
1
An Introduction to the Changing Economics of Technological Innovation in Medicine
ETHAN A. HALM and ANNETINE C. GELIJNS
The rapidly rising costs of health care became an increasingly urgent issue of policy concern during the 1980s, and they can be expected to remain so in the 1990s. Technological change generally is believed to be an important factor driving costs up, although, as Neumann and Weinstein argue in this volume (Chapter 2 ), significant conceptual and practical problems defy measurement of technology's precise contribution (1). In the 1970s “big-ticket” devices and procedures were singled out as the major culprit (2). Some states established certificate-of-need regulations to control their diffusion, but limitations of this approach soon emerged (3). It became apparent that the critical issue was not medical technology per se but a combination of economic, professional, and social incentives in our health care system that tend to diminish the importance of costs in decisions about patient care. For example, the establishment of Medicare and Medicaid in 1965 greatly increased the demand for medical services, thus increasing indirectly the demand for medical technology. At the same time, traditional systems of physician fee-for-service and hospital reimbursement according to retrospective charges insulated providers and patients from the immediate financial consequences of their decisions. These incentives applied to the acquisition of large, expensive devices as well as to routine clinical choices to order a test, prescribe a drug, or perform a procedure. Payment policies in these years could be generally characterized as encouraging innovation.1
Today the pendulum has started to swing the other way. Attention has shifted toward coverage and reimbursement as major instruments for promoting cost containment. This is illustrated by the adoption of the Prospective
Payment System (PPS) for Medicare based on diagnosis-related groups (DRGs), the rapid growth in health maintenance organizations (HMOs) and preferred provider organizations (PPOs), and the creation of a resource-based relative value scale (RBRVS) for physician services. These policy changes create very different incentives for providers to adopt and use medical technology. Less apparent, but also very important, they exert a strong indirect influence on investment in research and development (R&D).
In addition to payment, a wide range of public policies has evolved that foster or inhibit innovation. These policies attempt to accomplish a variety of different public goals (4). For example, policies to encourage a high level of public and private R&D include the federal support of biomedical research, tax credits, and legislation to protect intellectual property. Pre-marketing approval regulation of new health care products and liability statutes aim to prevent the diffusion of unsafe or inefficacious technologies. Trade policies attempt to encourage health-related exports, whereas anti-trust legislation intends to encourage competition.
Recently, concerns have been raised that various combinations of these policies may have unanticipated—and perhaps unwanted—effects on technological innovation in medicine. The main objective of this volume is to address these concerns and consider the complex interplay between innovation and public policy. The term “innovation,” in this context, refers to the development and introduction of new drugs, devices, and surgical procedures into clinical practice. Surgical procedures are included as an example of clinical procedures whose development does not necessarily depend on new health care products and may not involve the pharmaceutical or device industry. Within the broad range of policies that affect innovation, this volume focuses on the impact of United States regulatory and payment policies. In the final chapters, the policy environment for industrial innovation in the United States is compared with that in Europe and Japan.
A DYNAMIC MODEL OF TECHNOLOGICAL INNOVATION IN MEDICINE
Before we address the main theme of this volume, it seems useful to examine briefly the nature and dynamics of technological innovation. The increasing reliance of technology upon science during the twentieth century has given rise to a “linear model” of technological innovation, in which results were perceived to flow from basic research to applied research, product development, manufacturing and marketing, adoption, and use (see Figure 1.1 ). This is a supply-oriented model in which the critical need is assumed to be the provision of adequate funding for biomedical research. This model, however, has a number of theoretical and practical limitations. One of the more important limitations, from the perspective of this book, is
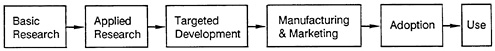
FIGURE 1.1 A linear model of the innovation chain
that it emphasizes advances in research as the main impetus to development and disregards the influence of demand considerations.
Empirical studies, undertaken primarily in the 1970s, began to question the primacy of research advances in stimulating technological change. These studies asserted instead that market demand was the major impetus to innovation. Following years of debate whether “science-push” or “demand-pull” factors were the governing influence on the innovation process, Mowery and Rosenberg decisively maintained that development is an iterative process in which an evolving scientific and engineering knowledge base and market demand interact to achieve a particular technology (5).
Nelson and Winter further defined the influence of market demand on R&D project selection (6). They observed that theories of innovation often have tried to make a neat distinction between R&D on the one hand and adoption on the other, with all uncertainty piled on the former. However, they acknowledged that not all uncertainty associated with a new technology can be resolved before its use in practice and that development does not end with the adoption of an innovation but continues for an extended period. Their premise is that the behavior of users in adopting or rejecting certain technologies over time provides important feedback signals as to the kind of development projects that firms subsequently will find profitable to undertake.
Most studies of innovation have focused on areas where preferences of users are expressed and mediated through market mechanisms. However, the “market” in medicine differs from markets in other sectors of the economy, where, in principle, consumer preferences determine what products are purchased (versus other products and their prices). Empirically, the following important differences can be discerned:
-
Market demand generally implies autonomous choice and realistic information of available alternatives by consumers. However, both autonomous choice and knowledge of the alternatives are often severely limited for patients, and health care professionals usually determine the kind and level of medical interventions needed. Although patients are the ultimate users, providers are the primary users for those that develop new medical technology.
-
In other sectors of the economy, the separation between developers on the one hand and users on the other is relatively clear. This is not the case
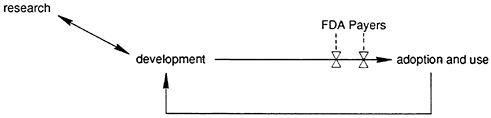
FIGURE 1.2 A dynamic representation of the innovation chain
-
in medicine. Clinicians, for example, have a dual role; they not only are the users of new technology, but also play an active role in its development. This duality is most visible in surgical innovation, especially when one considers the minor modifications in technique that occur in everyday surgical practice. It also exists in device and drug innovation, where the introduction of new products in clinical practice often leads to the unexpected discovery of new indications of use. For instance, after beta blockers were introduced for the treatment of cardiac arrhythmias and angina pectoris, physicians discovered their potential therapeutic value for more than 20 other conditions, not all of them cardiac (7).
-
New technologies—in addition to their benefits—may often entail a certain element of risk. However, the beneficial or adverse effects of medical technology are considered to be quintessentially different from those of many other technologies because, as Renee Fox observes, they affect “basic and transcendent axes of the human condition: life, conception and birth, body and mind, . . . and ultimately mortality and death” (8).
-
Finally, in other sectors of the economy new technologies generally are purchased by users. However, in medicine, technologies usually are paid for by public and private third-party payers and not by health care professionals or patients.
These idiosyncrasies of the health care market have prompted considerable government intervention in the development process. In view of their potential serious risks and because individual physicians cannot be expected to evaluate all emerging products, the Food and Drug Administration (FDA) exists to ensure the safety and efficacy of new drugs and certain classes of devices before they are marketed. Furthermore, with health care costs continuing to rise faster than the rate of inflation, public and private payers have emerged as important parties at interest in the innovation process. They have become increasingly critical as to what new technologies will be covered in their benefit package. Moreover, with the rapid growth in prepaid health plans and fee-for-service insurance programs with active utilization review and case management, incentives to use new technologies have changed. Thus, in addition to users, regulators and payers now influence the demand for new technology, and hence the incentives for R&D investment (see
Figure 1.2 ). In the following sections we more closely consider the impact of regulatory and payer policy and related decision making on drug, device, and surgical innovation.
THE DYNAMICS OF PHARMACEUTICAL INNOVATION
The papers in this volume on pharmaceuticals describe the ways in which the government affects the rate and direction of innovation. The economics of pharmaceutical innovation, however, need to be understood in terms of the interactive public policies that have evolved over time. A case in point is the strong interdependence that exists among pre-marketing approval, patent, and payment policies.
Regulatory policies for pre-marketing approval of drugs traditionally have exerted a strong influence on the dynamics of pharmaceutical innovation. Hutt (Appendix A ) makes explicit that two sources are responsible for current regulatory policies: statutes and administrative practices (9). The relevant statute (the Food, Drug, and Cosmetic Act) requires “substantial evidence . . . of safety and effectiveness . . . consisting of adequate and well-controlled investigations.” This broad mandate gives the FDA considerable discretion in determining the acceptable risk-benefit ratio for a marketing approval decision. Since the thalidomide tragedy of the early 1960s, there have been strong social and political pressures to reduce pharmaceutical risks to essentially zero. Under these pressures and because of the growing sophistication of animal toxicology and clinical research techniques, pre-marketing requirements have become increasingly detailed over time. The resulting system has provided important information on the safety and efficacy of new drugs, but it also has considerably lengthened the pre-marketing development process.
The tension between increasingly thorough pre-marketing evaluations and early product availability becomes urgent in the case of life-threatening disease. In the early 1980s this tension led the FDA to create a category of regulatory exceptions for cancer drugs (so-called group C drugs) to allow desperately ill patients to use promising but still experimental drugs before they are officially approved. However, it is only with the advent of the human immunodeficiency virus (HIV) epidemic and the activism of patient advocacy groups for acquired immune deficiency syndrome (AIDS) that the need to streamline the drug approval process for life-threatening diseases has come into the national spotlight and has further transformed regulatory practice. In 1987 the FDA created the “treatment IND” (investigational new drug) procedure for life-threatening and serious disorders. This procedure shortens the pre-marketing evaluation stage by merging Phase II and Phase III trials into more definitive Phase II trials, and it emphasizes more strongly the post-marketing evaluation stage (Phase IV) for providing safety and effectiveness information. More recently, the FDA has created the
“parallel track system” that may allow clinical use of an investigational drug prior to its release under a treatment IND, at least for those patients who cannot participate in clinical trials (10).
There are medical and economic reasons to expect that some of these changes will be extended to other disease categories. First, despite increasingly detailed pre-marketing trials, there are no zero-risk drug approval decisions. For example, the detection of delayed or rare adverse effects (less than 1 in 10,000) would require extremely long periods of testing or the exposure of many thousands of patients. Furthermore, valuable therapeutic information on the risks and benefits of a drug may emerge only after its diffusion into the often uncontrolled environment of general use; for example, side effects may be influenced by differing pharmacogenetic profiles of patients, comorbidities, environmental influences, and other factors. These side effects may go unnoticed in carefully controlled and selected pre-marketing studies; their detection requires post-marketing evaluation. Second, there is an economic argument that favors more emphasis on post-marketing research, especially if the pre-marketing evaluation period can be shortened. The increasing duration of pre-marketing development has made the process more costly and has reduced the effective patent life of new pharmaceuticals. Grabowski (Chapter 3 ) estimates that it takes an average of $231 million (in pre-tax 1987 dollars) to discover and develop a drug (11), and that by 1985 the effective patent life of new compounds had decreased to roughly 9 years 2 (12).
In view of the high-cost, high-risk nature of R&D and the relative ease with which new drugs can be copied by non-innovative firms, patents are crucial to R&D investment. The need to halt the continuing erosion of patent protection was recognized in the Drug Price Competition and Patent Term Restoration Act of 1984. Under its terms, manufacturers can have their patent term extended for up to 5 years to compensate for time that passes while waiting for approval.3 At the same time, however, the 1984 act makes it less time consuming and costly for generic drugs to get to market by making them eligible for an abbreviated new drug application (ANDA). Instead of the extensive animal and clinical data for a full NDA, this simplified application merely requires demonstrating that the active ingredient is bioequivalent to an already-approved drug whose patent is about to expire.4 Thus, the law has not lengthened the product life of innovative drugs, because when their patents expire, they lose their market share to generic drugs much more rapidly.
The loss of market share to much-lower-priced generic drugs has been exacerbated by the more stringent payment policies of the 1980s. Historically, drugs have been the least expensive of all medical technologies; with higher patient coinsurance rates and out-of-pocket fees, they were not main targets for cost savings. Except in the Medicare program (a major exception), prescriptions were generally reimbursed liberally. In the 1980s pay
ers came under increased pressures to contain costs, and with improvements in computerized insurance claims systems and utilization review, they began to scrutinize claims for medications more critically. As Mortenson indicates (Chapter 4 ), three issues permeate recent policy debates: (a) payment for experimental drugs and the care of patients in clinical trials, as well as payment for experimental drugs used outside of clinical trials (compassionate use); (b) payment for the use of approved drugs for non-approved purposes (off-label use); and (c) the coverage of new drugs under formularies and the passage of generic substitution laws (13).
Medical benefit contracts for nearly all payers, public and private, have standard provisions that specifically exclude coverage for “investigational or experimental” therapy. Until recently, this clause, especially with respect to medications, was not strictly enforced. Medications in Phase I, II, or III trials now are categorically denied reimbursement, however, and payers are starting to deny the associated costs of hospital care. The hesitancy to pay for the (often higher) costs of care associated with experimental drugs is understandable. One can question, however, whether, instead of refusing payment for all costs of care, payers should accede to the principle of “opportunity cost” (i.e., the cost insurers would otherwise have paid for patient care) when patients are involved in clinical research (14). Mortenson argues that this approach would be especially attractive to the treatment of solid tumors, where standard medical regimens are not very effective and experimental therapies offer the best chance for a clinical response (13).5
The second issue concerns reimbursement for drugs used for conditions that have not been specified on their label. As mentioned, drug development occurs not only before a drug is introduced into practice but also continues afterwards as additional indications emerge during its use in clinical practice. As Mortenson indicates, roughly 50 percent of chemotherapy regimens, which represent state-of-the-art oncological practice, are officially off-label (13). Reimbursement of off-label use has become controversial, for many payers interpreting their contracts restrictively have designated off-label drugs as investigational and excluded them from coverage. This would be less of a problem if FDA approval could be obtained rapidly for additional indications via so-called supplemental NDAs, but the FDA tends to give supplemental NDAs low priority for review. In addition, many new indications are found late in the life cycle of a drug, often near the time of generic competition, at which time manufacturers have little financial or marketing incentive to invest in further clinical trials. The use of such medical compendia as the U.S. Pharmacopeia Drug Information or the American Medical Association Drug Evaluations may provide a more valid basis for reimbursement than FDA approval.6 Some payers, such as members of the Health Insurance Association of America, have begun to revise their policies in this direction.
Finally, coverage policy for new FDA-approved drugs is undergoing pro-
found change. A case in point is the rapid growth of formularies in both the public and private sectors.7 One way that formularies can influence the economics of innovation is by introducing delays beyond those for FDA approval; for example, state Medicaid programs (which reimburse 15 percent of prescription drugs) may take 1 to 3 years to reach a formulary decision (15). At the same time payers have started to enforce generic and therapeutic substitution more stringently.
These changes in the regulatory and payment environment pose appreciable stresses for pharmaceutical R&D. According to Grabowski (11), the main strategic response of the industry to increasing R&D costs and shorter product life cycles has been to increase the prices of drugs sharply. The recent introduction of biotechnology drugs with thousand-dollar price tags has galvanized the HCFA and the U.S. Congress to look more closely into how manufacturers make pricing decisions. Grabowski presents new data on the private returns on pharmaceutical R&D (11). He finds that the average compound earns a real annual return of 9 percent. However, if prices had not increased during the 1980s, then an average drug would not have broken even within its expected market life.
Close scrutiny of drug pricing policies by government and an increase in the number of large, sophisticated institutional buyers will make it difficult for the drug industry to continue its price increases (16). Current trends will place a heavier burden on the industry to develop breakthrough drugs or second- and third-generation drugs that lead to important improvements in clinical outcomes and efficiency. For example, many innovators are focusing on drugs that require less frequent dosing and have less expensive costs of administration. Furthermore, because economic arguments have become central to the case for formulary acceptance, the development of new pharmaceuticals is increasingly supported by cost-effectiveness studies. Finally, as Spiegel states (Appendix B ), there is a strong trend toward industry mergers and consolidation to create economies of scale in research and marketing. Although the pharmaceutical industry generally has been very profitable and recent advances in research seem to present exciting opportunities for the development of new drugs, the papers in this volume underscore that the risks for pharmaceutical R&D have recently increased and may constitute impediments to drug development in the long run.
THE DYNAMICS OF DEVICE INNOVATION
The policy environments differ considerably for drugs and for devices, as can be expected given differences in the two industries and the nature of innovation. The device industry is younger, less concentrated, and comprises mostly smaller firms. There is much greater heterogeneity of medical devices in terms of design, purpose, and use and in the firms that manufac
ture them. Foote (Chapter 5 ) estimates the industry—7,000 manufacturers—to produce roughly 1,700 different types of medical devices (17).
In addition, as Kahn explains (Chapter 6 ), the nature of the R&D process is different (18). Because a device for a specific application often can be designed in a number of different ways, patents are less significant for device than for drug innovation. For example, the first lithotripter—invented and developed by Dornier—used shock waves generated by spark gap technology. However, Dornier's patent was not a significant entry barrier for competitors, who could easily design around the patent by generating shock waves electromagnetically, piezoelectrically, and by microexplosive technology (19). Except for complex and costly devices, such as lithotripters or imaging devices, medical device innovation does not require the large R&D investment required for drug development.
Furthermore, a high level of incremental innovation characterizes the development of new medical devices. As Kahn contends, “from the time the first preclinical testing is done to the time the product is introduced, and then for the first 6 months, a device is in state of flux” (18). Also, the product life of a device usually is much shorter than that for drugs; competitors may rapidly introduce a slightly modified version. Finally, the ultimate effectiveness and benefit of the device is often crucially dependent on the skills of the practitioner using or implanting the device. These elements of device heterogeneity, incremental innovation, and dependence on users can help explain some of the differences in the way medical devices are regulated.
The history of medical device regulation is shorter and less complicated than that of drugs. Although the FDA had some jurisdiction over medical devices as early as 1938, not until the 1976 Medical Device Amendments to the Food, Drug and Cosmetic Act were all medical devices required to be reviewed by the FDA before marketing. The law classifies devices in two ways: by level of risk (Class I, II, or III) and by descriptive category (pre-amendment, post-amendment, substantially equivalent, implant, custom, investigational, and transitional). The underlying principle is that the more potentially dangerous the device, the more stringent the regulatory scrutiny.8
Device manufacturers obtain FDA approval for marketing their products in two basic ways. The simplest, quickest, and most popular route is by designating that a device is the “substantial equivalent” to a pre-amendment device.9 In this case, under the 510(k) provision of the law, only pre-marketing notification is required. Since device innovation is predominantly incremental, this provision was intended to reduce the regulatory burden for technologies that were not significantly different from those already marketed. The other main route is more comparable to drug regulation and involves full pre-market testing and approval.
The differences between these two FDA approval routes are significant.
Because a 510(k) application requires much less time and effort on the part of a manufacturer, the average FDA response time to pre-marketing notification in one study took one-fifth the time taken to approve a pre-marketing application (PMA). This dramatic difference has not been overlooked by the industry—approximately 55 substantially equivalent 510(k)s are filed for each PMA filed (20).10
Foote reviews the trade-offs and conflicts among the various policies that affect device innovation (17). She refers to recent concerns about the implementation of the device amendments. For example, studies by the General Accounting Office (GAO) found weaknesses in pre-marketing review (broad use of 510(k) applications) as well as failures of post-marketing surveillance 11 (21-23). In response to these concerns, Congress enacted legislation in 1990 that “streamlines the device classification process, and expands FDA authority to track devices, recall defective products, and impose civil penalties on the industry. It also extends reporting requirements to hospitals and other facilities” (17). These reforms will address some of the weaknesses in current device evaluation. However, lessons from the pharmaceutical experience warn against creating a “device lag.” Foote recommends that Congress provide adequate resources to the FDA so that the agency can evaluate devices in a timely manner and make better use of post-marketing controls, as they are less restrictive for innovation than pre-marketing requirements. This recommendation is especially relevant, because the vast majority of device companies are small and do not have the resources (manpower, money, or time) to deal with an elaborate regulatory process.
Liability law also intends to deter the diffusion of unsafe products, as well as to permit compensation for injured users. Devices (like drugs) are caught up in an increasingly litigious society. The legal environment is complex and relatively unpredictable, and it can have serious negative effects on innovation. For example, a surge of liability suits, and the subsequent unavailability of insurance for manufacturers, was one of the major reasons that American industry virtually withdrew from contraceptive R&D (24). Consequently, there is a growing recognition that reform of the liability system is in order and that unnecessary overlap between regulation and liability in their pursuit of device safety should be eliminated (17).
Expensive medical devices were seen as the personification of rising costs in the 1970s; as a result, they have been the focus of cost-containment efforts for a longer time than have drugs.12 Implementation of Medicare's DRG-based PPS especially has affected expensive medical equipment. For a new device the increasingly restrictive payment circumstances often became manifest first in the form of the HCFA coverage decision for the Medicare program. Coverage decisions usually are made locally by HCFA fiscal intermediaries. Coverage issues of national importance are referred to HCFA's Office of Coverage Policy, which may request a formal technology assessment by the Office of Health Technology Assessment (OHTA) of the
Agency for Health Care Policy and Research in the U.S. Public Health Service. The statutory provision indicates that the coverage decision should be based on whether a device is considered “reasonable and necessary,” which has been translated to mean “accepted by the medical community as a safe and efficacious treatment for a particular condition.” Although it is reasonable for the HCFA to be wary of granting automatic coverage for all devices, given the leniency with which many get approved via simple 510(k) applications to the FDA, some observers have raised the question whether OHTA's review of the safety and effectiveness of FDA-approved devices is redundant given the FDA's mandate to ensure that new devices are safe and efficacious (25).
Following a Medicare coverage decision, a technology must be assigned to a certain DRG category. Although the price system is intended to be neutral under PPS, this is not always the case. For example, percutaneous transluminal coronary angioplasty (PTCA) was first assigned to a surgical DRG; this DRG provided a much higher level of reimbursement than the procedure cost, thus stimulating the adoption of PTCA. By contrast, cochlear implants were placed in a DRG that covered only a fraction of the cost of the device. This not only led to underdiffusion but also had adverse effects on subsequent R&D investment. For example, innovators developing second- and third-generation implants were unable to attract venture capital or to interest larger manufacturers to further pursue development.
In general, hospitals have a strong financial incentive to provide the least resource-intensive treatment under PPS (although competition, patient demand, and malpractice concerns may provide countervailing forces). The system promotes a significantly lower level of growth in service intensity than traditionally has been the case. Recalibration of DRGs could mitigate disincentives to use costly new technologies, but readjustments often considerably lag changes in medical practice (26). Furthermore, as Neumann and Weinstein point out (1), because PPS applies only to inpatient hospital services (Medicare Part A), hospitals have an incentive to provide more services in the outpatient setting (even if they could be provided more efficiently in inpatient care) and to use only those technologies that are cost effective over the short term of hospitalization. Hospitals may have little financial incentive to use technologies with long-term benefits, even though those technologies may ultimately have a greater impact on the efficiency of the system as a whole.
The incentives for capitated plans, such as HMOs and PPOs, to restrict utilization are somewhat different from those for hospitals under PPS. Because HMOs receive a fixed amount per enrollee and also deliver outpatient care, they have less incentive to (inappropriately) shift to outpatient care, and decision making is more likely to reflect concern with long-term cost effectiveness. This long-term perspective, however, may be tempered by the fact that HMOs may not pay for all patient services (such as long-term
nursing home care) as well as by the significant number of HMO enrollees who leave the system (1).
The change toward a more stringent payment policy seems to have exerted a strong effect on medical device innovation. For example, incentives to avoid restrictive Medicare Part A reimbursement controls have done more than change the locus of care; they have also stimulated the development of a whole range of new devices to be used exclusively in the outpatient setting. These include everything from smaller, lighter, and less expensive versions of hospital machines (which are more amenable to office use) to new, user-friendly, computerized infusion devices for the growing number of home health care applications. Moreover, the development of cost-effective technology has become an explicit R&D target, and device manufacturers are increasingly under pressure to demonstrate the cost utility of their innovations. A proposed HCFA rule change would require that device manufacturers have not only FDA approval attesting to safety and efficacy but also data showing that their technology improves outcomes or lowers resource use relative to existing alternatives. Yet the very essence of a new device is that its costs and benefits are uncertain until applied more widely and subject to considerable change thereafter. Foote discusses interim coverage as a more appropriate payment alternative —that is, covering costly devices for a designated period of time during which providers can gather information on costs and effectiveness.
THE DYNAMICS OF SURGICAL INNOVATION
Although surgical procedures typically involve the use of drugs and devices, their defining characteristics may be the special combination of surgical skills and abilities they entail. The dynamics for those procedures that do not center on a new product (e.g., the laser) are very different from those described above. According to Chang and Luft (Chapter 7 ), incremental innovation often occurs in everyday practice, but major surgical procedures generally are developed by specialists in academic centers (27). Whereas profit considerations play a role in the selection of drug and device R&D projects, in the case of surgical procedures return on investment must be defined broadly. Surgical procedures cannot be patented; although surgical innovators may gain higher fees and more patients, they receive no licensing fees for procedures performed by others. Motivating factors may include the need to offer patients improved surgical technology, the thrill of being first, and the attainment of national reputation and academic prestige. In comparison with drugs and devices, the costs of R& D are generally lower and less dependent on external funding.
Finally, if a new procedure is not centered on the use of a new product, no formal government regulatory system exists to evaluate it prior to diffusion. The realm of surgical procedure evaluation has been left to the medi
cal profession in the spirit of clinical autonomy. This takes place largely through peer review, the activities of medical societies, and Institutional Review Boards (IRBs). Although IRBS are responsible for reviewing university-based research, they are interested primarily in protecting the rights of human subjects and not in issues of evaluation. As a result, new surgical procedures generally are not systematically evaluated for safety and efficacy, and controlled clinical trials are often undertaken only after their diffusion.
In this context payment policies take on a quasi-regulatory rigor. Coverage and reimbursement decisions represent crucial determinations that limit or expedite the adoption and use of new procedures. Some commentators argue that they have become the rate-limiting step in diffusion—the true technological gatekeeper (28). The proliferation of managed care policies, such as surgical second opinion, pre-certification, concurrent review, and case management, signals a new level of scrutiny of surgery by payers. However, as mentioned, other factors affect the adoption of new technology as well. For example, Chang and Luft (27) call attention to the powerful influence that competitive forces can have on the adoption of new technologies. Sometimes, the presence of a new technology or a surgical team capable of performing an experimental operation has such cachet that it helps an institution portray itself as a modern facility that uses breakthrough technology to provide high-quality care. This technology/quality “halo” helps hospitals attract physicians and patients, and provides them a competitive advantage in their local and regional markets.
Changes occurring in the payment situation appear to affect surgical development and diffusion considerably, although empirical research is scarce. Chang and Luft point out that new procedures are identified chiefly through the coding system for insurance claims or hospital discharge summaries. In most cases an innovator seeks payment for an innovation by pursuing a new billing code or an additional code descriptor. Physicians must use the Current Procedural Terminology (CPT) system to describe their interventions to payers. Hospitals, under PPS, must categorize the world of care into specific DRGs. Payment systems are very sensitive to the experimental versus accepted status of a procedure just as they are for drugs and devices. Weaknesses in data and methods for assessing the value of new operations often are at the center of disputes about whether payers should reimburse practitioners or hospitals for a given procedure. More rigorous coverage criteria are forcing clinicians to improve their evaluation of the safety and efficacy of new procedures; however, the very nature of surgical innovation poses several challenges to traditional modes of evaluation that need to be addressed.
Chang and Luft describe how PPS has—and the implementation of the new RBRVS for physician payment will —affect the diffusion and development of surgical interventions (27).13 They extensively discuss the growth
of selective contracting as an attempt to both control costs and maintain quality. The underlying principle is that a good outcome from a complex, risky operation is highly dependent on the talents of a stable, experienced, well-run surgery team. As a prerequisite for bidding on a selective contract, an institution must demonstrate experience with the procedure, good success rates, and institutional commitment to maintaining the program in question. More recently, the nature of selective contracting has changed. It is more common to select only a few institutions among those that have met certain minimum standards. This has resulted in price competition among large institutions; for example, institutions have started to offer a package rate for coronary artery bypass grafting. Thus, selective contracting has become a way for large insurers to bargain for certain high-cost procedures with a center that has high-quality outcomes.
These payment changes are creating incentives to encourage the efficient use of resources, and they may limit the premature diffusion of costly surgical procedures. One drawback is that the mortality and morbidity data used to evaluate outcomes are relatively limited; insofar as this is true, selective contracting may focus on price because it has no adequate measure of quality. Changes in payment schemes also provide greater incentives for surgeons to develop more efficient variations of operations. Reducing operating room time is the most direct way of reducing the costs of surgery itself and of complications and their associated costs. Furthermore, interest is growing in developing less invasive operative procedures, such as laparoscopic gynecological surgery, cholecystectomy, and herniorrhaphy (31). The downside of payment reforms, however, may be to reduce the financial ability of hospitals to fund developmental activity and to evaluate and document the utility and costs of new procedures. Proposals that might remedy this situation include interim coverage and modifiable selective reimbursement; they require serious consideration if we want to continue incentives for innovation.
INTERNATIONAL COMPARISONS: EUROPE AND JAPAN
The United States, Europe, and Japan provide large-scale natural experiments as to the impact of public policies on medical innovation. In general, European governments are more heavily involved in the delivery, financing, and regulation of health care than is the United States, although considerable differences among European countries exist. Burstall (Chapter 8 ) provides an in-depth discussion of the European environment for pharmaceutical innovation (32). In comparison with American firms, European companies face several distinct disadvantages. First, Burstall argues that Europe cannot compete with the United States in basic science, partly because of Europe's smaller government support for academic and related research. Second, legislation to restore effective patent life has not yet been enacted in Europe.
Third, the majority of nations in the European Economic Community (EEC) regulate prices, and in some European countries' price levels are too low to provide adequate support for R&D. On the other hand, European firms enjoy certain advantages (32). In contrast to the United States, for example, generics are not as actively promoted; they account for no more than 5 percent of the market. Furthermore, the procedures to bring a drug through regulatory review seem to be more flexible and less time consuming in Europe. Finally, structural differences in the liability system contribute to making Europe significantly less litigious.
This situation will change with the creation of a truly common European market in 1992. EEC proposals to lengthen effective patent life will, if implemented, put Europe in an advantageous position relative to the United States. The harmonization of product-licensing procedures could either reduce or lengthen approval times, depending on the policies followed, and pricing issues still need to be resolved. Burstall surmises that American companies (as they are large and innovative) stand to benefit from the unification of the European market, but that it is difficult to be as optimistic about the European drug companies.
Hutton (Chapter 9 ) reviews the European policy environment for device innovation. In 1985 Europe was estimated to represent 25 percent of the world market for devices (33). Pre-marketing regulatory policies are less stringent in Europe; most nations tend to confine themselves to safety and technical performance criteria and do not include efficacy. Hutton contends that changes in payment policies, such as a growth in hospital budgeting systems, have had a greater impact on device innovation than regulatory policies. In addition to national policies, the EEC is expected increasingly to affect device innovation through its active anti-trust policy, support for R&D, and attempts to harmonize regulatory requirements among its member states.
Neimeth (Chapter 10 ) describes how the Japanese government used a combination of policies to rebuild a pharmaceutical industry whose manufacturing facilities were virtually destroyed during World War II (34). The government first enacted protectionist rules, established a patent law only for pharmaceutical processes and not for products, rewarded “me-too ” products similar to breakthrough products, and gave physicians financial incentives to prescribe and to dispense drugs. Furthermore, in 1967 a system of pre-marketing approval was enacted that had pre-clinical and clinical requirements different from those in other industrialized nations and that required clinical research to be undertaken in Japan.
In response to these patent and payment policies, Japanese firms focused on the development and manufacturing of me-too products. At the same time, the 1967 act established an entry barrier to foreign firms. Furthermore, the financial incentives embodied in the payment system formidably increased demand; by 1981, 40 percent of the overall health bill in Japan
was spent on drugs (by contrast, in the United States the share of national health care expenditures for drugs was 6 to 8 percent) (35). By that time, as Neimeth mentions, Japanese firms had acquired the basic technology, R&D capability, and financial strength to generate major new drugs, and the government then enacted a product patent law to protect these products. In the 1980s significant downward adjustment of prices occurred, and pre-marketing approval requirements were harmonized with international standards, allowing international competition. As a result of increased competition and decreased profitability, Japanese companies increased their investment in research and their globalization efforts.
Neimeth thus shows how patent, regulatory, and payment policies have combined to create a strong industry. This growth, however, has come at a price. First of all is the likely inappropriate use of drugs reflected in the immense share of total health care expenditures. Moreover, some observers believe that Japan's innovative capacity might have developed sooner if policies had allowed more foreign competition and if patent protection had been in place earlier.
CONCLUDING OBSERVATIONS
This society generally values technological innovation in medicine. Over time, a set of public policies has evolved to encourage the development of new medical technology. At the same time, in our pursuit of other policy objectives—such as enhancing safety, access, or cost-effective care—we may inhibit innovation. This volume discusses the complex interdependencies and trade-offs in public policy that affect the nature and rate of technological change.
In contrast to other sectors of the economy, research on the economics of innovation in medicine is just emerging, and this volume tends to pose more questions than it answers. This is especially true when one takes into account the influences that motivate innovation in the small device firm, the new biotechnology firm, or the surgeon innovator. Obviously, improved understanding of the basic mechanisms that underlie technological change is necessary if government interventions are to be successful in encouraging not only the diffusion, but also the development, of cost-effective technology. We hope that this volume stimulates much-needed empirical research on the economics of medical innovation and contributes to a better understanding of the critical issues in public policy during the 1990s.
NOTES
1. The term “payment policy” encompasses both coverage and reimbursement strategies and practices. Coverage refers to the decision to pay or not pay for a technology and under what circumstances. The reimbursement decision involves how much to pay for the technological intervention and how.
2. Patents provide a restricted period of monopoly power to an inventor to make, use, and sell an innovation. Effective patent life refers to the period between approval of the product and the patent expiration date. Officially, a U.S. patent entitles the holder to 17 years of legal protection, and it commences at the date of receiving the grant. In most European countries patent terms are 20 years, and they commence at time of filing.
3. According to the FDA, the mean approval time for new drugs in 1989 was 31.5 months or 2.7 years. Under patent term restoration, on average, 1.5 years will be added back to the patent “clock” (11).
4. Bioequivalence means that the generic compound must contain the same active ingredients as the brand-name counterpart and must be identical in strength, dosage form, and route of administration. A manufacturer demonstrates bioequivalence by showing in a small number of subjects (20 to 30) that the pharmacological absorption, availability, and excretion profile is within a 20 percent margin of variability to the brand-name product.
5. A related issue concerns experimental drugs that are unapproved for treatment of life-threatening diseases but are nonetheless used outside of the context of a clinical trial. Payers have been reluctant to pay for the use of these so-called compassionate use drugs. The Health Care Financing Administration (HCFA), in a ruling seemingly in conflict with its policy toward other investigational drugs, permits coverage for certain experimental cancer drugs (group C drugs). These inconsistencies need to be clarified.
6. These compendia provide a means by which physicians can ascertain the appropriate and effective indication of a drug referenced with up-to-date scientific literature the FDA may not have at the time of labeling. For example, the U.S. Pharmacopeia's drug information volume includes 25 percent more indications for drugs than are listed on the FDA label (K. Johnson, Director of Research, U.S. Pharmacopeia Drug Information, personal communication, September 1989).
7. A formulary is a list of drugs carried by a given institutional provider. Large organizations use formularies to buy drugs in bulk, as well as to limit the number of different drugs that are covered and/or that must be kept in stock. Choices about which drugs are carried usually are made by a hospital, HMO, or Medicaid pharmacy and therapeutics advisory committee. Decisions often are based upon assessments by committees of the relative safety, effectiveness, and cost-effectiveness compared to other formulary pharmaceuticals.
8. Class III devices, those that pose the most risk, are regulated the most restrictively. A manufacturer must demonstrate safety and effectiveness before receiving premarket approval. Class II devices must meet performance standards, and Class I devices, the least risky and regulated of the group, are subject only to general controls. In addition, all devices must meet these general control requirements, which include pre-market notification, reporting of adverse events, record keeping, labeling, and good manufacturing practices (17).
9. “Substantial equivalence” is not defined by the law, but it has been interpreted to mean modification of a previously approved or marketed pre-amendment device in a way that does not negatively affect its safety or effectiveness.
10. More recently, the FDA has sought to better ensure the safety and effectiveness of substantially equivalent devices, sometimes by requiring sponsors to submit performance and clinical data with their 510(k) application. This has been called a “mini-PMA” or “hybrid 510(k).”
11. One GAO survey found that 99 percent of the problems associated with the studied medical devices, including those that could or did cause injury, had not been reported to FDA (21). The study found failures of communication at every level, from device users to manufacturers and independent distributors.
12. An exception to this observation is that, unlike pharmaceutical firms, device manufacturers are allowed to charge investigators for the use of investigational devices, and they, in turn, can charge patients. The rationale behind this asymmetry is that device innovators usually are small firms that could not afford to run clinical studies unless they were able to recover costs during the development period. Devices also tend to cost much more than most experimental drugs. However, some observers have expressed concern about the commercialization of investigational studies for devices such as intraocular lenses, contact lenses, and the YAG laser (25).
13. Under a RBRVS system, payment is based on the estimated cost of resources, including amount of physician time and work effort; the costs of nonphysician personnel, office space, equipment, and supplies; and the cost of malpractice insurance. The fee schedule will also be adjusted for geographical variations in practice costs that partially reflect differences in cost of living (29,30).
REFERENCES
1. Neumann PJ , Weinstein MC. The diffusion of new technology: costs and benefits to health care . In this volume. Washington, D.C. : National Academy Press , 1991 .
2. Altman SH , Blendon RJ (eds) . Medical Technology: The Culprit Behind Health Care Costs? Washington, D.C. : DHEW Publication 79-3216 , 1979.
3. Hillman BJ. Government health policy and the diffusion of new medical devices . Health Services Research 1986 ; 21 : 681-711.
4. Maynard A , Hartley K. The regulation of the pharmaceutical industry. In Lindgren B (ed) . Pharmaceutical Economics . Stockholm : Liber Forlag , 1984 .
5. Mowery DC , Rosenberg N. The influence of market demand upon innovation: a critical review of some recent empirical studies . In Rosenberg N (ed). Inside the Black Box: Technology and Economics . Cambridge : Cambridge University Press , 1982 .
6. Nelson RR , Winter SG. In search of useful theory of innovation . Research Policy 1977 ; 6 : 36-76 .
7. Frishman WH. Clinical differences between beta-adrenergic agents: implications for therapeutic substitution . American Heart Journal 1987 ; 113 : 1190-1198 .
8. Fox RC. The cultural shaping of biomedical science and technology . A preface . International Journal on Technology Assessment in Health Care 1986 ; 2 : 189-194 .
9. Hutt PB. The impact of regulation and reimbursement on pharmaceutical innovation . In this volume . Washington, D.C. : National Academy Press , 1991 .
10. Rothman DJ , Edgar H. Drug approval and AIDS: benefits for the elderly . Health Affairs 1990 ; Fall : 123-131
11. Grabowski H. The changing economics of pharmaceutical research and development . In this volume . Washington, D.C. : National Academy Press , 1991 .
12. Grabowski H. Health Care Cost Containment and Pharmaceutical Innovation . Boston : Center for the Study of Drug Development , 1986 , Reprint RS S707 .
13. Mortenson L. Public policy and access to new drugs: the case of cancer chemotherapy . In this volume . Washington, D.C. : National Academy Press , 1991 .
14. Laubach GD. Clinical research and managed care: who should fund the cost of care in clinical investigation? In Institute of Medicine . Resources for Clinical Investigation . Washington, D.C. : National Academy Press , 1988 .
15. Wagner JL. Strategies for containing Medicaid prescription drug costs . Testimony for Senate Finance Committee . Washington, D.C. : .
16. Pollard MR. Managed care and a changing pharmaceutical industry . Health Affairs 1990 ; Fall : 55-65.
17. Foote SB. The impact of public policy on medical device innovation: a target of polyintervention . In this volume . Washington, D.C. : National Academy Press , 1991 .
18. Kahn A. The dynamics of medical device innovation: an innovator's perspective. In this volume . Washington, D.C. : National Academy Press , 1991 .
19. Gelijns AC. Innovation in Clinical Practice: The Dynamics of Technology Development . Washington, D.C. : National Academy Press , in press.
20. Food and Drug Administration , Office of Device Evaluation , Center for Devices and Radiological Health . Annual Report, FY 1985 . Rockville, MD : Food and Drug Administration , 1985 .
21. General Accounting Office . Medical Devices: The FDA's Implementation of the Medical Device Reporting Regulation . Washington, D.C. : Government Printing Office , 1989 (GAO/PEMD 89-10) .
22. General Accounting Office . Medical Device Recalls: An Overview and Analysis 1983-1988 . Washington, D.C. : Government Printing Office , August 1989 (GAO/PEMD 89-15BR) .
23. General Accounting Office . Medical Devices: Early Warning of Problems Is Hampered by Severe Underreporting . Washington, D.C. : Government Printing Office , (GAO/PEMD-87-1) .
24. Mastroianni L , Donaldson DJ , (eds) . Developing New Contraceptives . National Research Council and Institute of Medicine . Washington, D.C. : National Academy Press , 1990.
25. Kessler DA , Pape S , Sundwall DN. The federal regulation of medical devices . New England Journal of Medicine 1987 ; 317: 357-365 .
26. Anderson GF , Steinberg E. To buy or not to buy technology: acquisition under prospective payment . New England Journal of Medicine 1984 ; 311 : 182-185 .
27. Chang SW , Luft HS. Reimbursement and the dynamics of surgical procedure innovation . In this volume . Washington, D.C. : National Academy Press , 1991.
28. Halm EA. The payer as technological gatekeeper: methodological issues in technology assessment and payment policy . Paper presented at the Sixth Annual International Society of Technology Assessment in Health Care Meeting, Houston, Texas, May 21, 1990.
29. Hsiao WC , Braun P , Yntema D , Becker ER. Estimating physicians' work for a resource-based relative-value scale . New England Journal of Medicine 1988 ; 319 : 835-841 .
30. Physicians Payment Review Commission . Report to Congress . Washington, D.C. : Physician Payment Review Commission , 1990 .
31. Goldsmith, MF. Some new twists to one of the most common procedures in U.S. general surgery . The Journal of the American Medical Association 1989 ; 262 : 3248-3249 .
32. Burstall ML. European policies influencing pharmaceutical innovation . In this volume . Washington, D.C. : National Academy Press , 1991.
33. Hutton J. Medical device innovation and public policy in the European Economic Community . In this volume . Washington, D.C. : National Academy Press , 1991 .
34. Neimeth R. Japan's pharmaceutical industry postwar evolution . In this volume . Washington, D.C. : National Academy Press , 1991 .
35. Spilker B , Cuatrecasas P. Inside the Drug Industry . Barcelona : Prous Science Publishers , 1990 .




















