 STP), and the seawater chlorine/ sodium ratio is generally mirrored closely in marine aerosols, at least in the larger particles (diameter over 1 Em). Smaller particles often show a deficit of chlorine compared to Cl/Na in seawater. Generally, the C1- con- tent of aerosol particles decreases with distance from oceans; isolated observations of higher chloride in conti- nental aerosol particles have been attributed to large intrusions of marine air or to pollution sources. In rain- fall, there is generally more chloride (up to 10 mg/1) in marine precipitation than over continents (where C1- is as low as 0. 1 mg/1~. The total annual precipitation of C1- is probably the largest sink of atmospheric halogen, but much of this represents removal of short-lived marine boundary layer chloride. Few data~are available for C1- in snow. Organic bromine gases, Brg, are much less studied and are apparently present at much lower concentra- tions than Clg. Total Brg concentrations of 14 to 68 ng/m3 STP have been observed in clean tropospheric air; marine air generally contains more Brg than conti- nental air. Only five species of Brg have been measured specifically CH3Br, CH2Br2, CHBr3, C2H4Br2, and CF3Br; there are few, if any, southern hemispheric data; and some of these are from the Antarctic. CF2Br2 has been used as an air-trajectory tracer in regionally pol- luted air. Thus, although there are indications of higher Br°g concentrations over oceans than continents, there are no data available on interhemispheric differences of total Br°g or individual species. Recently, it has been shown that there is an annual springtime peal: in gas- eous and particulate bromine in the Arctic atmosphere. It is likely that most of these elevated gaseous levels (perhaps 20 times normal) are Br°g species. Hi. 129 Inorganic bromine gases, Brg, have been measured collectively through methods similar to those for total Clg. Very little information is available on latitudinal variations, and no individual Brg species have been detected with chemical or spectroscopic specificity. Available data suggest that Brg concentrations exceed those of Bra by at least a factor of 2. Also, diurnal varia- tions of Brg have been observed in the marine atmo- sphere. As with gaseous bromine, there are fewer data avail- able for bromide in aerosol particles than for chloride. Typical concentrations of particulate Br~ are 5 to 10 ng/ m3 STP in marine air. As with chloride, most of this bromide mass resides on large-diameter particles. Total gaseous bromine always exceeds total particulate bro- mine, usually by a factor of 5 to 20. There is some evidence for loss of bromine from aerosol for particles with time and for diurnal variations in particulate Br- levels. The springtime Arctic bromine bloom men- tioned above was observed first for particulate bromide. Approximately between mid-February and mid-May, particulate bromide levels are about 100 ng/m3 STP compared to about 6 ng/m3 STP for the rest of the year in the Arctic atmosphere. Br- in precipitation has not been measured extensively, but several studies of marine precipitation suggest that the ratio of Br- to C1- tends to exceed that for seawater. Organic iodine gases, Ig, have been measured in the same way as Clg, but with much less extensive efforts. Total gaseous iodine values of 5 to 30 ng/m3 STP have been observed with elemental analysis methods; it is thought that most of this I is organically bound. CH3I has been measured with good latitude coverage by two groups. Concentrations generally range from 1 to 6 pptv, with values between 10 and 20 pptv in the marine boundary layer over biologically active oceans. There appears to be slightly more CH3I in the northern hemi- sphere. While CH3I is the only Ig species detected specif- ically to date, it is not safe to say that it is the dominant species-relatively little effort has been expended so far. No inorganic iodine gases, Ig, have been detected with specificity, but attempts have been made to measure total Ig. Values around 10 ng/m3 STP have been reported, but it is likely that Ig species contributed to the apparent Ig values. Despite the fact that only one Ig and zero Ig species have been identified, it is clear that total gaseous I exceeds total particulate I (by a factor of 2 to 4~. In turn, iodine in marine aerosols (both I- and IO3 ~ is greatly enriched over the value expected from the iodine/chlorine ratio of seawater. Iodine enrichments of 100 to 1000 have been observed. Accordingly, it is alittle surprising that gaseous I concentrations exceed those of particulate I. No global or extended-area studies of I in precipitation have been performed.
130 Organic fluorine gases, Fg, have been studied fairly extensively, at least the chlorofluorocarbon class of Fg species (because of interest in the sources of stratospheric chlorine). The dominant Fg species by concentration is CC12F2. Total Fg values are about 1 ,ug/m3 STP, and this total is growing with time because of increasing anthro- pogenic input and the long residence time of Fg species. There are very few known occurrences of C-F bonds in natural products, marine or terrestrial; FIT species are entirely anthropogenic. For Fg, there are almost no data in nonurban air. Tropospheric observations have been limited to SF6 and to fluoride-based analyses (probably but not necessarily of HF) in polluted air. Fluoride in particles is likely due to fluorine-contain- ing contaminants released in industrial processes. This current view is based mostly on post-1977 data that have shown lower F- levels in precipitation and particles than were seen earlier with cruder analytical methods. Although the question is not settled, present indications are that observed distributions of F- in rain and parti- cles are mostly from continental sources. In the Element Cycle Matrices section of this docu- ment (Appendix C), a brief summary appears for the state of knowledge of distributions of halogens in the troposphere. SOURCES OF ATMOSPHERIC HALOGENS As in our discussion of the distributions of halogens, here we summarize briefly the sources of each halogen element, roughly in order of decreasing available knowl- edge. Organic chlorine gases have natural and industrial sources. The most prevalent species, CH3C1, is calcu- lated to be furnished to the atmosphere at a rate of about 2 x 106 metric tons annually; less than 5 percent of this source is industrial. Almost all other Clg species are anthropogenic, or mostly so. Based on knowledge from marine natural-products chemistry, it would not be sur- prising to find natural sources of CHC13, or even CC14. For CH3C1, it is suspected that natural sources include marine microbial processes and biomass burning. It is clear that there are no in situ atmospheric sources of Cod. R-C1 molecules, where R is an organic group such as CH3, are not synthesized in the open air of the earth's oxidizing atmosphere. The principal source of chlorocarbons and chloro- fluorocarbons is from the escape of these substances from their usages as solvents and degreasers and in foam-blowing processes and refrigeration units, their release from aerosol spray cans, and their use in a vari- ety of specialized processes in electronics, medicine, and manufacturing. PART II ASSESSMENTS OF CURRENT UNDERSTANDING Very little direct and verified information is available on sources of Clg species such as HC1. Although its vertical and latitudinal distributions are not known, HC1 is probably most concentrated (1 to 2 ppbv) in the marine boundary layer, where its residence time is per- haps 4 days. A global source of 108 metric tons of HC1 per year would be required to maintain this concentra- tion of HC1. Independently, it has been estimated that 3 to 20 percent of the annual input of sea-salt chloride is liberated from these particles as gaseous species, proba- bly HC1. If so, 2-12 x 108 tons/yrofHC1 is so produced. Also, volcanoes and combustion are thought to emit perhaps 6 x lo6 and 3 x 106 tons/yr, respectively, glob- ally. Although small in comparison with the global input from volatilization of sea-salt chloride, these latter sources could dominate regionally. All of these sources are represented schematically in Figure 7.6, an outline of tropospheric halogen cycles. Processes such as the reactions of sea-salt aerosols with polluted continental air masses could also release NO2C1, NOC1, C12, or even gaseous NaCI. Particulate chloride in the marine atmosphere results from sea-salt aerosol production. As sketched in Figure 7.6, these particles become airborne as a result of breaking waves, whitecap bubble-bursting, and impact of precipitation drops on the sea surface. Gas-to-particle conversion can also produce particulate chloride. Over continents, there are volcanic and com- bustion sources of HC! and particulate C1- . Organic bromine sources are much less well under- stood, especially in light of the springtime Arctic bloom mentioned above. Neither the sources of the back- ground or seasonally perturbed Brg levels are clear. It is known that usage of the agricultural fumigant, CH3Br, can inject some volatile CH3Br into the atmosphere, but quantities are uncertain. Also, it is likely that some ofthe observed atmospheric C2H4Br2 is from combustion of automobile and truck fuel additives. Also, bromoform (CHBr3) has been observed in the Arctic Ocean surface waters. Many bromine-containing marine natural products have been identified, and further investiga- tions are needed. A few other anthropogenic bromocar- bons are also of interest. Sources of inorganic bromine gases have not been explored at all. Clearly, the tropo- spheric oxidation of Brg species, largely by tropospheric OH, must produce some Brg in situ. Sources of bromide in aerosol particles and precipitation are probably an incorporation of sea-salt bromide and scavenging of Brg by clouds, rain, and aerosol particles. Sources of tropospheric iodine have also been explored only crudely. For Ig species, only CH3I has been studied. There are indications that biogenic CH3I from the oceans, possibly from biological methylation of seawater I-, is an important source. The direct emission of I' from seawater has been suggested from certain laboratory experiments In which O3 was allowed to react
TROPOSPHERIC CHEMICAL CYCLES 8 km (26K')-H IGH LATITUDES TR OPOSPH E R E , and/or Fog Aqueous ~ \' Chemistry ~I | Cloud ~N Ha ~ H NO3, NO2, SO2, ~ Cycl ing | ( Particulate X ) Washout/Rainout , .. ... . - ~\ \ I ( HX (gases), RX, XO2?, XNO? >~ CH3CI, CC12F2, CH3Br, CH31 J RX Vapor (CH3 1, ?) ~X2, HX ! Bubble & Sea Spray from: ~ ~ hi' (H 1, 12, ? ~, 1 Breaking Waves / \K ,: __ 3. Precipitations _~_ ~SEA (71.2% of total surface area of earth) _ ~.:: LAND (28.8% of total surface area of earth) 131 Dry Deposition ~\ RX from Land (?) ~HX from Land (?) FIGURE 7.6 Schematic diagram to show processes and to exemplify key species in global tropospheric halogen cycles. X denotes F. C1, Br, or I. with dissolved I-. Particulate I- (and/or IO3 ~ is highly enriched with respect to C1- in marine aerosols. Com- pared to the seawater ratios, I-/C1- is usually 100 or even 1000 times enhanced, especially on small particles. Clearly, some fractionation process is at work at the air- sea interface as particles are injected into the atmo- sphere. The involvement of iodine-rich organic films has been suggested. Aged aerosol particles do gather gaseous iodine to increase the I-/C1- ratio further, and if so, what are the sources of Ig that allow this? Fluorine sources, especially those for Fg, are similar to those for chlorine. In addition to the chlorofluorocar- bons discussed above, a few pure fluorocarbons are also of interest. One of these, CF4, is probably from alumi- num ore processing, but possibly also from various car- bon-electrode processes. Certain perfluoroethanes and perfluorocylohexanes are also entering the atmosphere now from a variety of specialized usages, often as inad- vertent emissions. Fg species such as HE are known to be pollutants from industrial processes such as aluminum refining and cement production. Also, one can imagine that gaseous HF is released from fluoride-containing aerosol particles as these particles become drier and acidified (HF is a weak acid compared to H2SO4 and HOOD. Sources of F- in particles and precipitation indude sea-salt input and industrial airborne particles. REACTIONS AND TRANSFORMATIONS OF HALOGENS Considering the many and complex reactions homogeneous gas phase, heterogeneous (gas-particle) and homogeneous liquid phase that are possible with halogens in the troposphere, research on them to date is very sparse. Consequently, very little is known about the mechanisms of halogen reactions and transformations. By contrast, stratospheric halogen reactions are limited to those in the gas phase, and these are known to be important. Organo-halogen gases, R-X, oflow molecular weighs are generally volatile and not very soluble in water. Pho- tochemical reactivity increases from fluorine to chlorine to bromine to iodine, that is, as halogens replace hydro- gen atoms in compounds; C-F bonds are stronger than C-CT, C-Br, or C-I bonds. Perfluorocarbons and per- chlorocarbons are generally stable in the troposphere and are not susceptible to attack by 03, OH, or tropo- spheric photons. Instead, they decompose only in the stratosphere and above when attacked by vacuum ultra- violet and electronically excited oxygen atoms. Other R-X species exhibit a wide range of photochemical reac- tivity. Some are photolyzed in the troposphere (e.g., CH3I), and most are dissociated by OH attack to form inorganic halogen species.
132 PART II ASSESSMENTS OF CURRENT UNDERSTANDING Inorganic halogen gases are potentially important in chloride into the atmosphere is around 6 x 109 tons/yr, tropospheric photochemical cycles, although no major but much ofthis chloride is airborne for a day or less. By specific role has yet been proven. Volatile species exist contrast, the annual input of CC12F2 is only about 3 x for each of the halogens, and a great variety of species 105 tons/yr of chlorine, but it is transported to the strato need to be considered. For chlorine and fluorine, the sphere, and its residence time is 100 years. Accordingly, stability of HC1 and HF greatly slows regeneration of C] the global atmospheric cycles of halogens and the sinks and F atoms. Hydrogen donors, R-H, react readily with for atmospheric halogens are made up of terms that are C1 and F to form HC1 and HF, so the free atoms and their difficult or meaningless to compare. Oxides are not thought to be very prevalent. Longer The largest single sink for atmospheric halogens is chain lengths for gas-phase catalytic processes are possi- represented by precipitation. Perhaps two-thirds of the ble for bromine andiodine because HBr and HIareless large sea-salt particles that carry the most mass are stable once formed. Possible roles for Ig species in removed by precipitation, and one-third by gravita destroying O3 and affecting other tropospheric photo- tional settling. Ninety percent of the removal occurs chemical cycles have been proposed, but great uncer- over oceans. Although these values have been deduced tainties exist. Examples include lack of information on for chlorine, they are probably similar for bromine and the levels of Ig concentrations, unavailability of certain iodine, whose sources are predominantly marine. Fluo rine sinks are probably dissimilar in their distribution. As is indicated in Figure 7.6, dry deposition removes halogen gases and particles. Surface-active species like HC1, HF, or HOC1 are probably most affected by dry deposition; the least-affected species are probably the organo-halogen gases. Finally, those portions of the atmospheric halogen cycles that penetrate the strato sphere, for example, perhalocarbons like CC12F2, have their upward flows counterbalanced by downward return flows of HC1 and HF in precipitation and dry deposition, at least in a steady state. chemical kinetic data, and possible interferences by het- erogeneous processes. Until there are some field data on specific halogen-containing inorganic species, little pro- gress can be expected. Stratospheric investigations have provided some guidance, especially for chlorine and bromine, but direct tropospheric studies are needed. Heterogeneous reactions and transformations need attention, but have received little. There is strong evi- dence that heterogeneous reactions are responsible in great part for the very existence oftropospheric HCl, yet few, if any, mechanistic studies have been performed. There is some evidence that particulate bromine con- centrations increase at night and decrease by day and that gaseous bromine exhibits opposite diurnal behav- ior, but no studies of possible mechanisms are available. Similarly, the processes that lead to loss and/or uptake of halogens from marine aerosols of various sizes have not yet been investigated, nor have analogous processes in precipitation been studied. Such investigations are greatly hampered by a dearth of field data and of funda- mental kinetic and photochemical data from the labora- tory. Equilibrium-type data such as vapor pressures, even when available, are not necessarily valid when complicated multiphase, multiconstituent mixtures are to be considered. Homogeneous aqueous-phase reac- tions and transformations are potentially very impor- tant in clouds, rain, and water-coated aerosol particles, for example, with halogens as oxidizing agents, but vir- tually no research has been performed on this topic. REMOVAL PROCESSES FOR HALOGENS A very large spectrum of time constants exists for the residence times of various halogen-containing gases and particles, and the global atmospheric cycles of the halo- gens encompass both large and small reservoirs and transfer rates. For example, the annual input of sea-salt BIBLIOGRAPHY Barnard, W. R., and D. K. Nordstrom (1982~. Fluoride in precipi- tation: II. Implications for the geochemical cycling of fluorine. Atmos. Erwiron. 16:105-1 1 1. Berg, W. W., P. D. Sperry, K. A. Rahn, and E. S. Gladney (~1983~. Atmospheric bromine in the Arctic. J. Geophys. Res. 88:6719- 6736. Chameides, W. L., and D. D. Davis (1980~. Iodine: its possible role in tropospheric photochemistry. J. Geophys. Res. 85: 7383-7398. Cicerone, R. J. (~1981~. Halogens in the atmosphere. Rev. Geophys. Space Phys. 19: 123- 139. Duce, R. A., J. W. Winchester, and R. VanNahl (~1965~. Iodine, bromine and chlorine in the Hawaiian marine atmosphere. I. Geophys. Res. 70: 1775-1799. Eriksson, E. (1959~. The yearly circulation of chloride and sulfur in nature: meteorological, geochemical and pedological implica- tions, 1. Tellusll:375-403. Eriksson, E. (~1960~. The yearly circulation of chloride and sulfilr in nature: meteorological, geochemical and pedological implica- tions, 2. Tellus 12: 63 - 109. Rasmussen, R. A., M. A. K. Khalil, R. Gunawardena, and S. D. Hoyt (~1982~. Atmospheric methyl iodide (CH3I). J. Geophys. Res. 87:3086-3090. Singh, H. B., L. J. Salas, and R. E. Stiles (~1983~. Methyl halides in and over the eastern Pacific. i. Geophys. Res. 88:3684-3690. World Meteorological Organization (~1981~. The Stratosph~e 1981. WMO Global Ozone Research and Monitoring Project Report No. 11. 503 pp.
133 TRACE ELEMENTS BY R. DUCK CURRENT ISSUES The cycles of most trace elements in the troposphere have received relatively little attention from atmo- spheric chemists for several reasons. Trace elements, i.e., all elements except C, N. O. S. H. and the halo- gens, are present in such low concentrations that they have little impact on the overall photochemistry of the troposphere, its acid-base characteristics, or on climate. Many, if not most, trace elements are present entirely in the particulate phase and are not directly involved in gas-particle conversion processes or other aspects of gas- phase tropospheric chemistry. Being primarily present in aerosol particles, their tropospheric residence times are of the order of days to a few weeks, and there has been relatively little effort to evaluate global-scale changes in their distribution caused by human activity. The tropospheric chemistry of many trace elements is an important part of the present-day overall biogeo- chemical cycles of these elements, but in most cases the tropospheric part of these cycles is poorly known. For example, the mobilization of Hg in the environment, whether it be from natural or pollution sources, is pri- marily through the troposphere in the gas phase, but very few data are available on the Hg concentrations in the remote troposphere, and even less is known about its chemical speciation and primary sources. Phosphorus is one of the primary nutrients in both the terrestrial and marine biosphere, and there is some evidence that tro- pospheric transport of P to the ocean may be significant in certain regions. However, the understanding of the spatial and temporal distribution of P in the tropo- sphere, of its chemical forms and sources, and even of whether a long-surmised gaseous species exists is . . . extreme y primitive. Trace elements can most conveniently be separated into two groups: Group A includes those elements that almost certainly spend their entire tropospheric lifetime on aerosol particles. Group B includes those elements for which a vapor phase, or likelihood of a vapor phase, exists. Group A includes such lithophilic elements as Al, Fe, Na, Ca, Mg, Si, V, Cr. Cu. Mn, and the rare earths. Group B includes such elements as B. Hg, Se, As, Sb, Cd, Pb, and possibly Zn and P. SOURCES AND TRANSPORT Trace element distribution patterns in aerosol parti- cles are of considerable use in determining sources, transport paths, and deposition for the particles them- selves. This is quite valuable since aerosol particles are an important end product for virtually all tropospheric cycles through heterogeneous and homogeneous reac- tions, and they play a major role in weather and climate. Through the use of interelement ratios, it is often possi- ble to determine the sources for aerosol particles. For example, Al/Sc ratios on aerosol particles similar to that present in the earth's crust are an indication of a crustal weathering source, whereas Na/Mg ratios similar to that in the ocean suggest a marine source. The use of such "reference" elements as Al or Sc for the crust, Na or Mg for the ocean, and noncrustal V (i.e., that vana d~um present on aerosol particles that Is not derived from the earth's crust) for combustion of residual fuel oil or Pb for the combustion of gasoline containing tetraethyllead has proven quite useful. There are many sources that have not been so easily tagged, but efforts to determine appropriate trace element signatures are con- tinuing. For example, B is being examined as a signa- ture for coal burning, and As for smelter operations. This approach is potentially useful for identifying other specific sources of primary aerosols, including the ter- restrial biosphere, volcanism, extraterrestrial particles, and a number of specific pollution sources. Recent efforts to identify regional source areas of aerosol parti- cles through the use of a number of trace elements also show considerable promise. Trace elements used have included Se, Sb, As, Zn, In, noncrustal V, noncrustal Mn, and their interelemental ratios. There is growing evidence that anthropogenic proc- esses, followed by long-range tropospheric transport, can result in significant changes in the tropospheric and oceanic concentrations of certain toxic and essential trace elements on the near-global scale. For example, concentrations of Pb, both in the marine troposphere and in the surface waters of the Atlantic and Pacific Oceans, particularly in the northern hemisphere, are considerably elevated as a result of the burning of gaso- line containing tetraethyllead on the continents and its subsequent tropospheric transport over and deposition to the oceans. The mobilization of other toxic elements such as Hg and Se by fossil fuel combustion and As by smelters and in herbicides and defoliants may be equiva- lent to or greater than mobilization by natural sources. In fact, as is the case for most cycles, one can make much more accurate estimates of the global source strengths from pollution sources for trace elements than from such natural sources as volcanism, the oceans, and the bio- sphere. In particular, there is virtually no information on the production of vapor-phase trace elements or direct pro- duction of aerosol particles containing trace elements by
134 the terrestrial biosphere. The apparent increased vola- tilization of Hg by higher plants (relative to release from unvegetated soils of comparable concentration) has been explained as a "detoxification" process, although it may simply be an expression of the ion-concentrating processes of plants combined with the reducing poten- tials created by the charge separation processes of metabolism. In any case, vegetation appears to have a major role in the cycling of Hg. For a number of other elements, including As, Sn, Se, Pb, and Sb, biological methylation has been observed in the laboratory, and methylated forms of many of these elements have been observed in highly polluted areas. For a few elements, ionic methyl compounds have been observed in uncon- taminated regions. A further understanding of the bio- logical production of methylated metals requires the development of specific detection capabilities for these species. The entire area oftrace element release from the biosphere requires considerable effort in the future. DISTRIBUTION A growing data base is developing on the trace ele- ment composition of aerosol particles in remote regions. Reasonably good data are available over short time periods from the boundary layer in both polar regions and over the Atlantic and Pacific Oceans. Virtually no data are available on the vertical distribution of trace elements in these regions, however, and this information is critical to evaluate sources and fluxes of the trace elements. Information on the mass-size distribution of trace elements on aerosol particles is of considerable value in ascertaining sources and source processes for these elements. Many additional data of this type are required. Very little is Mown about the chemical form of the trace elements in the vapor phase, but for elements in Group B. a vapor phase does, or is expected to, exist. Mercury apparently exists primarily as a gas in the form of elemental Hg, with evidence emerging for some organic forms as well, probably methylated. Mercury is one of the few metals whose ions can be reduced to the metallic state at reduction potentials frequently found in biological systems. The metal has an appreciable vapor pressure at 25°C (~1 x 10-3 mm Hg3; thus the pres- ence of gaseous elemental Hg in the troposphere is not . . surprlslng. Although the vapor phase apparently dominates tro- pospheric B and B(OH)3 has been suggested as the pri- mary vapor phase, no measurements have corroborated the presence of B(OH)3. Although there are a number of volatile borane derivatives, their formation requires much more reducing conditions than those apparently achieved by microorganisms under anoxic conditions. Measurement of specific B species in the troposphere is PART II ASSESSMENTS OF CURRENT UNDERSTANDING clearly required before even a rudimentary understand- ing ofthe B cycle is possible. In many respects, Se parallels S in geochemical behavior. Vapor-phase Se has been observed in remote and urban regions, where it may account for about 25 percent of the total Se present. However, except for dimethylselenide and dimethyldiselenide measured in urban and near-urban areas in Belgium, the chemical form of the vapor phase of Se is unknown. Approximately 10 percent of the As in the marine boundary layer is apparently present in the vapor phase. The form of the vapor-phase As is also unknown, although dimethyl arsinic acid has been observed in the oxic marine environment and trimethyl arsine is known to be produced by certain fungi. Methylated forms of Se, Sb, Pb, and other trace ele- ments have been observed in other compartments of the environment (e.g., the ocean and plants). In the case of Sb, Hg, and Pb, it is unclear whether methylation can occur in oxic regions or only under anaerobic condi- tions, where microorganisms are probably of considera- ble importance. For all these trace elements in the vapor phase, the data base from remote regions is extremely small, in some cases being as few as 5 to 10 samples. TRANSFORMATIONS ED SINKS Very little information is available on transformation reactions involving trace elements in the troposphere. The residence time for the vapor phase of many trace elements in the troposphere may be very short, perhaps only minutes or hours, as is probably the case for certain As and Pb species. From mass balance considerations, the residence time of total (vapor plus particle) As in the global troposphere has been estimated as ~ 10 days. Evidence suggests that the residence time of vapor- phase As is considerably shorter, however. For some trace elements the vapor phase probably has a longer residence time than the particulate phase and is the dominant phase in the troposphere, as is likely the case for Hg and perhaps B. The residence time for elemental Hg may be as long as several months. Estimates of the residence time of vapor-phase Se, B. etc., compounds have not been made. It is likely that the removal of these trace elements is primarily governed by precipitation processes. For all these elements, woefully little is known about the specific chemical species present. Information on the chemical form of these trace elements in the vapor phase and details of their tropospheric reaction paths and rates are required before the importance oftransfor- mation reactions to their tropospheric cycles can be eval- uated. Certain trace elements, particularly transition metals, may be important as catalysts for reactions in cloud and rain droplets. For example, the oxidation of
TROPOSPHERIC CHEMICAL CYCLES SO2 to sulfate in solution is enhanced by the presence ot Mn ions. The importance of transition metals as cata- lysts depends on a number of factors, including abun- dance, chemical form, stable oxidation states, bonding properties, and solubility. Copper, manganese, and per- haps vanadium may be important as homogeneous cat- alysts in solution. For heterogeneous catalysis, solubility is not important; and the metals above, as well as Fe, Ti, and perhaps Cr. are of potential importance. The role played by trace element catalysis in aqueous atmo- spheric chemical reactions is largely unknown at present, but potentially very important and should . . . receive 1ncreasec . attention. BIBLIOGRAPHY Braman, R. S., and M. A. Tompkins (1979~. Separation and determination of nanogram amounts of inorganic tin and methyltin compounds in the environment. Anal. Chem. 51:12- 19. Brinckman, F. E., G. i. Olson, and W. P. Iverson (1982~. The production and fate of volatile molecular species in the environ- ment: metals and metalloids, in Atmospheric Chemistry, E. D. Goldberg, ed. Springer-Verlag, Berlin, pp. 231-249. Cunningham, W. C ., and W. H. Zoller ~ 1 98 1 ). The chemical composition of remote area aerosols.~. Aerosol Sci. 12: 367-384. Duce, R. A., R. Arimoto, B. I. Ray, C. K. Unni, and P. I. Harder (1983~. Atmospheric trace elements at Enewetak atoll: I. Concentration, sources, and temporal variability.~. Geophys. Res. 88:5321-5342. Fitzgerald, W. F., G. A. Gill, and A. D. Hewitt (1983~. Air/sea exchange of mercury, in Trace Metals in Seawater, C. S. Wong, E. Boyle, K. W. Bruland, I. D. Burton, and E. D. Goldberg, eds. Plenum, New York, pp. 297-315. 135 Fogg, T. R., R. A. Duce, and J. L. Fasching (1983~. Sampling and determination of boron in the atmosphere. Anal. Chem. 55:21 79-2184. Galloway, I. N., I. D. Thornton, S. A. Norton, H. L. Volchok, and R. A. N. McLean (1982~. Trace metals in atmospheric deposition: a review and assessment. Aimos. Environ. 16:1677- 1700. Graham, W. F., and R. A. Duce (1979~. Atmospheric pathways of the phosphorus cycle. Geochim. Cosmochim. Acta 43: 1195- 1208. Harrison, R. M. and P. H. Laxen (1978~. Natural sources of tetraalkyllead compounds in the atmosphere. Nature 275: 738- 739. Jiang, S., H. Robberecht, and F. Adams (1983~. Identification and determination of alkyl selenide compounds in environ- mental air. Atmos. Environ. 17: 1 1 1-1 14. Lantzy, R. L., and F. T. Mackenzie (1979~. Global cycles and assessment of man's impact. Geochim. Cosmochim. Acta 43:511- 515. Maenhaut, W., H. Raemdonck, A. Selen, R. Van Grieken, and I. W. Winchester (1983~. Characterization of the atmospheric aerosol over the eastern equatorial Pacific. i. Geophys. Res. 88:5353-5364. Mosher, B. W., and R. A. Duce (1983~. Vapor phase and particu- late selenium in the marine atmosphere. ]. Geophys. Res. 88:6761 -6768. Nriagu, J. O. (1979~. Global inventory of natural and anthropo- genic emissions of trace metals to the atmosphere. Nature 279:409-411. Rahn, K. A., and D. H. Lowenthal (1984~. Elemental tracers of distant pollution aerosols. Science 223: 132- 1 39. Settle, D. M., and C. C. Patterson (1982~. Magnitudes and sources of precipitation and dry deposition fluxes of industrial and natural leads to the North Pacific at Enewetak. ]. Geophys. Res. 87:8857-8869. Slemr, F., W. Seller, and G. Schuster ~ 1 98 1 ). Latitudinal distribu- tion of mercury over the Atlantic Ocean. J. Geophys. Res. 86:1159-1166. Walsh, P. R., R. A. Duce, and J. L. Fasching (1979~. Consider- ation of the enrichment, sources, and flux of arsenic in the troposphere. J. Geophys. Res. 84: 1719-1726.
136 AEROSOL PARTICLES BY I. M. PROSPERO An aerosol is defined as a suspension of fine liquid and/or solid particles in a gas. In the case of interest to us, that gas is the atmosphere. Although in the strict sense of the definition the word aerosol refers to the particle and gas phases as a system, the term is often used to refer to the particle phase alone. The subject of aerosol particles is somewhat anoma- lous in the general context of this section on cycles since all the other subjects focus on specific chemical species. Indeed, in most cases, the atmospheric aerosol is the end product of many of the chemical processes acting on the aforementioned species. Many of these reaction prod- ucts are relatively unreactive in the aerosol phase. Because of this unreactivity and because of the relatively short residence time of aerosols in the troposphere (on the order of a week or two), the aerosol phase can be considered to be, in effect, the sink for many gas-phase species (see Figure 7. 7~. Aerosol particles are treated as a separate cycle because they can have an impact on a number of impor- tant physical processes in the atmosphere. The relation- ship between aerosol properties and atmospheric proc- esses is depicted in Part 1, Figure 2.2. For example, aerosols play an important role in the hydrological cycle they can affect cloud microphysics, which, in turn, can affect the types, amounts, and distribution of FIGURE 7.7 Aerosols as an end product of atmospheric reactions. Major reaction pathways for gas-phase constituents are depicted by solid lines. Interactions between chemical families are indicated by dashed lines. Heavy (double) arrows show key heterogeneous pathways involving aerosols (A) and precipitation (P) (Turco et al., 1982). rainfall. Clouds play a critical role in fixing the albedo of the earth; thus, if aerosols affect the amount, type, and distribution of clouds, then changes in aerosol concen- tration and properties could have the effect of changing the albedo. Aerosol particles can affect light as it passes through the atmosphere by the mechanisms of scattering and absorption. The most obvious radiative consequence of airborne particles is the appearance of haze and degra- dation of visibility. Less obvious, but more important, are the possible effects of these same particles on the heat balance ofthe earth. Particles can cause a decrease in the amount of radiation reaching the ground, can increase or decrease the albedo, and, if the aerosol absorbs light, can cause atmospheric heating. In order to understand these optical effects, it is necessary to know the chemical composition of the aerosol and its size, because these characteristics will determine the aerosol scattering and absorption properties. Also, the composition of aerosols is often size dependent; thus a specific physically (or chemically) active species could be concentrated in a limited portion of the particle size spectrum. Finally, there is the concern about the impact of many pollutants on health. The vector for many ofthese harm- ful species is the aerosol particle. However, the ability of aerosol particles to penetrate to the lung is dependent on A HO2 ~ HNO2 ~ p NO ~ ''ti ~ HNO3: P. A A, p ~ ~02 - HO2NO2 I \ \ ~ ~ A' ,, ---~ NH NH3 ~ \ ~ a' A, P t He' - ~(CH3)2S C - ;: I :~HSO3: CH3OOH CHO) CS2 H2S H2SO4 ~ cod ~ P,A A,P
TROPOSPHERIC CHEMICAL CYCLES the aerosol size and its chemical properties as a function ~ OI SlZe. . ,, SOURCES From the standpoint of production processes, aerosol particles can be categorized as being either primary or secondary. Primary aerosols are those directly emitted as a solid or a liquid, while secondary aerosols are derived from materials initially emitted as gases. Impor- tant natural primary aerosols are the salt residue from sea spray, wind-blown mineral dust, ash from volcanic eruptions, and organic particles from biota (pollen, spores, debris, etch. A major class of anthropogenic primary aerosol is smoke particles. However, the burn- ing of land biota is another major source of smoke; this source can be either natural or man-made, depending on how the fire was started. There are a number of major sources of secondary aerosols. As described in preceding sections, many bio- logical systems emit gaseous species that are eventually converted to aerosols. Finally, many of man's activities release gases that are aerosol precursors. Estimates of annual aerosol mass production rates are summarized in Table 7.8. The quality of these data leaves much to be desired. The wide range of estimates for some species is a reflection of the poor state of knowl- edge of their sources. Nonetheless, it is clear that knowl- edge of the input rates from anthropogenic sources is much better than that of the rates from natural sources. In the category of primary particles, natural sources (predominantly sea salt and soil dust) far outweigh anthropogenic sources. However, it must be borne in mind that the anthropogenic sources are concentrated in a relatively small area and hence will be much more significant on a local and regional scale. Obviously, the major source of sea-salt aerosol is the ocean. Although a fair amount of research has focused on the sea-salt aero- sol production mechanism and especially on the rate of production as a function of various environmental parameters, there is still considerable debate on the results of such work and their interpretation. The rates of production and the physical and chemical characteris- tics of sea-salt aerosol are important for a number of reasons. For example, salt spray may play an important role in transporting trace metals and organic materials from the ocean into the atmosphere and in absorbing or reacting with gaseous species in the marine boundary layer. Because the oceans are so large and the mass of sea-salt aerosol is so great, these processes must be understood in order to understand global chemical cycles in general. The major sources of soil aerosols are arid regions. Clearly, much soil dust, such as that from the Sahara, is 137 generated by purely natural processes. However, in some cases, the distinction between natural and anthro- pogenic sources is not so clear. For example, recent work has shown that large quantities of soil dust are being transported out of Asia far into the North Pacific. Much of this material is believed to have been deflated from agricultural regions in China in the spring after the soils have been ploughed for planting. Also, in the United States, the primary standard for total suspended partic- ulate materials, as defined in the Clean Air Act, is most widely violated in agricultural areas because of the mobilization of soil dust by farming activity. It is clear from Table 7.8 that there is considerable uncertainty in the rates of mobilization of soil dust. However, it is even more uncertain as to what fraction is derived from natu- ral sources as a consequence of natural processes. The range of estimates for production rates from vol- canoes and biomass burning is extremely large. This uncertainty is a consequence of the difficulty in obtain- ing data on sources that are sporadic, widely dispersed, and relatively inaccessible. TRANSPORT Aerosols, whether primary or secondary, can be transported great distances in the atmosphere. As previ- ously stated, large quantities of soil aerosols are rou- tinely transported thousands of kilometers over the oceans from their sources in continental regions. Simi- larly, pollutant aerosols can be transported great dis- tances. For example, of the acid species in aerosols over the northeast United States and Canada, a large fraction is derived from sources in the central United States. On a larger scale, the trace metal composition of aerosols in Arctic haze episodes suggests that the sulfate-rich parti- cles are adverted primarily from sources in Europe and Asia. These interpretations are supported by meteoro- logical studies and trajectory computations. Aerosol species such as sea salt and soil dust are rela- tively inert, and their principal physical and chemical properties remain essentially unchanged during trans- port in the atmosphere. Because of their relatively con- servative nature, these species can serve as tracers for atmospheric transport and removal processes. Some research along these lines has already begun with some success. Given a sufficiently large data base, measure- ments of these species could be used to validate atmo- spheric transport models that are currently under devel- opment. Unreactive species are most useful for such validation because it is not necessary to make any assumptions about in situ chemistry during transport. An advantage of using aerosols as tropospheric tracers is that their lifetime is of the same order as the lifetime of a typical synoptic meteorological event, about a week.
Source After Peterson and Junge (1971),a <5,um After Hidy and Brock (1971)a 138 PART II ASSESSMENTS OF CURRENT UNDERSTANDING TABLE 7.8 Estimates of Global Particle Production from Natural and Man-Made Sources ~ ~ o6 tons/yr) After Study of Marl 's Impact on Climate ( 1 9 7 ~ ja Other < 20,um < 6 lamb Estimatesa Man-Made Direct particle production Transportation 1.8 Stationary fuel sources 9.6 Industrial processes 12.4 Solid waste disposal 0.4 Miscellaneous 5.4 Subtotal 29.6 37-110 10-90 6-54 54 126 Particles formed from gases Converted sulfates 200 110 130-200 Converted nitrates 35 23 30-35 Converted hydrocarbons 15 27 15-90 Subtotal 250 160 175-325 270 Total man-made 280 269 185-415 396 Natural Direct particle production Sea salt Windblown dust Volcanic emissions Meteoric debris 500 250 25 o 1095 300 180 60-360 100-500 60-300 4 0.02-0.2 25-150 15-90 Forest fires 5 146 3-150 Subtotal 780 1610 428- 1100 1730 Particles formed from gases Converted sulfates 335 37-365 130-200 Converted nitrates 60 600-620 140-700 160 Converted hydrocarbons 75 182-1095 75-200 154 Subtotal 470 2080 345-1100 1319 Total natural -1250 3690 773-2200 3049 Grand Total - ~1530 3959 958-2615 3445 1000-2000 70 60-360 128 + 64 200 + 100 4.2 1-10 0.02-0.2 aFor references see Bach (1976) or Prospero et al. (1983). hValues are for particles < 6 ,um as recomputed byJaenicke ( 1980). SOURCE: Bach, 1976. TRANSFORMATIONS Gases can react in the atmosphere to produce nonvol- atile products that end up in the aerosol phase. It is clear from Table 7.8 that for many species the quantities of aerosol produced in this manner equal or exceed the quantities emitted directly as particles. This is true for sulfate and nitrate species that are currently of interest because of their role in the formation of acid rain. Clouds play a dominant role in the formation, modifi- cation, and removal of aerosols. The condensation of water vapor on particles, and the phoretic, diffusive, or inertial capture of particles by droplets lead to the incor poration of particles within the aqueous phase. Solution reactions, including those with dissolved gases, become possible, and transformations can occur. If subsequent droplet growth leads to precipitation, the aerosol is removed from the atmosphere. However, if the droplet reevaporates, as it does in over 90 percent of the cases, then the aerosol is regenerated, but its size and composi- tion are changed. Cloud cycling is probably the major mechanism for modifying the atmospheric aerosol in the lower troposphere. In contrast to the processes of parti- cle interactions and coagulation, which are reasonably well understood, the cloud cycling aspects of aerosol- hydrometeor-gas interaction are poorly understood.
TROPOSPHERIC CHEMICAL CYCLES REMOVAL PROCESSES Precipitation is the major mechanism for the removal of aerosols from the atmosphere. For example, it has been shown that 80 to 90 percent of the radioactive fallout deposited on the earth's surface was brought down in precipitation. In turn, the composition of pre- cipitation is determined to a considerable extent by the composition ofthe aerosol phase. Gravitational removal is important only for relatively large particles (i.e., those larger than about 10-pm diameter). Sedimentation will be important for soil aerosols close to the source area and for sea-salt aerosols. Sedimentation is generally not important for anthropo- genic materials. Pollution control measures have sharply reduced the rates of emission of large particles. On the other hand, the size of secondary aerosols is less than 1-,um diameter, and consequently, these aerosols have a very small settling velocity. DISTRIBUTION Because of the relatively short residence time of aero- sols in the atmosphere, their distribution will be closely linked to the distribution and activity of sources and to the controlling meteorological phenomena. Thus, in order to characterize any trends in the concentration and distribution of aerosols, it will be necessary to sam- ple frequently on a broad spatial scale that encompasses the suspected major source regions and the dominant meteorological systems. Some well-conceived regional sampling programs are currently in place. However, on a larger scale, with a few exceptions, current sampling efforts leave much to be desired from the standpoint of the species studied, the quality ofthe data, the frequency of sampling, and the location of stations. CONCLUSIONS We can identify a number of areas that warrant fur- ther research on aerosols: 1. The role of aerosols in geochemical transport and anthropogenic impacts. The atmosphere is an impor- tant mode of transport for many species. For example, the anthropogenic emissions of sulfur, mercury, and lead to the atmosphere already exceed the stream loads for these elements, while the emissions of copper, arsenic, zinc, tin, selenium, molybdenum, antimony, and silver are within a factor of 10 of stream fluxes. 2. Gas-particle processes. The photochemical and chemical reactions that initially transform gases into sec- ondary reaction products are extremely complex and 139 not yet fully understood. The particles formed by these mechanisms are mostly in the "fine particle" size range (i.e., submicrometer). It is estimated that several hun- dred million tons of fine particles are formed every year as a consequence of the emission and subsequent reac- tion of natural and anthropogenic gaseous species. The role of organics in aerosols is poorly understood. Yet, the concentration of particulate organic carbon in the atmosphere is quite high. For example, in most ocean areas, the mean value is comparable to that of mineral aerosols and non-sea-salt sulfate and nitrate. Gas-to-particle conversion appears to be a major mech- anism for the production of fine-particle carbon over the oceans and also over the continents. Unfortunately, there are very few concurrent measurements available for both the vapor and the particulate phases. 3. The role of aerosols in the hydrological cycle. It is clear that clouds play a major role in the formation and removal of aerosols. As stated earlier, any process that acts on clouds could have an impact on weather and climate. There is now sufficient evidence to conclude that anthropogenic emissions, especially of sulfur and nitrogen species, do indeed have an impact on cloud microphysics. Given the importance of sulfur in the hydrological cycle and bearing in mind that about half of the global flux of sulfur has an anthropogenic origin, there is good cause for concern that man may be altering weather on a larger scale. Of particular interest are the possible effects on the urban and regional scale where the magnitude of the anthropogenic sulfur sources is dramatically higher for example, in the eastern United States, where it is 10 times that of natural sources. The assessment of the impact of man on weather or climate is difficult for a number of reasons. One has to do with the fact that climate is subject to variations that are completely natural in origin. Thus, until there is a better understanding of the mechanisms that determine climate, it will be difficult to ascertain the ways in which it has been changed, or might be changed, by anthropo- genic activities. Therefore, research efforts directed at elucidating impacts must be balanced by efforts directed at gaining an understanding of basic processes. In the case of aerosols, one of the basic processes of great importance is the role of aerosols in cloud physics. Although the impact of aerosols on the hydrological cycle from the standpoint of precipitation quantity and distribution cannot be quantitatively assessed with cer- tainty, it can be stated with certainty that there has been a very marked impact on precipitation quality, most notably in the increased acidity of rain. 4. Radiative transfer. There are a number of differ- ent aerosol types that are important and that could play
140 a major role in climate by altering the radiation budget of the earth: a. Volcanic debris. Most important in this cate- gory are the sulfur species that are injected into the stratosphere, where they are oxidized to sulfuric acid droplets, which have a residence time of years. b. Soil dust. About one-third of the surface of the continents is arid and a potential source of soil dust. Here the impact of man on soil mobilization is a major concern. c. Elemental carbon (soot). This material is highly efficient absorber of radiation; thus it is impor- tant that its abundance and distribution be measured. However, scientists have a very poor idea of the global budget of carbon because much carbon is produced in remote regions by slash-and-burn agricultural practices and because carbon aerosols are difficult to measure with current analytical techniques. 5. Characterization of temporal and spatial trends. A recurring conclusion in the assessments of the possible impact of aerosols on climate is that there is a serious lack of information about the composition, concentration, and physical properties of aerosols and their temporal and spatial variability. Despite the undeniable evidence that anthropogenic materials are being transported over great distances, there is no evidence that the particles have significantly reduced atmospheric transmission in remote regions. For example, there is no evidence of any long-term decrease in transmission in the data from the Manna Loa Observatory or from the Smithsonian Astronomi- cal Observatories. However, we do not mean to mini- mize the possibility that such increases may be in the process of occurring; we merely emphasize that, with current measurement techniques and the length of the records on hand, one cannot separate any trends, if they exist, from the natural variability of the atmospheric aerosol. Indeed, it is essential that the natural processes be understood before any anthropogenic effects can be identified. BIBLIOGRAPHY Bach, W. (1976~. Global air pollution and climatic change. Rev. Geophys. Space Phys. 14:429-474. Barry, R. G., A. D. Hecht, J. E. Kutzbach, W. D. Sellers, T. PART II ASSESSMENTS OF CURRENT UNDERSTANDING Webb, and P. B. Wright (1979). Climatic change. Rev. Geophys. Space Phys. 17:1803-1813. Blanchard, D. C. (1984~. The production, distribution and bacte- rial enrichment of the sea-salt aerosol, in The Air-Sea Exchange of Gases and Particles, W. G. N. Slinn and P. Liss, eds. D. Reidel, Boston, Mass., pp. 407-454. Charlson, R. I., and H. Rodhe (1982~. Factors controlling the acidity of natural rainwater. Nature 295:683-685. Dockery, D. W., and J. D. Spengler ( 1981~. Indoor-outdoor rela- tionships of respirable sulfates and particles. Atmos. Environ. 15:335-343. Edmonds, R. L., ed. (1979~. Aero biology, The Ecological Systems Approach, US/IBPSynthesis Series, Vol. 10. Dowden, Hutchinson and Ross, Stroudsburg, Pa., 386 pp. Friedlander, S. K. (1978~. Aerosol dynamics and gas-to-particle conversion, in Recent Developments irz aerosol Science, D. T. Shaw, ed. Wiley, New York, pp. 1-24. Galloway, J. N., G. E. Likens, W. C. Keene, and J. M. Miller (1982~. The composition of precipitation in remote areas of the world.~. Geophys. Res. 87:8771-8786. Hinds, W. C. (1982~. Aerosol Technology. Wiley, New York, 424 pp. Holland, W. W., A. E. Bennett, I. R. Cameron, C. du V. Florey, S. R. Leeder, R. S. F. Schilling, A. V. Swan, and R. E. Wailer (1979~. Health effects of particulate pollution: reappraising the evidence. J. Epidemiol. 110:525-659. Jaenicke, R. (1980~. Atmospheric aerosols and global climate. J. AerosolSci. 11:577-588. Lodge, Jr., J. P., A. P. Waggoner, D. T. Klodt, and C. N. Crain (19813. Non-health effects of airborne particulate matter. Atmos. Environ. 15:431 -48-2. Pewe, T. L., ed. (1981~. Desert dust: origin, characteristics and effect on man. Geol. Soc. Amer. Spec. Pap. 186, 303 pp. Podzimek, J. (1980~. Advances in marine aerosol research. J. Res. Atmos. 14:35-61. Prospero, J. M. (1981~. Aeolian transport to the world ocean, in The Sea, Vol. 7, The Oceanic Lithosphere, C . Emiliani, ed. Wiley Interscience, New York, pp. 801-874. Prospero, I. M., V. Mohnen, R. .iaenicke, R. Charlson, A. C. Delany, l. Moyers, W. Zoller, and K. Rahn (1983~. The atmo- spheric aerosol system: an overview. Rev. Geophys. Space Phys. 21: 1607-1629. Rahn, K. A. (1981~. Relative importance of North America and Eurasia as sources of Arctic aerosol. Atmos. Er~virorz. 15:1447- 1456. Seller, W., and P. I. Crutzen (1980~. Estimates of gross and net fluxes of carbon between the biosphere and the atmosphere from biomass burning. Clim. Change2:207-247. Subcommittee on Airborne Particles (SAP) (1979~. AirborneParti- cles. National Research Council. University Park Press, Balti- more, Md.,343 pp. Toon, O. B., and J. B. Pollack (1980~. Atmospheric aerosols and climate. Amer. Sci. 68: 268-278. Turco, R. P., O. B. Toon, R. C. Whitten, R. G. Keesee, and P. Hamill (1982~. Importance of heterogeneous processes to tropospheric chemistry: Studies with a one-dimensional model, in HeterogeneousAtmospheric Chemistry. Geophysical Mon- ograph Series, Vol. 26, R. Schryer, ed. American Geophysical Union, Washington, D.C., pp. 231-240.
