
2
The Mechanistic Basis of Radon-Induced Lung Cancer
INTRODUCTION
This chapter summarizes the current state of knowledge of the various processes that are presently considered to be involved in the induction of cancer by radon progeny. Inclusion of the chapter was motivated by the desirability of providing, where possible, a biological and mechanistic framework for epidemiologic analysis of risk models. Various components of the process of radon-induced carcinogenesis are understood to some degree, but we do not yet have a complete mechanism-based understanding of the entire process. In particular, although our understanding of the various radiation-related components (such as the effects of dose and dose rate) is at least semiquantitative, our knowledge of the various steps in the carcinogenic process (particularly at the genetic level) is at best qualitative in spite of important research findings since the publication of the report of the 4th Committee on the Biological Effects of Ionizing Radiations, BEIR IV (NRC 1988).
Consequently, systematic quantitative mechanism-based (biophysical) modeling of the entire process of lung-cancer induction by radon progeny is beyond present capabilities. However, some elements, such as dose-rate effects, can be modeled on the basis of specific assumptions and used to guide epidemiologic analyses and risk modeling.
In broad terms, the types of information available on radon carcinogenesis can be characterized as molecular, cellular, animal, and human. All contribute to our current understanding of the mechanistic basis of alpha-particle induction of lung-cancer.
A principal justification for studying cancer cells in vitro, abstracted from the entire organism, is that a neoplasm is usually considered as arising from a single cell that has undergone a critical change. Evidence of that includes the fact that some malignancies can be propagated by a single cell, and many, but not all, tumors have been shown to be monoclonal in origin, in that every cell carries the same biochemical marker (for example, Pathak 1990). It is important to note that many steps are involved from the malignant transformation of a single cell to the development of an overt neoplasm, including tissue response and potential immunological factors (Nagarkatti and others 1996), and care must be taken in directly extrapolating exposure and dose-response relationships for cells exposed to low doses of high-LET particles to risk for the development of cancer.
Cellular and molecular research generally focuses on early changes induced by radon and attempts to understand the mechanisms involved in production and repair of these changes. Such mechanistic understanding is essential to evaluate the response of cells to environmental radon exposure in which only a small fraction of the cell population interacts with the alpha particles. However, the role and progression of these cellular and molecular changes in the development of disease also can be addressed with experimental-animal studies. It should be noted that although many of the studies discussed in this chapter used interactions of bronchial tissue with alpha particles as the experimental model, there is a much greater base of information on interactions of x rays with other target tissues. When appropriate, we draw inferences from these other experimental models, but such conclusions will inevitably be less certain than those derived from experiments with alpha particles and bronchial tissue, the target tissue for radon-induced damage.
As the cells of a cancer grow and divide, progressive stages, or steps, from preneoplasia to malignancy can be identified. Those steps have been described as initiation, promotion, and progression. The progressive nature of carcinogenesis has been known for many years; it was first described in phenomenologic terms for skin cancer in animals. With sputum cytology, it has been possible to use histologic changes in lung cells as a predictive measure of bronchogenic cancer (Saccomanno and others 1988). The progressive cellular changes suggest a multistage process during the development of radon-induced lung-cancer. More-recent evidence of the multistep nature of cancer has come from studies of the clinical progression of colorectal cancer from polyp to metastatic cancer (see, for example, Fearon and others 1990). Those studies have demonstrated an association between the clinical progression of the cancer — from a benign state, through nonmalignant adenomas, to full-blown cancer — and the activation of oncogenes, the loss of antioncogenes, and other chromosomal changes.
Although the multistep nature of radiation carcinogenesis is almost certainly true, it is as yet only a qualitative observation. Our current state of knowledge precludes systematic quantitative understanding of all the various steps from early subcellular lesions to observed malignancy, and of the potential influence
that these multiple steps can have on the shape of the dose-response relationship at low doses.
RADIATION AND ONCOGENES
The identification of oncogenes and findings on their role in human cancer have made it possible to understand why agents as diverse as retroviruses, ionizing radiation, and chemicals can result in tumors that are indistinguishable from one another (Bishop 1983; Bishop and Varmus 1984). A retrovirus can insert a gene into a cell, and radiation and chemicals can produce a mutation in a gene that is already in the cell; all can activate oncogenes.
A central feature of oncogenes is that they act in a dominant fashion. The presence of a single copy of an activated oncogene in a cell is sufficient to produce a transformed phenotype, even in the presence of a normal copy of the gene (Lee and others 1987). Cells that are already immortal, such as NIH 3T3 mouse cells, can be transformed to a malignant state by transfection with a ras oncogene. Primary rat embryo fibroblasts, which are short-term cultured cells, are not transformed by the ras gene alone or by the myc gene alone, but can be transformed by transfection of the cells with both myc and ras (Land and others 1983). That is interpreted to mean that the myc gene confers immortality, whereas the ras gene produces the change reflected in morphology (Land and others 1983). Generally, at least 2 activated oncogenes in cooperation are needed to convert a primary cell to a tumorigenic line (Hunter 1991). Oncogene products that act in the nucleus cooperate most efficiently with products that act in the cytoplasm, as exemplified by the combination of ras and myc.
Over 100 oncogenes have been identified in human cancer; most belonging to the ras family. However, activated oncogenes are associated with 10–15% of human cancers and tend to be found more commonly in the leukemias and lymphomas and less commonly in solid tumors. Oncogenes have been shown to be activated by a range of genetic changes, for example by point mutations, as in ras (Bos 1990); deletions, as in Nmo-1 (Petersen and others 1989); reciprocal translocations, as in myc (Dalla-Favera and others 1983); and gene amplification, as in myc (Brodeur and others 1984).
Ionizing radiation, including alpha radiation, is not particularly efficient at producing point mutations, but it does produce large interstitial deletions and reciprocal translocations with high efficiency (for example Evans 1991; Metting and others 1992; Searle and others 1976). Consequently, in assessment of the predominant initial radiation damage — the first of the many steps by which alpha particles can induce cancer — deletions or translocations seem to be the most likely candidates for the first changes.
Numerous experimental and epidemiologic studies have demonstrated that radiation can cause cancer (Martland 1931; Court-Brown and Doll 1958; Beebe and others 1962). That it does so via direct or indirect alterations to DNA is clear
in in vitro studies, such as those of Borek and others (1987) in which DNA isolated from radiation-transformed C3H10T1/2 cells was shown to transform recipient cells after transfection.
The molecular mechanisms of radiation-induced transformation are unknown. Several studies have used indirect methods to attempt to identify oncogenes in radiation-transformed cells (Guerrero and others 1984; Shuin and others 1986; Hall and Hei 1990). One approach has been to search DNA isolated from radiation-transformed cells for mutations in known oncogenes. In that way, K-ras and N-ras were shown to be activated in some of the mouse lymphomas induced by gamma radiation (Newcomb and others 1988); it is not known, however, whether these are the initial radiation-induced changes. Another approach has been to determine whether any known oncogenes are overexpressed in transformed cells. This requires measuring mRNA in known oncogenes. Two studies used the method to examine gamma-irradiated C3H10T1/2 cells (Schwab and others 1983; Krolewski and Little 1989). Each used several overexpressed, cloned oncogenes as probes, but they could not identify an oncogene; both speculated on the possibility that gamma radiation could activate an as-yet-unidentified oncogene. A more-recent direct approach to the question has been to isolate the oncogenes present in the transformed cells. Such an approach was used in an attempt to isolate an oncogene from gamma-irradiated C3H10T1/2 cells (Hall and Freyer 1991). Many cloned oncogenes have been tested by hybridization and were negative so the gene has not yet been identified. Later experiments by Hei and colleagues (1994b) showed that a single small dose of alpha particles (30 cGy of absorbed dose), corresponding to an average of a few particles per cell nucleus, can cause human bronchoepithelial cells to become tumorigenic. A dominant gene is involved, inasmuch as the phenotype can be transmitted by transfection. Again, no known oncogene has been identified. The data support the speculation that one or more as-yet-unknown oncogenes can be involved in radiation-induced transformation.
TUMOR-SUPPRESSOR GENES
Suppressor genes act recessively: both copies must be lost or inactivated for the cell to express the malignant phenotype. Stanbridge (1976) showed that if a hybrid was made by fusing a normal human fibroblast to a malignant HeLa cell, the normal cell suppressed the expression of malignancy by the HeLa cell. It was shown further that if during the repeated subculture of the hybrid cells, chromosome 11 was lost, the malignant phenotype was restored. It was inferred that chromosome 11 in the normal human fibroblast contains a gene capable of suppressing the malignant phenotype. In later experiments, Saxon and colleagues (1986) injected microcells containing a single human chromosome 11 into HeLa cells and found that it suppressed their malignant phenotype; if chromosome 11 was lost from the cell, the malignant phenotype was restored.
The importance of suppressor genes became evident from the work of Knudson (1971) with retinoblastoma. A familial form of retinoblastoma occurs at a high rate and a sporadic form at a very low rate. Knudson argued that in the familial form 1 mutant allele with lost function is inherited from the affected parent. A somatic event during embryogenesis inactivates the normal allele inherited from the unaffected parent. Almost all the children of such pairs of parents exhibit bilateral retinoblastoma. In sporadic retinoblastoma, 2 somatic mutations are necessary, the second in a descendant of a cell that suffered the first. Those double events are much less likely than a single event, so the incidence of the sporadic form of retinoblastoma is much lower. Knudson elaborated the "2-hit hypothesis" in the early 1970s (Knudson 1971). By the middle 1970s, the location of the relevant gene was identified on chromosome 13 (Cavanee and others 1985); in the 1980s, the Rb gene was cloned and sequenced (Lee and others 1987). The Rb gene is present in all cases of retinoblastoma and associated sarcomas; it is sometimes present in cases of other tumors, such as small-cell lung-cancer, bladder cancer, and mammary cancer.
The action of radiation is a potential mechanism for deleting a suppressor gene. Alpha particles are particularly efficient at producing large deletions (for example, Metting and others 1992). Two radiation-induced breaks in the same arm of a chromosome can readily result in a deletion. Studies with defined restriction cuts in cellular and plasmid DNA have indicated that small deletions can also result from processing of single sites of DNA damage (Thacker 1994).
A suppressor gene acts recessively, so the deletion would have to occur in both chromosomes of a pair; this would be a very low-frequency event. In practice, the loss of the pair of suppressor genes often occurs by the process of somatic homozygosity (Cavanee 1989). One chromosome of a pair is lost, a deletion occurs in the other chromosome, and then the second chromosome and the deletion are replicated. Consequently, the cells in the tumor have 2 chromosomes that originated from the same parent. That has been shown to be a mechanism in retinoblastoma, small-cell lung-cancer, and glioblastoma; the case of glioblastoma is particularly interesting, inasmuch as somatic homozygosity must occur in 2 different chromosomes for this high-grade tumor to be produced (Cavanee 1989).
The list of suppressor genes whose location and function are known is growing steadily. The 2 most common and most intensively studied are the Rb gene and the p53 gene, both of which are directly involved in cell-cycle checkpoint control (Kasten and others 1991; Smith and others 1994).
GENOMIC INSTABILITY
The multistage nature of cancer is one of the most pervasive hypotheses in cancer research. The idea is over 60 years old and continues to derive support from research findings, such as recently from the work of Vogelstein and col-
leagues (Vogelstein 1990; Fearon and others 1990) with hereditary colon cancer. The progression from normal epithelium to metastatic cancer appears to involve a number of mutations in different oncogenes and tumor-suppressor genes and multiple chromosomal changes.
In the multistage formation of radiation-induced carcinogenesis, it is unclear as to how a single relatively small dose of radiation could result in mutations in so many different genes. The induction of multiple mutations seems highly unlikely, but data from the Japanese atomic-bomb survivors clearly show that a modest dose of radiation can induce many types of solid tumors, including those in the digestive tract. A more likely possibility is that radiation causes mutations in a gene responsible for the stability of the genome, which leads to a mutator phenotype. The multiple mutations and chromosomal changes follow as a cascade because of the induced instability as described below.
Both densely ionizing and sparsely ionizing radiation have been shown to induce chromosomal and mutational changes that appear in the progeny of exposed cells many generations after the initial exposure (Morgan and others 1996). The changes can occur in a high proportion of the surviving irradiated cells even after doses that give an average of only 1 alpha-particle traversal per cell. Examples of radiation-induced changes that are used as indicators of genomic instability are chromosomal aberrations, gene mutations, and even tumor induction in animal-model systems (Kennedy and Little 1984; Seymour and others 1986; Gorgojo and Little 1989; Chang and Little 1992; Kadhim and others 1992, 1994, 1995; Sabatier and others 1992; Martins and others 1993; Marder and Morgan 1993; Selvanayagam and others 1995). The high proportion of initially irradiated cells that transmit the instability phenotype and the variety of events observed suggest that this is not the result of a targeted effect of the initial radiation damage of specific genes, but rather a consequence of more-generalized damage to the cell; whether the initial damage is genetic or epigenetic is an unresolved question. Induced genomic instability is transmissible to progeny cells and can persist for multiple generations. Although this is an attractive hypothesis to account for carcinogenesis by low doses of high linear-energy-transfer radiation, typified by single-particle traversals, the case is far from proved.
INDIVIDUAL AND GENETIC SUSCEPTIBILITY
There is much published evidence that many cancer-predisposing genes are present in the human genome (Sankaranarayanan and Chakraborty 1995). For some tumor types, changes in these genes are responsible for a large fraction of the total cancer frequency. For example, 40% of children with retinoblastoma carry a germ-line mutation in the RB1 gene (Vogel 1979; Cowell and Hogg 1992). The tumor-suppressor gene p53 has been associated either directly or indirectly with at least 50% of human cancers (Hollestein and others 1991), although a causal link is less clear.
There have been substantial breakthroughs in the molecular biology and mechanisms involved in the genetics of breast cancer and about 5–10% of breast cancers might be inherited (Newman and others 1988). The BRCA1 gene has been located on chromosome 17 (Hall and others 1990) and cloned (Miki and others 1994). About half the inherited breast cancer and more than half the ovarian cancers are thought to be associated with mutations in the BRCA1 gene. With linkage analysis, a second gene (BRCA2) involved in breast cancer has been identified (Wooster and others 1994). It has been suggested that 1 or both of those 2 genes might be responsible for up to 90% of all familial breast cancer cases (Sankaranarayanan and Charkraborty 1995). While the risk to the individual carrying the mutated gene is very high, such mutations account for only about 4% of the total breast cancer patients. A larger proportion, perhaps 9 to 18% of all breast cancer, is associated with carriers of the ATM gene (Swift and others 1991).
Another common cancer related to genetic breakthroughs is hereditary non-polypotic colon cancer (Bodmer and others 1994). Genes associated with certain rare diseases such as ataxia telangiectasia (Shiloh 1995) and xeroderma pigmentosum (Kaur and Athwal 1989), have also been identified. Some suggestive evidence links lung-cancer with several genes affecting carcinogen metabolism (such as CYP2D6, CYP1A1, and GSTM1), but the links are quite speculative (Caporaso and others 1995).
There is now considerable evidence that a substantial fraction of spontaneous cancers have a genetic basis (Cavenee and White 1995), and it has been estimated that the prevalence of cancer-predisposing disorders is about 16 per 1,000 live births (Sankaranarayanan and Chakraborty 1995). Some evidence has been presented (for example, Swift and others 1991; Lavin and others 1994) that ionizing radiation might interact with the genetic predisposition to increase the frequency of radiation-induced cancer. The current evidence for that hypothesis is still relatively weak (for example, Hall and others 1992), but if a radiation-sensitive subpopulation did account for most of the radiation-induced tumors of a specific type, this would profoundly influence risk estimates. Ataxia telangiectasia heterozygotes, who probably constitute more than 1% of the U.S. population (Swift and others 1986), are an example of such a relatively large subpopulation that could, at least in principle, be at increased risk for radiation-induced carcinogenesis (Swift and others 1991; Hall and others 1992; Lavin and others 1994).
The role of genetic susceptibility in the induction of cancer by environmental insults, including low-LET radiation and radon exposure, has been reviewed by Cox (1994a,b). There is evidence from transgenic mice that cancer predisposition increases the frequency and decreases the latency of cancer formation initiated by low-LET radiation (Kemp and others 1994). In addition, a study of patterns of inheritance in mice (Franko and others 1996) suggested a genetic component to radiation-induced pulmonary fibrosis. No such strong evidence has yet been found in human populations. For lung tumors, altered phenotypes
and genotypes in several genes, such as the CYP family and GSTM1, have been associated with tobacco-related cancers (Anttila and others 1994; Kihara and others 1995), but the available data do not support a causal association between these markers and cancer risk (Alexandrie and others 1994; Raunio and others 1995; Caporaso and others 1995).
Even though there is ample evidence that many cancers have a strong genetic basis, the evidence that cells isolated from persons with cancer-predisposed genotypes are more sensitive to radiation than are normal cells seems to be mixed (Sanford and others 1989; Scott and others 1996). In addition, the current evidence that people with a cancer predisposition are at higher risk for radiation-induced cancers is limited. However, current knowledge of the functions of the cancer-predisposing genes, and of the consequences of their mutations constitutes sufficient grounds for assuming that among the genotypes of those predisposed to cancer there are some that also convey increased risk for radiation-induced cancers. There is also sufficient rationale for attempting to estimate quantitatively the effect of genotype-dependent differences in cancer predisposition on sensitivity to radiation-induced cancer (Sankaranarayanan and Chakraborty 1995). There is clear evidence of the existence of genes that are related to susceptibility to many forms of spontaneous cancer, and these genes could also be markers of an increase in susceptibility to radiation-induced cancer. This hypothesis remains to be proved. As genes such as the ATM gene for ataxia telangectasia are identified and sequenced, much attention will be focused on the possibility that some persons have a genetically based susceptibility to radiation-induced cancer (Sankaranarayanan and Chakraborty 1995), possibly including lung-cancer induced by alpha particles. Present risk models do not include individual susceptibility.
Further insights into the role of genetic predisposition can be gained from comparison of the effect of radon in various animal species; there are marked species differences in the responsiveness of experimental animals to radon. Early studies in dogs (Cross and others 1986), mice (Morken 1973; Palmer and others 1973), and Syrian hamsters (Palmer and others 1973; Cross and others 1981) exposed to very high exposures of radon resulted in few lung tumors. The tumor incidence was 21% in dogs, zero in mice, and 1.3% in Syrian hamsters. In hamsters, there were no tumors at exposures below 108 Jhm-3 [30,000 working-level months (WLM)]. Many of the exposures were high enough to result in marked life-shortening which can decrease tumor frequency and short-term pathologic changes. In contrast with dogs, mice, and Syrian hamsters, rats have a high incidence of respiratory-tract tumors after exposure to radon (Chameaud and others 1982; Cross and others 1984, 1986; Cross 1994a,b; Gray and others 1986).
The mechanistic bases of these interspecies differences are important to define, but the current evidence suggests that prima facie species-to-species extrapolations of absolute risk are unlikely to be useful since there are differences in response observed following the same insult delivered to different species. Thus,
direct extrapolation of animal data to humans cannot be used to predict absolute risk. Data derived in humans can produce patterns of risk which might well be of use (Brenner and others 1995), in that the endpoint remains the same but only the radiation dose/dose rate/quality changes.
Research has been conducted to determine whether the resistance to radon in Syrian hamsters relative to that in rats was related to delivered dose or induced damage at the same level of exposure (Khan and others 1995). Rats and Syrian hamsters were exposed at the same time, which resulted in exposure to the same radon level and dose, and the frequency of micronuclei as an indicator of radiation dose was measured in deep-lung fibroblasts. It was determined that the exposure-response relationship for radon-induced micronuclei per Jhm-3 (WLM) was higher in the Syrian hamster than in the rat. That suggests that the dose and damage to the lung cells were similar in the 2 species and that the amount of chromosomal damage initially induced might not be related directly to the differences in species sensitivity for the induction of lung-cancer. Combining research on cellular and molecular changes with whole-animal exposures could provide some understanding of the basis of species and strain differences; these differences eventually might be related to individual changes in sensitivity for the induction of cancer.
CELL-CYCLE EFFECTS
It is well established that ionizing radiation in general and alpha particles in particular produce a dose-dependent delay in progression through both the G2 and the G1 stages of the cell cycle (for example, Lucke-Huhle and others 1982; Kasten and others 1991). The G2 delay has been postulated to give the cell time to repair damage before entering into mitosis (Maity and others 1994). The G1 delay has been shown to depend on the function of the tumor-suppressor protein p53 (Kasten and others 1991) and to be controlled to some degree by Rb gene expression (White 1994). Tumor cells without p53 or with a mutated p53 have lost their ability to respond to cell-cycle arrest after exposure to gamma rays (White 1994). The molecular mechanisms associated with radiation-induced cell-cycle delay have been reviewed (Murnane 1995; Rowley 1996). Cell-cycle progression and delay constitute a multistep process that involves well-defined temporal and spatial changes in expression, phosphorylation, and complex interactions between the level and structure of proteins (Metting and Little 1995; Murnane 1995; Rowley 1996). The importance of DNA damage in producing cell-cycle delay response and the importance of the delay in repair of genetic and lethal damage have been demonstrated and reviewed for dividing mammalian cells (Murnane 1995).
The information available on the response of cells to high-LET radiation damage delivered in G0/G1 cells and in the role of cell-cycle delay in these cells as they move from G0 into a cycling stage is far from complete. Consequently,
the role of cell-cycle delay in altering response or affecting risk associated with indoor exposure to radon is not clear. However, most respiratory tract epithelial cells have rather long cell-turnover times of about 30 days (Adamson 1985), and spend only a small fraction of the total time in stages of the cell cycle that are most radiation sensitive. Inasmuch as the dose rate and number of traversals per cell are very low in the respiratory tract, the probability of alpha-particle traversal in a cycling cell is very low. In addition, the efficiency of cell killing by alpha particles might also decrease the relevance of cell-cycle delay to a risk assessment model. Those considerations make it likely, although not certain, that cell-cycle delay produced by environmental radon exposure plays a minor role in changing potential response or risk.
APOPTOSIS
After exposure to ionizing radiation, mammalian cells die by one of 2 distinct processes. The classic form of death has been called ''mitotic death"; cells die in attempting to divide as a consequence largely of complex chromosomal aberrations (Carrano and Heddel 1973). An alternative mode of death is by "apoptosis," or programmed cell death (Stewart 1994), which involves a characteristic progression of phenotypic changes, including induction of DNA fragmentation and the cell finally being phagocytosed by its neighbors. The relative importance of the 2 modes of cell death varies widely. For some cell types, apoptosis dominates; for others, apoptosis is seldom seen; in yet others, they are about equal. In most self-renewal tissues, apoptosis is a common mechanism to remove damaged or unwanted cells. Radiation-damaged cells are no exception. Failure of processes that lead to apoptotic death and removal of the damaged cells presents an alternative pathway to carcinogenesis for a radiation-damaged cell (Thompson 1995). While apoptosis is generally associated with doses significantly higher than doses usually attributed to radon progeny, apoptosis might be present at low doses.
RADIATION-INDUCED PERTURBATIONS OF CELLULAR PROLIFERATION
It has been demonstrated that changes in regulation of cell proliferation play an important role in the development of cancer (Brooks and others 1982; Cohen and others 1992), and it has been suggested that changes in cellular proliferation can be used in risk assessment of exposures to carcinogens (Clayson and others 1989; Clifton and others 1991; Goldsworthy and others 1991). It has also been established that an increase in cell turnover in the upper and lower respiratory tract follows experimental inhalation of radon (Taya and others 1994) and that, in the nose and upper respiratory tract, this increase is related to the areas with the highest radiation dose (Atencio 1994).
The cell types and normal turnover rate in respiratory tract cells vary by the region of the respiratory tract and the cell type involved (Adamson 1985). Changes in cell kinetics in the respiratory tract have been demonstrated after internal deposition of radioactive materials (Sanders and others 1989), external radiation exposure (Adamson 1985), and inhalation of radon (Atencio 1994; Bisson and others 1994; Taya and others 1994).
Taya and colleagues (1994) demonstrated an increase in the labeling index (which reflects the proportion of cells synthesizing DNA) as a function of exposure at 0.42–3.465 Jhm-3 (120–990 WLM) in rat alveolar, bronchiolar, bronchial, and tracheal epithelial cells over a range of times after exposure. The maximal increase in proliferation was at 14 days for all 4 regions of the respiratory tract. In studies of the nose and upper respiratory tract, Atencio (1994) demonstrated a similar time-dependent increase in cell proliferation after exposure to 0.595 Jhm-3 (170 WLM) of radon progeny. The labeling index increased after the end of the exposure to a peak between 14 and 50 days and then returned nearly to background levels. The increase was observed only in the trachea, the nasal septum, and the middle section of the larynx. Several of these regions were calculated to have high deposition for vapors (Kimbell and others 1993) and small particles (James 1994). In rats, the bronchial region, which is calculated to be at greatest risk for cancer induction, also receives the highest dose and responds with the highest cell-proliferation response to inhaled radon. Overall, the findings suggest a relationship between initial dose and changes in cell proliferation.
However, in considering these results, it is important to recognize that overall exposure rates differ widely; in studies of rats, a few weeks to months, in miners, a few years to about half the lifetime, and in residential exposures, a lifetime.
Radon-induced tissue damage and cell killing increases cell turnover to replace damaged cells (Taya and others 1994). This radon-induced increase in cell proliferation can result in repair of tissue damage. Apoptosis can eliminate damaged cells directly and normal and enhanced cell proliferation can also eliminate damaged cells at mitosis (Carrano and Heddle 1973) potentially reducing the risk for cell transformation and cancer. On the other hand, changes in cell kinetics have the potential to increase clonal expansion of altered or mutated cells increasing risk. Cell proliferation is a required step during cancer induction without which cancer cannot form, thus, enhanced cell proliferation can be viewed as a mechanism of either tissue repair or promotion of the cancer process.
CELLS AT RISK
To determine the dose, energy distribution, and cellular processes essential for radon-induced carcinogenesis, it is important to identify the respiratory tract cells at risk from radon exposure. In radon-inhalation studies, the cells of the
respiratory tract receive the highest radiation dose and are presumably at the highest risk (NRC 1988).
The differences in tumor incidence at different locations in the respiratory tract might have a purely dosimetric explanation or might imply that some cells are more sensitive to transformation by radiation than others. For example, no tracheal tumors were reported in rats exposed to radon, even though it has been estimated that the tracheal epithelium receives a larger dose for a particular exposure than do fibroblasts in the deep lung (Brooks and others 1990b; Khan and others 1994). It also has been demonstrated in tracheal epithelial cells that radon inhalation can cause the induction of early stages of cell transformation by producing enhanced-growth variants—cells that continue to grow in selective media (Thomassen and others 1990); this suggests that tracheal epithelial cells have the potential to produce cancers. The lack of tumors in the trachea suggests differential cell- and tissue-specific responses to alpha particles.
Tumors in humans arise primarily in the segmental and subsegmental airways (Saccomanno and others 1996), in contrast with tumors in rats, which are found deeper in the lung. Identifying the cells at risk for the induction of cancer in humans is important from both a biologic and a dosimetric standpoint; but it is difficult, and our understanding is still uncertain (Masse and Cross 1989). Recent human studies (Saccomanno and others 1996) have examined the spatial distribution of lung-cancer in ever-smokers that mined or did not mine uranium. A major observation was that the frequency of small-cell lung-cancers in the central region of the lungs of uranium miners that smoked cigarettes was higher (30.8% of the total) than observed in the same region of nonminers (10.6%). This region receives the highest radiation dose to lung epithelial cells (appendix B).
Histogenesis of tumors in the tracheobronchial region suggests a common epithelial progenitor cell. It has long been assumed that the basal cell is most at risk (Ford and Terzaghi-Howe 1992a) because its role in repopulation and differentiation is seen as similar to that of the basal cells in other tissues, such as the skin. Ford and Terzaghi-Howe (1992a,b) showed that isolated basal cells from the rat trachea are the cells that are capable of growth in vitro and in vivo; that suggests that they are the precursor cells for the induction of tumors. Electron microscopy and immunohistochemistry have revealed that individual tumor cells coexpress features associated with several different tumor types (McDowell and Trump 1984). Uses of transfection techniques with oncogenes also has suggested that there is a common cell of origin for the induction of tumors in the respiratory tract (Pfeifer and others 1991; Amstad and others 1988).
Other lines of research have suggested that the major airway epithelial cell type at risk for radon-induced cancer is the secretory cell (Johnson and Hubbs 1990; Johnson 1995). It has been demonstrated that in rat trachea, secretory cells constitute a major progenitor cell compartment. Secretory cells proliferate in response to physical or chemical trauma and are involved in repair and maintenance of the tracheobronchial lining (Keenan and others 1983). Studies with
denuded tracheal grafts showed that secretory cells that were isolated with a cell sorter could reestablish an epithelium composed of basal, secretory, and ciliated cells. When pure populations of basal cells were used, only basal and ciliated cells were found in the repopulated graft. Those findings suggest that secretory cells can differentiate to form all the cell types in the trachea, whereas basal cells have more-limited capacity to differentiate. The observations that both cell types can divide and differentiate, point to the potential role of the secretory cell in radon-induced cancer induction.
The cells involved in radiation-induced tumors in the pulmonary parenchyma, as opposed to the airways, also are unidentified. Adenocarcinomas are thought to arise in the peripheral lung and display both mucous and serous cell differentiation. Bronchioloalveolar tumors, however, possess features of both Clara cells and alveolar type II cells. Clearly, cells can share common differentiation pathways during insult and the progression to cancer. These observations indicate that using tumor-cell structure as an indicator of the cells at risk in the peripheral lung might be misleading.
Robbins and Meyers (1995) have conducted extensive studies to define the cell populations in the human respiratory tract that are capable of cell division and thus might serve as the progenitor cells for respiratory tract cancer. They have determined that both basal and suprabasal cells are dividing in human tracheobronchial mucosa and that the suprabasal cells proliferate more frequently than the basal cells. From that observation, they developed a model that suggests two different stem-cell populations, which they call reserve stem cells (basal cells) and transient-amplifying stem cells. The transient-amplifying stem cells can, as demonstrated by Johnson and Hubbs (1990), give rise to all the different cell types, whereas the reserve stem cells renew the transient-amplifying stem-cell population and give rise to a narrower range of cell types.
TARGET SIZE
A growing, although still small body of scientific literature suggests that the number of cells that respond to alpha-particle radiation is greater than the number of cells traversed by alpha particles. Hickman and colleagues (1994) found that exposure of lung-epithelial cells to small doses of alpha particles from a 238Pu source caused an increase in expression of the p53 gene in many more cells than were calculated to have been traversed. They concluded that the hit cells had communicated with the cell population and caused the remainder of the cells to respond. Thus, the target for interaction with alpha particles could be much larger than the cell nucleus.
It also has been demonstrated in both human and rodent cells that after exposure of cells to low doses of alpha particles, the number of cells with an increase in the frequency of sister-chromatid exchange (SCE) aberrations was larger than the number of cell nuclei traversed by alpha particles (Nagasawa and
others 1990; Nagasawa and Little 1992; Deshpande and others 1996). Again, both intercellular communication and tissue-culture factors could be involved in these changes. It is important to note here, however, that SCE formation is unlikely to be relevant in carcinogenesis.
Brooks and his colleagues (1990a) have demonstrated that when cells were exposed simultaneously to very low doses of alpha particles (6 cGy) and x rays, the response to the x rays with respect to both cell killing and induction of micronuclei was markedly increased. That suggests that the alpha particles change the responsiveness of the whole cell population, even though only a small percentage of the total cell population was traversed by alpha particles. The studies were conducted with confluent lung epithelial cells, so the damaged cells could potentially transmit information to neighboring cells. Clastogenic factors have also been isolated from the blood of Chernobyl accident victims; these factors remained in the blood for years after the exposure (Emerit and others 1995).
The significance of cellular responses in "bystander" cells in the development of radiation-related disease and risk cannot be defined at this time. However, since cellular changes can be induced in more cells than are traversed by alpha particles, it might be necessary to redefine target size for alpha particles to include a much larger volume than the cell nucleus through which the radiation traverses.
Thus, the long-accepted assumption that causing a heritable effect, such as cancer, requires a track to pass through the nucleus of affected cells is no longer unequivocal, in that now there is limited evidence that an ionizing event in 1 cell can affect its neighbor(s). The data are intriguing, but there is no real evidence that cells that are adjacent to traversed cells are damaged in such a way as to be transformed to a malignant state, nor has a cellular or molecular mechanism been proposed for how this could occur. In any case, the so-called bystander effect is unlikely to affect the concept of linearity. For a low dose-rate alpha-particle exposure, the number of cells traversed, say per year, is a small fraction of the total and doubling the dose approximately doubles the number of cells traversed by a track (rather than doubling the number of tracks in each cell, as with x rays). Consequently, the linear relationship between dose and effect is still the most tenable, whether the carcinogenic event results from a direct effect on the cell concerned or from an indirect effect from damage to a neighboring cell.
THE SPECIAL NATURE OF BIOLOGIC DAMAGE INDUCED BY ALPHA PARTICLES
Alpha particles, which produce a high density of ionizations along their path (high-LET), differ greatly from sparsely ionizing (low-LET) radiations, such as x rays in their microscopic, spatial, and temporal patterns of interactions with cells,
subcellular structures, and molecules (see Figure 2-1). Insult from alpha particles is concentrated in a relatively small number of densely ionizing tracks that each traverse a cell in less than 10-12s and deliver to it a large localized energy (about 10–50 cGy). In addition, in the case of alpha particles, relatively more of the individual damage is due to direct ionizations in the DNA rather than to hydroxyl-radical reaction, and it is more clustered on the scale of DNA and chromatin. For any radon exposures of practical relevance, a given cell in the lung is likely either to be unirradiated, to receive an instantaneous large energy deposition from a single alpha particle, or to receive a small number of such large energy depositions, each well separated in time by months or years.
For example, even for a miner in the highest-exposure cohort receiving 2.8 Jhm-3 (800 WLM) over 5 years, a cell nucleus in the bronchial epithelium is estimated to receive on the average only about 1–5 alpha-particle traversals per year (Table 2-1). In contrast, a similar absorbed dose to the lung from gamma rays would imply that each cell nucleus had received a very large number (about 104) of low-LET electron tracks, at the rate of about ten per day.
To calculate the dose to individual cells in the respiratory tract it is necessary to know the traversal number and the distance of the cells from the surface of the airways. The anatomy of the airway is discussed in appendix D on comparative dosimetry to illustrate the location of the cells of interest. To calculate the traversal number it is essential to define the location of the cell relative to the surface and the thickness of the mucus on the airway. Average values of mucus thickness at each of the anatomical locations were considered in the dose calculations. The hit numbers for different cellular locations and cell types are illustrated in Table 2-1. The variation of mucus thickness would be greater in the upper respiratory tract, nose and trachea, than in bronchus or bronchioles. Since the major site of cancers are in the smaller bronchus and bronchioles, the variation in mucus thickness was not considered as a major source of uncertainty in the dose calculations.
At the DNA level, also, there is a substantial difference between the spectrum of initial ionization damage from alpha particles and from gamma rays. Alpha particles produce more large clusters of multiple ionizations within the DNA and in adjacent molecules than do gamma rays (Goodhead and Nikjoo 1989). Considerations of track structure and radiation chemistry lead to the expectation that this will result in severe locally damaged sites (clustered damage) that the cell is less likely to be able to repair (Goodhead 1994; Ward 1994). Although the chemical nature of the individual single-strand breaks and base damages might not depend on the radiation type, their repairability could be influenced by the presence of the additional damage. The measured gross yields per Gy of initial DNA double-strand breaks (DSBs), irrespective of complexity, are about the same for alpha particles and gamma rays, according to recent measurements (Prise and others 1987; Charlton and others 1989; Brenner and Ward 1992; Jenner and others 1993; Prise 1994).
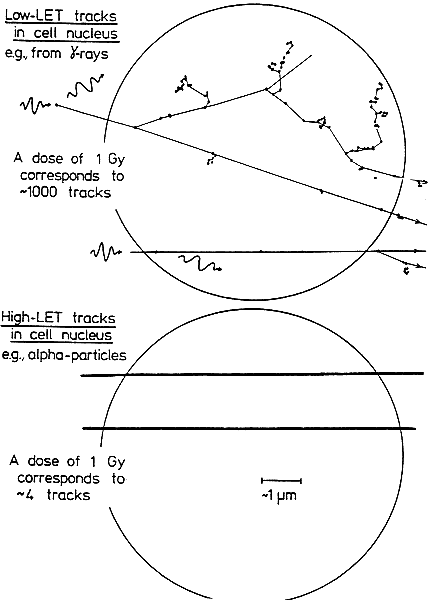
TABLE 2-1 Summary of computed alpha-particle hitsa for lung cells in different target locations
|
|
2.8 Jhm-3 (800 WLM) over 5 years in mineb |
0.0267 Jhm-3 (7.6 WLM) over 1.1 years in minec |
50 Bqm-3 for 70 years in homed |
150 Bqm-3 for 70 years in homee |
||||
|
Target location |
Mean number hits |
Mean hits per year |
Mean number hits |
Mean hits per year |
Mean number hits |
Mean hits per year |
Mean number hits |
Mean hits per year |
|
Bronchial Basal cell nuclei |
6.7 |
1.3 |
0.063 |
0.058 |
0.15 |
0.0022 |
0.46 |
0.0066 |
|
Bronchial Basal cell cytoplasm |
19 |
3.8 |
0.18 |
0.17 |
0.43 |
0.0062 |
1.3 |
0.19 |
|
Bronchial Secretory cell nuclei |
26 |
5.3 |
0.25 |
0.23 |
0.60 |
0.0086 |
1.8 |
0.026 |
|
Bronchial Secretory cell cytoplasm |
384 |
77 |
3.65 |
3.32 |
8.4 |
0.12 |
25.3 |
0.36 |
|
Bronchiolar Secretory cell nuclei |
15 |
3.1 |
0.15 |
0.13 |
0.22 |
0.0031 |
0.65 |
0.0093 |
|
Bronchiolar Secretory cell cytoplasm |
130 |
26 |
1.2 |
1.1 |
1.79 |
0.026 |
5.4 |
0.077 |
|
a These hit numbers represent the mean number of alpha-particle hits at a given cell (or nucleus) location in the lung during the specified time period. In general, many cells and nuclei will have occupied each location during the extended time period. The mean number of hits per cell cycle may be estimated as about 1/12 of the mean number of hits per year on the basis of a turnover time of about 30 days for the majority of respiratory tract epithelial cells (Adamson 1985). The hit numbers were evaluated at the request of the BEIR VI committee by A.C. James based on the lung model and reference value of ICRP 66 and the BEIR VI committee's reference work pattern and aerosol conditions for miners and reference aerosol conditions for homes (without cigarette smoke). b Typical for highest exposure miner cohort (Colorado)—see appendix D. c Typical for lowest exposure miner cohort (Radium Hill)—see appendix D. d Approximate average concentration for homes in USA. e Approximate USA action level concentration. |
||||||||
Reduced cellular repair of initial DNA DSBs caused by alpha particles and other high-LET radiations, relative to x rays has been observed (Ritter and others 1977; Coquerelle and others 1987; Blocher 1988; Fox and McNally 1990; Jenner and others 1993). Greater complexity and reduced repairability of alpha-particle induced breaks have been indicated also by studies with radical scavengers in mammalian cells (de Lara and others 1995) and in plasmid systems (Jones and others 1993; Hodgkins and others 1996). Association between separate DSBs produced by the same particle track can also occur because of the higher-order DNA-chromatin structure. It has been suggested that the high-LET particles can therefore yield an excess of DNA fragments of 0.1–2 kilobases (Holley and Chatterjee 1996) and also very large deletions, up to about 200 kb, have been observed (Löbrich and others 1996). Such associations might make repair more difficult and slower (Löbrich and others 1994).
Overall, even low-level exposure to alpha particles presents the "irradiated" proportion of the exposed cells with very large instantaneous burdens of damage, including severe clustered damage at the DNA level and multiple spatially correlated damage sites from the same alpha-particle track within the cell. Therefore, it is possible that biologic consequences of alpha-particle exposure differ from those of low-LET irradiation in both qualitative and quantitative respects (Goodhead 1988).
Low-dose alpha-particle induced radiation damage differs from damage caused by x rays in that the potential to transmit viable mutations or aberrations can be substantially reduced by cell death induced by the same single alpha particle. This single-track survival probability depends heavily on cell type and geometry, varying from essentially zero to over 80%. Such large variability makes it difficult to extrapolate relative mutagenic or carcinogenic effectiveness of alpha particles from effects of other kinds of radiation and from cell type to cell type.
It can be inferred from the average number of alpha-particle traversals per lethal event that the single-track survival probability for a given cell type depends on the shape of the nucleus at the time of exposure. The amount of energy deposited per nucleus and the extent of the biologic response depend on both cell and nuclear size and shape (Geard 1985; Raju and others 1993). For a variety of fibroblast cell lines, it was found that, for an alpha-particle of given energy, the incidence of a lethal event was roughly constant per unit track length through the cell nucleus at about 0.03–0.08 lethal events per micrometer (Goodhead and others 1980; Roberts and Goodhead 1987; Raju and others 1993). However, the probability is greater for radiosensitive lines and some other cell types, including hematopoietic cells (Lorimore and others 1993), and can in some cases lead to a negligible single-track survival probability (Griffiths and others 1994). Before the advent of microbeam irradiators (described below), it was not possible to measure directly the probability of cell survival after exposure to exactly 1 alpha particle.
Cellular and molecular studies have been conducted to define the nature of radon-induced damage. In many cases, other alpha-emitting radionuclide sources or particle accelerators have been used to expose cells to high-LET particles. A recent development has been the construction of alpha-particle microbeams capable of delivering exactly 1 alpha particle to each cell in a dish (Geard and others 1991; Braby 1992; Nelson and others 1996). Such particle research makes it possible to define the influence of LET and particle fluence on the induction of a wide variety of end points (Cox and others 1977b; Lloyd and others 1979, 1984; NCRP 1990; Goodhead and others 1991; Miller and others 1995). It has been well established that high-LET radiation, such as slow alpha particles, produces greater amounts of biologic damage per unit of dose or energy absorbed than low-LET radiation (Barendsen and others 1966; Brooks 1975; Goodhead and others 1980; Thacker and others 1982; Schwartz and others 1992; Evans 1993; Jostes and others 1993; Piao and Hei 1993; Brooks and others 1994; Jostes 1996; Simmons and others 1996). The derived values of relative biologic effectiveness (RBE) depend on the biologic effect under study, cell types, cell growth, and conditions of irradiation. A useful summary of the relationship between LET and RBE has been published by the National Council on Radiation Protection and Measurements (NCRP 1990). In comparison with normal cells, the RBE for cell inactivation by alpha particles is usually lower for cells that are very radiosensitive because of a genetic defect (Cox 1982).
A review of studies on chromosomal aberrations demonstrated a linear dose-response relationship for the induction of cytogenetic damage by high-LET radiation, including alpha particles, and a linear-quadratic function was needed to describe the induction of aberrations by acute exposure to low-LET radiation (Bender and others 1988; NCRP 1990). Dose fractionation or dose protraction had little effect on the induction of chromosomal aberrations following exposure to high-LET radiation (Brooks 1975; Bender and others 1989). At the chromosomal level, alpha particles produce initial DNA breaks that are more closely associated with each other than those produced by x rays. Those differences would be expected to result in increased yields of alpha-particle-induced aberrations of the exchange type, such as translocations, which are indeed observed (for example, Searle and others 1976). In addition, the spectrum of aberrations, including the occurrence of a high proportion of complex aberrations involving more than 2 chromosomes, can be substantially altered even after few alpha-particle traversals (Griffin and others 1994, 1995).
Several investigators (Bedford and Goodhead 1989; Cornforth and Goodwin 1991; Goodwin and Cornforth 1994; Loucas and Geard 1994) have used the premature-chromosome-condensation technique to investigate the initial yields of chromosomal damage after the passage of alpha particles and the kinetics of damage repair that produces "final" chromosomal damage. Showing trends similar to those in the data on DSB yields and repair, these experiments demonstrated that alpha particles produced no more than about twice the initial chromosomal
breakage (about 5 min after exposure) as x rays. However, the yield of remaining damage measured after about 1 h was considerably larger after alpha-particle exposure.
A wide array of studies have been conducted to determine the frequency and nature of mutations induced by high-and low-LET radiation. In general, it has been found that alpha particles and other high-LET radiation produce greater frequencies of mutants per survivor per unit dose than does low-LET radiation and that RBE values tend to be greater for mutation induction than for cell inactivation (Thacker and others 1979, 1982; Chen and others 1984; Iliakis 1984; Metting and others 1992; Cox and Masson 1994b; Griffiths and others 1994; Bao and others 1995). The RBE for mutation by alpha particles, such as from radon progeny, is in the LET region of maximal effectiveness (Cox and Masson 1979).
In an in vivo study, a statistically significant correlation was reported between household radon levels and HPRT mutants in peripheral blood lymphocytes of residents (Bridges and others 1991). The observation that a mutant yield could be detected with significance relative to background implied a much greater effectiveness than had been found for alpha particles in high-dose in vitro experiments. However, the correlation was not confirmed in a larger follow-up study by the same research group (Cole and others 1996), and a negative correlation was reported in a separate smaller study (Albering and others 1994).
Conventional chromosome analyses for unstable aberrations in blood lymphocytes of persons living continuously in houses with very high radon concentrations have shown a significant increase in dicentrics and rings (Bauchinger and others 1994). Applying fluorescence in situ hybridization (FISH) techniques to these same samples showed slightly, but not significantly (p<0.1), raised overall frequencies of symmetric translocations, including significantly raised levels in males alone (Bauchinger and others 1996). The two sets of results are consistent and the authors suggest that the FISH measurements are less sensitive to discriminate radon exposure because control frequencies of translocations are much higher than those of dicentrics and radon doses to bone marrow, that should contribute most to stable aberrations, are lower than to mature lymphocytes. For more information see chapter 4.
The spectrum of mutants produced by ionizing radiation is wide and can depend on the gene, the host cell, the radiation quality, and the dose. Ionizing radiation in general and alpha particles in particular are most efficient at producing large genetic changes, deletions, and rearrangements, rather than point mutations. Several studies have suggested that densely ionizing radiations produce a higher proportion of gene mutations involving large deletions than do x rays (Chen and others 1990; Evans 1991; Hei and others 1994c; Schwartz and others 1994; Bao and others 1995; Kronenberg and others 1995; Zhu and others 1996). It has also been suggested that the frequency of large deletions increases as a function of dose for high-LET radiation (Hei and others 1993; Zhu and others 1996). Other investigators found no statistically significant difference between
high-and low-LET radiation-induced mutagenesis in the fraction of mutations that lost the entire active gene (Thacker 1986; Kronenberg and Little 1989; Evans 1991, 1993; Jostes and others 1994), whereas in an episomal shuttle-vector model, deletions were observed to be more frequent after alpha-particle exposure than x-ray exposure but were of comparable size (Lutze and others 1992, 1994).
Those observed differences in the patterns of mutation induced by high-LET radiation in different cell lines seem to be partially resolved by the observation that in cell systems in which genes that were critical to cell survival were close to the marker gene, a rather low frequency of mutations was induced by deletions. In cell systems in which the marker gene was not near a critical gene and the marker gene was not involved in cell survival, a larger fraction of the total mutations were produced by large deletions after alpha-particle exposure than after low-LET radiation exposure (Evans 1991; Bao and others 1995).
A variety of additional differences have been reported between the spectra of deletions caused by high-and low-LET radiation. These include a higher proportion of smaller but more complex deletions and rearrangements caused by alpha particles (Bao and others 1995; Jin and others 1995; Thacker 1995; Chaudhry and others 1996) and differences in growth characteristics (Metting and others 1992; Chaudhry and others 1996; Amundson and others 1996). Hence, radiation produces a spectrum of mutations different from those which arise spontaneously or are caused by other agents, and the spectrum can depend on radiation quality.
Research at the sequence level is continuing in a number of laboratories to evaluate the molecular nature of mutations induced by alpha particles from radon (Jostes 1996). It should add to the understanding of the nature of the mutagenic lesions induced by high-LET radiation. One of the objectives of such research is to determine whether unique changes associated with high-LET damage can be used as a ''signature" of alpha-particle-induced damage. Such a signature could help to identify environmental agents that are responsible for observed mutagenic damage (Schwartz and others 1994). Other studies also have proposed signatures for alpha-particle-induced biologic damage in the form of the induction of specific point mutations in the tumors of uranium miners (Taylor and others 1994; Vahakangas and others 1992); however, the results are not consistent, nor have they been confirmed by larger-scale animal studies (McDonald and others 1995; Kelly and others 1995).
BIOLOGIC EFFECTS OF LOW EXPOSURE LEVELS TO ALPHA PARTICLES
The primary approach to radon risk estimation involves epidemiologic studies of underground miners whose mean exposure was typically much larger than average residential exposures. For example, the average radon-progeny exposure of the Colorado miner cohort was about 2.8 Jhm-3 (800 WLM) over an average duration of 5 y. That implies that an average of about 7–26 alpha particles would
have traversed each target-cell nucleus location in the segmental bronchial epithelium over this time (Table 2-1). Thus, very few target cells and their progeny in miners in this cohort are anticipated to have been traversed by only one particle. In the pooled miner data (Lubin and others 1994a), the average exposure was 0.567 Jhm-3 (162 WLM), which would correspond to about 1–5 traversals per cell nucleus.
It follows that past risk estimates based on all the miner data (Lubin and others 1994a) have been driven by data on miners in whom multiple traversals either dominated or were comparable in number with single traversals. For an average indoor exposure at a concentration of about 50 Bqm-3 (1.25 pCiL-1) or an exposure of about 0.0007 Jhm-3/yr (0.2 WLM/yr), less than 1 in 400 basal cell nuclei (or less than 1 in 100 secretory cell nuclei) will be traversed per year by a single alpha particle, and less than 1 in 104 cells will be traversed by more than one alpha particle (Table 2-1). Therefore, in extrapolating from miner exposures to environmental exposures, previous models, based primarily on highly exposed miners, translated effects of multiple traversals to the effects of single traversals. In chapter 3 we attempt to avoid that problem by focusing on the low-exposure miners.
However, the prima facie approach of performing alpha-particle experiments at both high and low doses to guide the extrapolation does not allow for a direct assessment of the effects of a single alpha particle. That is apparent if we consider an in vitro experiment designed to measure the effects of single alpha particles traversing, say, C3HI0T1/2 cells growing as a monolayer with large projected nuclear areas. Assuming an LET of 150 keV/µm, and a low practical dose of about 0.1 Gy, on the average each cell nucleus will be traversed by a single alpha particle. However, because the number of traversals of a given cell is Poisson-distributed, about 37% of the cell nuclei will not be traversed by alpha particles, about 26% will be traversed by more than 1 particle, about 8% by more than 2, and about 2% by more than 3; this precludes a direct assessment of the effects of exactly 1 alpha particle.
Two experimental approaches permit the effects of exactly 1 alpha particle to be investigated. One way is to unfold the Poisson distribution from experimental dose-response data mathematically (Brenner 1989), provided that the cell geometries and sensitivities are sufficiently uniform. A more-direct approach to the problem of single-particle traversals is to design experiments in which exactly one alpha particle is delivered to each of many cells. This microbeam-irradiator approach, previously mentioned, is being pursued in several laboratories around the world, including those at Columbia and Texas A & M universities and Gray Laboratory (Geard and others 1991; Braby 1992; Folkard and others 1995; Nelson and others 1996). The basic notion for this approach is that the locations of cells are determined and recorded by a computerized image-analysis system; then a highly collimated beam of low-intensity alpha particles is directed at each cell in turn, and a radiation detector is used to determine when one or any given number
of alpha particles have passed through the cell. The dish is then moved so that the next cell is under the collimated beam. True single-particle irradiation should allow measurement of the effects of exactly 1 alpha-particle traversal relative to multiple traversals. These techniques should also allow evaluation of the effects of cytoplasmic traversals and traversals through nearby cells—the potential "bystander" effect discussed earlier.
BIOLOGIC EFFECTS OF ALPHA PARTICLES AT LOW EXPOSURE RATES
In the last few years, it has become increasingly clear that densely ionizing radiation such as alpha particles can exhibit an inverse dose-rate effect for carcinogenesis (for example, Miller and others 1993); that is, for a given dose or cumulative exposure, as the dose rate is lowered, the probability of carcinogenesis increases. The phenomenon has come to be known as the inverse dose-rate effect because it is in marked contrast to the situation for sparsely ionizing radiation, which with protraction in delivery of a given dose, either by fractionation or by low dose rate, usually results in a decreased biologic effect.
The extent and consistency of published reports on the in vitro and in vivo inverse dose-rate effects, leave little doubt that such effects are real (Charles and others 1990; Brenner and Hall 1992). Of interest here is that the inverse dose-rate effect has been clearly demonstrated in miners exposed to radon-progeny alpha particles at different exposure rates. From comparisons of epidemiologic studies involving different average radon-progeny exposure rates, Darby and Doll (1990) inferred the existence of an inverse dose-rate effect. On the basis of epidemiologic studies, an inverse dose-rate effect was reported by Hornung and Meinhardt (1987) in Colorado uranium miners, by Sevc and colleagues (1988) and Tomásek and colleagues (1994a) in Czech uranium miners, and by Xuan and colleagues (1993) in Chinese tin miners. In a recent joint analysis of 11 cohorts of miners exposed to radon progeny, Lubin and colleagues (1995a) clearly demonstrated the existence of a significant inverse dose-rate effect.
Irrespective of the detailed mechanisms involved, and provided that they are confined to single independent cells, basic biophysical arguments imply that if a target cell or its progeny is hit by 1 or 0 alpha particles, it cannot show a dose-rate effect of any kind. Mechanistically (see, for example, Barendsen 1985; Goodhead 1988; Curtis 1989; Brenner 1994), a cell traversed only once by an alpha-particle cannot "know" or respond to any changes in dose rate. Thus, no inverse dose-rate effect would be expected at very low exposures, but such effects would be possible as the cumulative exposure increased to a point where multiple traversals of the targets become significant. The resulting overall effect therefore will be the result of an interplay between cumulative exposure and exposure rate (Brenner 1994). These considerations are summarized in Figure 2-2, which depicts a protraction effect that increases with increasing exposure and decreases with
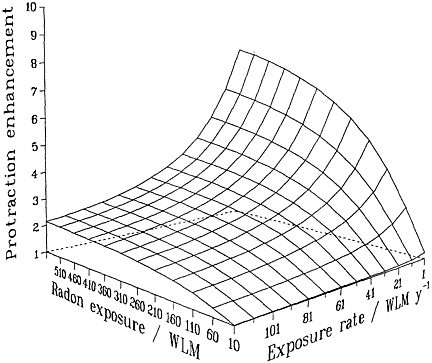
FIGURE 2-2 Schematic illustration of the relative effect of protraction, illustrating the interplay between exposure and exposure rate. Modified from NRC (1994).
increasing exposure rate. The particular quantitative values of the surface in the figure depend on the model and parameters, but its general features are likely to be largely independent of the model and have been shown to be consistent with the miner data (Lubin and others 1995a).
Possible mechanisms by which an inverse dose-rate effect could arise from exposures to single cells include
-
High-dose-rate saturation of effect from multiple track traversals in a particularly sensitive phase of the cell cycle (Rossi and Kellerer 1986; Brenner and others 1993).
-
Killing of initiated cells before they multiply.
-
Enhancement of cellular repair (Burch and Chesters 1986).
-
A correlation across the cell cycle between cell killing and oncogenic transformation induction (Elkind 1994; Brenner and others 1996).
Additional mechanisms in tissues for a low-dose-rate increase in effect could include
-
Promotion of the transformation process or enhanced misrepair (Hill and others 1984).
-
Enhancement of cellular proliferation (Moolgavkar 1993).
-
Age-dependent host variations in sensitivity or second mutations in expanding initiated clones (Leenhouts and Chadwick 1994).
An average lifetime exposure from an indoor radon concentration of 200 Bqm-3 (5.41 pCiL-1), which is about 4 times the average indoor exposure, would result in on average about 1 alpha-particle traversal per bronchial epithelial cell nucleus location (0.6 for the location associated with the bronchial basal nuclei or 2–4 for the location of bronchial secretory cell nuclei; see Table 2-1). In most indoor-exposure situations, protraction would be expected to have little effect on risk unless there are large additional spatial and temporal factors, such as persisting long-range cell signaling or clonal expansion. In contrast, in the miner studies, even though exposure rates are higher, the higher exposures result in a statistically significant reduction in risk per unit of exposure. That conclusion, which is consistent with the results from miner studies (see Figure 2-3 and Lubin and others 1995a), depends essentially on the notion that a dose-rate effect of any kind requires that autonomous target cells be exposed to multiple alpha-particle traversals. It should also be noted that in tissues, cells may die and be replaced many times during a lifetime. On the basis of these considerations, data on lower-exposure subset of the miner cohorts would be expected to yield the most applicable estimate of residential risk.
Although the miner data show an inverse dose-rate effect, indoor-exposure data will probably show none, because of the low probability of multiple alpha-particle traversals in the low-exposure situation of a residence. That is in accord with results on lung-cancer induction by radon in rats. Specifically, results for high cumulative exposures over 3.5 Jhm-3 (1,000 WLM) show a statistically significant inverse dose-rate effect: for the same cumulative exposure, irradiation over longer periods resulted in significantly higher lung-cancer rates than irradiation over shorter periods (Cross 1992; Gilbert and others 1996). As the exposure was decreased, no significant inverse dose-rate effect was observed (Gilbert and others 1996). At exposures corresponding to less than 1 alpha-particle traversal per cell, 0.0875 Jhm-3 (25 WLM), no increase in lung-cancer incidence was observed as the exposure rate was decreased (Morlier and others 1994). All this is consistent with the pattern presented in Figure 2-3.
In contrast with the high-exposure studies, some of the low-exposure studies in rats yielded evidence of a decrease in lung-cancer incidence with decreasing exposure rate (the "conventional" dose-rate effect). Specifically, when 0.0875 Jhm-3 (25 WLM) was protracted over 18 m, rather than over 4–6 m, a decrease in lung tumor incidence was observed (Morlier and others 1994), although the statistical significance was marginal (p = 0.056). Those studies suggest that biologic variables, such as fraction of the life-span during the exposure and age at
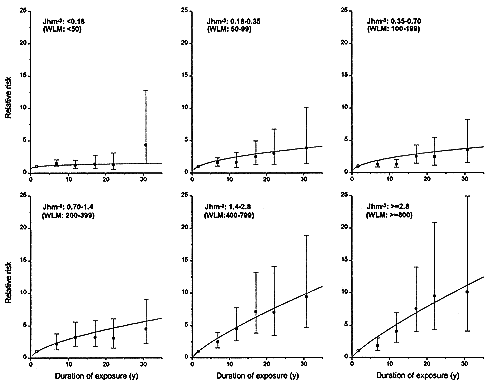
FIGURE 2-3 Relative risks of lung-cancer, by duration of exposure, in 11 miner cohorts analyzed by Lubin and others (1995a). Each panel represents a different total exposure. For miners with the highest exposures of > 1.4 Jhm-3 (> 400 WLM), there is a marked inverse dose-rate effect. The inverse dose-rate effect is less apparent for miner exposures between 0. 18 and 1.4 Jhm-3 (50 and 400 WLM) and it is essentially undetectable for exposures under 0. 18 Jhm-3 (50 WLM).
exposure, play an important role in the development of lung-cancer, at least in experimental animals (Cross 1994a,b).
INTERACTIONS BETWEEN LUNG CARCINOGENS
Radon is only one of the respiratory carcinogens to which humans are exposed. Tobacco-smoking is an extremely important risk factor for lung-cancer in miners, as well as in the general population; and other lung carcinogens, such as arsenic, are also present in mines. A brief review of the in vitro and in vivo studies related to the issue of interactions between lung carcinogens follows.
Using an in vitro assay for oncogenic transforming C3H10T1/2 cells, Piao and Hei (1993) applied cigarette-smoke condensate (CSC) and observed a dose-dependent increase in the incidence of both cytotoxicity and oncogenic transformation. The frequency was significantly increased if the CSC was combined with a dose of either gamma rays or alpha particles. The transformation frequencies in cells treated with a combination of CSC and 0.5 Gy of alpha particles with energies selected to simulate radon-progeny alpha particles was consistent with the 2 agents acting in an additive manner, not a multiplicative manner.
The report of the BEIR IV committee (NRC 1988) reviewed the animal studies that included exposure to both radon progeny and cigarette smoke. The relevant studies included experiments involving rats conducted by the Compagnie Generale des Matieres Nucleaires (COGEMA) in France and experiments involving dogs conducted by Pacific Northwest Laboratories (PNL) in the United States. The report noted that the COGEMA experiments showed synergism (greater-than-additive effects) if the exposure to cigarette smoke followed the exposure to radon progeny but not if the smoke exposure preceded the radon-progeny exposure. In the PNL experiments, lung tumor incidence was decreased if the animals were exposed to radon progeny and cigarette smoke on the same day, as opposed to sequentially.
Since the BEIR IV report, there have been several additional reports from COGEMA (Monchaux and others 1994) and PNL (Cross 1992). Cross and coworkers have reviewed the newer studies (Cross 1992, 1994a,b). The PNL group conducted initiation-promotion-initiation experiments with cigarette smoke and radon exposure (Cross 1992). Those experiments involved various sequences of exposure to smoking and radon progeny and splitting the dose of radon progeny. Only preliminary findings on lung tumors are available and the number of cancers has been very small. The findings of the COGEMA studies have been summarized recently (Monchaux and others 1994; Yao and others 1994). The extent to which lung-cancer incidence was increased by cigarette-smoke exposure after radon exposure was shown to depend on the duration of exposure to smoke. Decreasing duration was associated with decreasing lung-cancer incidence.
In spite of long-term research by 2 groups of investigators, the animal ex-
periments on smoking and radon progeny have not yielded strong evidence on the combined effects of the 2 exposures. The findings are inconsistent and dependent on the sequence of exposures. In the residential setting, exposure to cigarette smoke and exposure to radon progeny occur essentially simultaneously throughout adulthood. Among the miners, smoking and radon exposure can take place simultaneously or radon exposure can begin before or after smoking has started (Thomas and others 1994). The unique pattern of sustained smoking by humans, which has not been replicated in the animal experiments, is an additional barrier to extending the findings of the animal studies to humans.
THE DOSIMETRIC APPROACH TO RADON RISK ESTIMATION
The approach to domestic radon risk estimation taken in this report involves epidemiologic studies of people who have been exposed to radon. A different, and possibly complementary, approach is to estimate radon risks on the basis of people exposed primarily to sparsely ionizing radiation—largely the Japanese atomic-bomb survivor cohorts. This so-called dosimetric approach to radon risk assessment using ICRP quantities (ICRP 1991) has the following logic:
-
Use physical models to estimate a bronchoepithelial dose per Jhm-3 (WLM).
-
Convert that lung dose, with the specified radiation weighting factor (wR) (ICRP 1991), to an equivalent dose for radon-progeny alpha particles in the bronchial epithelium.
-
Convert the equivalent dose to an effective dose, with the appropriate tissue weighting factor for lung.
-
Use the best estimate for the lifetime-fatality probability coefficient per unit of effective dose to estimate the lifetime risk per Jhm-3 (WLM).
A more direct dosimetric approach could be to apply an appropriate alpha-particle RBE factor specifically for lung, rather than the general radiologic protection quantity wR (or "quality factor," Q). Then one could estimate the lifetime risk per Jhm-3 (WLM) from the lung-cancer-fatality probability coefficient per unit of absorbed dose of atomic-bomb survivor data extrapolated to low dose rate.
Assuming a quality factor of 20, Burchall and James (1994) used a dosimetric approach to estimate a risk from residential radon exposure and found the risk to be larger than estimated from the miner data (for example, Lubin and others 1994a) by a factor of 4–5. However, it is difficult to interpret the difference between the two approaches. In light of the uncertainties in many of the steps involved in arriving at both types of risk estimates, the difference is modest. One of the major uncertainties in the dosimetric approach is related to the current impossibility of estimating RBE directly in any realistic quantitative sense for relevant in vivo end points. Consequently, the rationale usually adopted is to
estimate its values for a variety of in vitro end points that are considered to be relevant to cancer induction and to be adequately quantifiable and then to define from these data sets a single value that is judged to be applicable to human cancers overall. The use of in vitro oncogenic-transformation data as a basis for risk estimates for more complex end points, such as carcinogenesis in general in humans, has been discussed elsewhere (ICRU 1986). Essentially, the rationale, other than the pragmatic issue of quantifiability, is that the radiation weighting factor is used for predicting only relative risks (compared with risks associated with gamma rays or x rays) of one kind of radiation relative to another, rather than absolute risks. However, many data on in vitro effects or carcinogenesis in animals show that the RBEs for the same kind of radiation depend substantially on the biologic system and cancer type under study. The RBE for induction of lung-cancer by radon-progeny alpha particles remains uncertain.
On the basis of in vitro data on the C3H10T1/2 oncogenic-transformation system of Brenner and colleagues (1995) and data on the induction of micronuclei in rat-lung fibroblasts and CHO cells by radon and gamma rays of Brooks and colleagues (1994), a quality factor of 10 seems appropriate for cells at depth in the bronchial epithelium. That is half the currently recommended radiation weighting factor (ICRP 1991). It would result in partial reconciliation of dosimetrically and epidemiologically based radon risk estimates, although it would probably be misleading to over-interpret the resulting level of agreement, because many assumptions are involved in both approaches.
MECHANISTIC CONSIDERATIONS IN ASSESSING RISKS ASSOCIATED WITH RADON
The mechanistic considerations discussed above must be incorporated into the design of epidemiologic analyses to estimate radon risks. We summarize the main issues of relevance to the estimation of risks associated with radon progeny.
Biologically-Based Risk Models
A biologically-based risk model is a formalism that potentially provides realistic quantification of all the relevant steps from energy deposition to the appearance of cancer. If such a model were available, epidemiologic data could be fitted to it, and the resulting parameter estimates could be used to quantify the different mechanistic steps in radiation carcinogenesis. Low-dose extrapolation could then be conducted with more confidence than for a situation in which data are fitted with a purely empirical formalism. However, epidemiologic data usually include only incidence and mortality. Biologic models need cell proliferation rates and other factors, and such information is not usually available. Such approaches to radiation risk estimation have been proposed and critically dis-
cussed by various authors (for example, Moolgavkar and others 1993; Crump 1994a,b; Little and others 1994; Moolgavkar 1994; Goddard and Krewski 1995; Little 1995).
Those approaches must be considered desirable, in the long term, as a framework for interpreting the radiation-epidemiologic data. Today, however, it is important to recognize the complexity of the processes involved in radiation carcinogenesis and the many gaps in our knowledge of the most-basic relevant processes. Although the use of biologically-based models provides valuable insights into the carcinogenic process, the models are not sufficiently well developed to be used for quantitative risk estimation; indeed, their use might lend more credibility to the resulting risk estimates than is warranted.
Although all the steps leading from the deposition of radiation energy to the development of cancer are not understood, some general trends have emerged from the considerations in this chapter, which can be used to guide specific assumptions of the epidemiologically based analysis of radon-induced lung-cancer. These trends are discussed in the remaining part of this chapter.
Extrapolation from High to Low Radon-Progeny Exposures
The challenge is to guide the extrapolation of risks from radon-progeny exposures at which effects can readily be observed and risks quantified down to lower exposures at which events might occur with probabilities too small to measure with sufficient precision in any human population. Low exposures and doses correspond to the traversal of cells by single alpha particles. As the dose is further decreased, the insult to cells that are traversed by an alpha particle remains the same, but the number of traversed cells decreases proportionately. There is good evidence that a single alpha particle can cause a substantial change in a cell. For example, a single traversal by an alpha particle with an LET of 120 keV/µm can result typically in about 10–20 double-strand breaks in a cell deduced from measured yields of about 30–40 dsb/Gy for low-LET radiation (Ward 1988; Stenerlow and others 1996), similar relative yields for alpha particles (Jenner and others 1993; Prise 1994), and a dose of about 0.2–0.5 Gy to a cell traversed by a single alpha particle, depending on cellular geometry. Even allowing for the substantial degree of repair that is known to take place, the passage of the particle most likely causes some irreparable damage or permanent change. Direct evidence of such clastogenic changes, based on single alpha-particle microbeam irradiation (Geard and others 1991; Braby 1992; Nelson and others 1996) has been reported. There is also convincing evidence that alpha particles are efficient at inducing genomic instability (Kadhim and others 1992, 1994, 1995; Sabatier and others 1994), so traversal by a single particle can potentially initiate a cascade of events that can lead to chromosomal aberrations or delayed mutations many generations later. The later effects can be in cells adjacent to those actually traversed.
Those observations, taken separately or together, provide a mechanistic basis for a linear relationship between alpha-particle dose and biologic effect at dose levels that correspond to 1 particle traversal per cell or less. At those exposures, varying the dose proportionately varies the number of cells traversed by alpha particles, but does not alter the level of damage sustained by cells that are traversed. That is the situation for exposure to alpha particles from radon progeny in a domestic environment where it is unlikely that any cell at risk in the bronchial epithelium is traversed by more than 1 alpha particle in a lifetime. As an example, an exposure of 0.0175–0.07 Jhm-3 (5–20 WLM) would result in an extremely low probability that any cell would be traversed by more than 1 alpha particle. Within and below that exposure range, linearity is thus a reasonable assumption with the implication of no threshold in dose. At the minimum, this linearity refers to effects in single cells since varying the dose merely changes the number of cells traversed but does not alter the level of damage per cell. Of course, the development of a tumor involves many steps beyond oncogenic transformation in a single cell, but the weight of evidence for the clonal origin of most tumors (Wainscoat and Fey 1990) suggests that this argument also applies to tumor induction by radon. Indeed, over this range, linearity and a threshold seem to be mutually exclusive. It is important to note that we have considered only low-dose extrapolation of the effects of alpha particles, and these arguments do not necessarily apply to the effects of sparsely ionizing radiation, such as x or gamma rays.
At higher doses of densely ionizing radiation, various processes can, in principle, result in a nonlinear relation, as has often been observed (for example, Ullrich and others 1976; Ullrich 1983; Fry and others 1985; Chmelevsky and others 1984; NRC 1988; Furuse and others 1992). These high-dose effects include the following:
-
Damage resulting from the interaction of chromosomal breaks from different alpha particles would be expected to result in a component of response that is quadratically related to dose.
-
Decreased efficiency of repair of alpha-particle damage, which, in principle, could be reduced at very high doses through saturation.
-
Cell killing and alterations in cell proliferation (from radiation and tobacco exposure) which are likely to influence the development of an initiated cell into a tumor.
-
Production of diffusible clastogenic factors at high doses which might be able to contribute to generalized tissue response remote from the sites of individual alpha-particle decays.
-
Inverse dose-rate effects which could change the slope of the dose-response relation at high exposure levels.
Although those factors could all potentially affect linearity, they require substantial doses. They might be important in the high-exposure miner data, in which
multiple cell traversals occur with significant probabilities, but are unlikely to be important at low exposure levels that correspond to 1 particle or less per cell.
In summary, the weight of evidence from cellular and molecular studies strongly supports the concept of linearity with dose in cellular systems including cell lethality, mutation, or transformation with no threshold for low-dose alpha-particle irradiation but leaves open the possibility of a change in slope or a departure from linearity for cancer induction at higher doses. The overwhelming evidence for the monoclonal origin of most cancers suggests linearity without threshold would also apply to low-dose radon-induced carcinogenesis. This observation emphasizes the desirability of extrapolating to typical indoor exposures from the lowest exposure range that is practical in the miner data set.
Effect of Changing Exposure Rate
Extrapolation from higher to lower radon exposures is also affected by the inverse dose-rate effect (Brenner 1994). In vivo and in vitro experiments have shown an inverse dose-rate effect for alpha-particle irradiation. Specifically, protracting a given total dose (experimentally, at least 0. 10 Gy) of densely ionizing radiation, such as alpha particles, can increase oncogenic transformation in vitro or carcinogenesis in vivo. This dose-rate effect, whatever its underlying mechanism, operates at doses corresponding to multiple particle traversals per cell but is likely to disappear at low doses corresponding to an average of much less than 1 traversal per cell (Figure 2-2). A similar dependence of effect on exposure is clearly evident in the miner data (Figure 2-3).
Extrapolating radon risk from the full miner data to the low-exposure domestic situation involves extrapolating from a situation in which multiple traversals are common to one in which they are rare; consequently, such an extrapolation would be from a situation in which the inverse dose-rate effect might well be important to one in which it is likely to be unimportant. That presents a problem for the committee's risk assessment in that the mechanisms whereby inverse dose-rate effects operate are not yet established, although several mechanisms have been hypothesized. However, given that both experimental evidence and fundamental biophysical evidence suggest that the inverse dose-rate effect should be of little importance below about 0.35 Jhm-3 (100 WLM), these considerations again underline the importance of assessing risks of radon in homes on the basis of miner data corresponding to as low an exposure as possible.
Interaction of Radon Progeny with Other Agents
Experiments with a combination of alpha-particle and tobacco-smoke condensate exposure, using oncogenic transformation in vitro as a test assay, show that effects of the 2 agents are consistent with a purely additive interaction (Piao and Hei 1993). With such in vitro systems, large-scale experiments are possible
that yield unequivocal results. However, such experiments might well be good models only for the initiation part of the carcinogenic process. Although the data are hard to interpret in animal experiments, alpha particles and tobacco smoke often appear to produce effects that are larger than additive; this observation can be understood in that tobacco smoke, as well as being a carcinogen, contains irritants that stimulate cell proliferation which is a known factor in oncogenesis. The experimental data are consistent with the supra-additive model which appears to be most useful in the human data (see chapter 3).
Biologic Signatures of Alpha-Particle Cancers
Early attempts (Vahakangas and others 1992; Taylor and others 1994) to identify a molecular ''signature" of prior alpha-particle damage through the identification of unusual point mutations have not yet proved useful (Rossi 1991; Hei and others 1994a; McDonald and others 1995; Bartsch and others 1995; Hollstein and others 1997). More mechanistic approaches based on larger-scale genomic alterations (for example, Brenner and Sachs 1994; Griffin and others 1995; Savage 1996) are more promising. The newer techniques currently require further experimental validation but offer hope for future molecular epidemiologic approaches to the radon problem.
Individual Susceptibility
Animal experiments show significant but unexplained differences among species in susceptibility to lung-cancer from radon progeny. For example, rats are susceptible to radon-induced lung-cancer, but Syrian hamsters are extremely resistant. In addition, within a given species, different inbred strains show variations in susceptibility to ionizing radiation or chemical carcinogens. Cell lines or animals with specific repair deficiencies also show increased susceptibility to radiation-induced malignant or premalignant changes, although there have been few experiments specifically with alpha particles from radon progeny.
The sum of available evidence leads to the conclusions that for an outbred human population a broad spectrum of susceptibility to alpha-particle-induced carcinogenesis would be expected and that there could be a marked increase in susceptibility in people suffering from a genetic deficiency. Evidence of genes related to susceptibility for many forms of cancer is emerging, although there is as yet no convincing evidence of a gene that confers sensitivity to radiation-induced cancer on a heterozygotic population.
Much research is being focused on the possibility of subpopulations that might have a genetically based increased susceptibility to radiation-induced cancer. The existence of subpopulations that are highly sensitive to alpha-particle-induced lung-cancer could substantially affect individual risk estimates but may have only minor impact on population risk estimates.

































