
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and Virus Vectors,” organized by Bernard Roizman and Peter Palase (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
A deletion mutant in the human cytomegalovirus gene encoding IE1491aais replication defective due to a failure in autoregulation
EDWARD S. MOCARSKI*†, GEORGE W. KEMBLE‡, JOHN M. LYLE*, AND RICHARD F. GREAVES*§
*Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA 94305–5402; and ‡Aviron, Mountain View, CA 94043
ABSTRACT Human cytomegalovirus (CMV) replication begins with the expression of two regulatory proteins, IE1491aaand IE2579aa, produced from differentially spliced transcripts under control of the ie1/ie2 promoter-enhancer. A deletion mutation removing all 406 IE1491aa-specific amino acids was engineered into the viral genome and this mutant (RC303ΔAcc) was propagated on an IE1491aa-expressing human fibroblast cell line (ihfie1.3). RC303ΔAcc failed to replicate on normal human fibroblasts at low multiplicities of infection (mois). At mois >3 plaque-forming units per cell, virus replication and production of progeny were comparable to wild type. However, at mois between 0.01 and 1, mutant virus replicated slowly on normal fibroblasts, a pattern that suggested initiation of productive infection required multiple hits. Replication of RC303ΔAcc correlated with the ability to express IE2579aa, consistent with a role for IE1491aain positive autoregulation of the ie1/ie2 promoter-enhancer and with data suggesting that virion transactivators compensate for the lack of IE1491aaunder high moi conditions, ie1-deficient CMV should be completely avirulent, suggesting its utility as a gene therapy vector for hematopoietic progenitors that are normal sites of CMV latency.
Human cytomegalovirus (CMV), a herpesvirus with a very large, 235-kbp genome and a coding capacity that exceeds 220 ORFs (1, 2), is recognized for its ability to cause acute disease in the immunocompromised host and in the developing fetus (3). Productive replication is believed to depend upon expression of two nuclear phosphoproteins, IE1491aa and IE2579aa, expressed prominently as α (IE) gene products during infection. Both proteins share 85 amino-terminal amino acids (aa) due to alternative splicing and polyadenylylation (4–6) of transcripts initiating at a strong promoter-enhancer (7, 8). Based on functional assays, both are believed to play key transcriptional regulatory roles during replication (for review, see ref. 9). IE2579aa is likely to be responsible for the switch from α to β gene expression (10–12) as well as for the shut-off of the ie1/ie2 promoter via a specific cis-repression signal to which it binds (13, 14).
IE1491aa was originally identified as the “major immediate early” protein (15) and appears to play a role in activation of viral and cellular gene expression, alone or in combination with IE2579aa (10, 11, 16, 17). IE1491aa has been assigned an autoregulatory role during replication, exhibiting a capacity to transactivate expression from the ie1/ie2 promoter-enhancer via NF-κB sites (16, 17). Evidence has supported a role for IE1491aa in the transactivation of other cellular promoter elements, including the E2F (18) and NF-βA (19) binding sites, the CCAAT box (20), and additional less-defined sites (21, 22). IE1491aa also exhibits the capacity to augment transactivation in combination with the strong heterologous transactivator, IE2579aa, without having any effect by itself, on a number of different simple promoters (10, 11, 21, 23). Although not necessarily related to its role in gene regulation, IE1491aa has also been reported to localize to metaphase chromatin (24) and to block adenovirus E1a-induced apoptosis (25).
Direct investigation of the role that IE1491aa plays during viral replication requires ie1-deficient viral mutants. Here, a deletion that removes all IE1491aa-specific coding sequences is used to demonstrate an important role for this protein during viral replication in culture. The phenotype we observe is consistent with a failure of ie1-deficient virus to activate expression from the ie1/ie2 promoter-enhancer and thereby ensure sufficient levels of IE2579aa for initiation of productive replication. While critical at low multiplicities of infection (mois), infection at intermediate to high mois compensates for the lack of IE1491aa, suggesting that transactivators in the virion tegument (26, 27) can function in place of this α gene product. The growth characteristics of mutant virus suggests potential use as a replication-impaired human-specific vector that can deliver genes to hematopoietic progenitor cells.
MATERIALS AND METHODS
Viruses and Cells. The Towne strain of CMV was propagated in human fibroblasts (HFs) prepared from newborn foreskin and maintained in DMEM (GIBCO/BRL) supplemented with 10% NuSerum (Collaborative Research), amino acids and antibiotics as previously described (28). IE1491aa− expressing ihfie1.3 cells were derived from HFs by transduction with a murine retrovirus vector N2/CMV-IE (29) and selecting cells in medium containing Geneticin (GIBCO/ BRL) at 400 μg/ml followed by immortalization with the retroviral vector LXSN16E6E7 (30) as previously described (28). Following outgrowth, ihfie1.3 cells were carried in the absence of Geneticin, but continued to express constitutive levels of IE1491aa based on antibody-detection methods (unpublished observations). Stocks of recombinant viruses RC303ΔAcc and Towne/Tol11 1.1 were propagated and plaque assayed on ihfie1.3 and HF cells, respectively, except where noted in the text. All mois of mutant virus were calculated from stock titers on ihfie1.3 cells and all virus stocks were prepared from infected cells exhibiting maximal cytopathic effects by sonication in a 1:1 mixture of cells in medium (at 106 cells per ml) and autoclaved nonfat milk.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CMV, cytomegalovirus; moi, multiplicity of infection; wt, wild type; HF, human fibroblast; hpi, hours postinfection.
|
† |
To whom reprint requests should be addressed. e-mail: mocarski@leland.stanford.edu. |
|
§ |
Present address: Department of Medicine, Cambridge University, Cambridge CB2 2QQ, United Kingdom. |
Transfection. PacI-digested cosmids Tn23, Tn26, Tn44, Tn45, Tn46, Tn47, Tn51, and Tol11 (10 μm each) (31) were mixed with 2 μg of Sal-digested pON303ΔAcc (or pON303) (16, 32). To construct RC303Acc or Towne/Tol11 1–1, 10 μg of the pON303ΔAcc- or pON303-containing cosmid mixtures were used to transfect 106 ihfie1.3 cells by the calcium phosphate precipitation technique as described (31).
DNA Blot Hybridization and Immunoblot Analyses. Total infected cell DNA was isolated (33) from infected HF or ihfie1.3 cells; 1 μg was digested with EcoRI, ClaI, or BamHI (New England Biolabs) using the manufacturer’s recommended conditions and was transferred to Hybond-N+ (Amersham) before hybridization with [32P]dCTP (Amersham) random-primed radiolabeled pON303 or pON2307 (an exon 4-specific probe) (34). Immunoblot analysis was performed according to published protocols using human CMV-seropositive antiserum, murine monoclonal antibodies CH443 (specific for exon 4; unpublished results) and CH160 (specific for exon 2; ref. 35), renamed 1203 (Goodwin Institute, Ft. Lauderdale, FL), or rabbit polyclonal antibody raised against the glutathione S-transferase fusion protein carrying aa 232– 400 of IE1491aa from exon 4. Horseradish peroxidase-conjugated goat anti-human IgG, goat anti-rabbit IgG, and goat anti-mouse IgG (Vector Laboratories) were used as secondary antibodies and blots were developed using the Enhanced Chemiluminescence System (Amersham) according to the manufacturer’s protocol.
RESULTS
Construction of RC303ΔAcc. Recombinant viruses were constructed from seven Towne strain cosmids (Tn23, Tn26, Tn44, Tn45, Tn46, Tn47, and Tn51) and one Toledo strain cosmid (Tol11) (31) that formed an overlapping set except for a 15-bp gap between Tn51 and Tol11 (170,506–170,520) as depicted in Fig. 1. This gap was bridged with either pON303ΔAcc (16) or pON303 (32), resulting in the production of either ie1-deficient RC303ΔAcc or wild-type (wt) Towne/ Tol11 1.1 (Fig. 1). We used established transfection conditions for generation of recombinant CMV (31). SalI-linearized plasmid was added in a molar amount equivalent to an individual of PacI-linearized cosmids and cotransfection of this mixture was carried out on ihfie1.3 cells to complement IE1491aa in trans. When the genome structure of the resulting recombinants was analyzed, two of five independent pools arising from the pON303ΔAcc-containing set carried a uniform population of the expected mutant, and two pools made with pON303 contained the expected Towne/Toledo wt chimera (31). Blot hybridization of EcoRI-digested DNA from mutant RC303ΔAcc and wt Towne/Tol11 1.1 transfection pools and from plaque pure RC303ΔAcc produced the expected patterns (Fig. 2). Hybridization with pON303 probe revealed a 10-kbp EcoRI fragment in the wt virus, which was reduced to 8.6 kbp in the mutant (Fig. 2A). An ie1 exon 4 probe (pON2307) hybridized with the 10-kbp fragment in Towne/ Tol11 1.1 but failed to hybridize with any fragment in the mutant. A 3.9-kbp ClaI fragment containing ie1 was reduced to 2.5 kbp as a result of the deletion (Fig. 2B). After evaluation of BamHI digests (data not shown) and EcoRI and ClaI digests, we concluded that the two independent RC303ΔAcc isolates and two independent Towne/Tol11 1.1 isolates exhibited no differences outside of the 1.4-kbp deletion that had been engineered into the mutant virus.
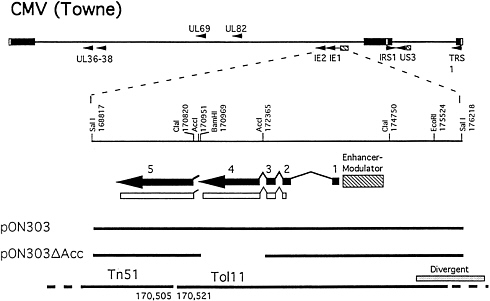
FIG. 1. Construction of ie1-deficient CMV, RC303ΔAcc. (Upper) CMV genome depicted with viral regulatory gene (9) loci represented by small arrows. Expanded below is a partial restriction site map of the SalI fragment (nt 168,817–176,218) containing the ie1/ie2 region depicted as five exons (solid thick lines and arrows) that give rise to the predominant α proteins, IE1491aa (exons 1–4) and IE2579aa (exons 1–3 and 5) via alternative splicing and polyadenylylation site usage. Protein-coding sequences are depicted as open boxes and enhancer regions are depicted as a hatched boxes. (Lower) The plasmids and cosmids used in construction of recombinant viruses RC303ΔAcc and Towne/Tol11 1.1. Plasmids, pON303 (32), and pON303ΔAcc (16) as well as sequence endpoints of cosmids, Tn51 and Tol11, are depicted. The region of sequence divergence between strains Towne and Toledo (2) is indicated by stippling above cosmid Tol11. All nucleotide sequence coordinates are based on the strain AD169 genome sequence (1).
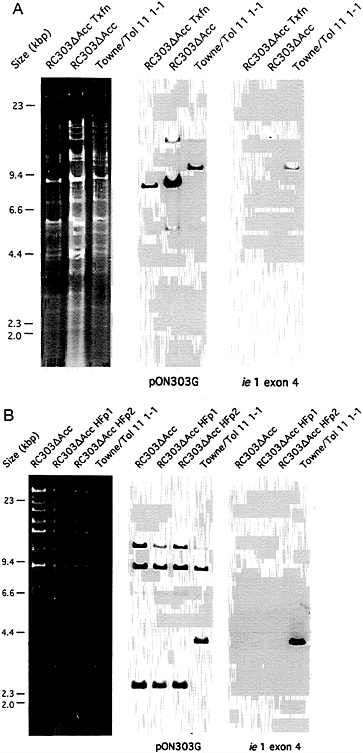
FIG. 2. Analysis of RC303ΔAcc genome structure and stability. (A) Restriction digest and DNA blot hybridization analysis of mutant following transfection (RC303ΔAcc Tfxn) and plaque-purification (RC303ΔAcc) compared with wild-type (Towne/Tol11 1–1) transfection stock. The left three lanes show ethidium bromide-stained EcoRI digests of products resolved in a 0.5% agarose gel. The middle and right three lanes show an autoradiogram of a DNA blot of EcoRI digests hybridized with an ie1/ie2 region probe, pON303G (16), and an ie1 exon 4 specific probe, pON2307 (34), respectively. (B) Restriction digest and blot hybridization analysis of mutant DNA from complementing ihfie1.3 cells (RC303ΔAcc) or DNA made from noncomplementing HF cells following one (RC303ΔAcc HFp1) or two (RC303ΔAcc HFp2) successive passages. The left four lanes show ethidium bromide-stained ClaI digests resolved in a 0.5% agarose gel. The middle and right four lanes show an autoradiogram of a DNA blot of ClaI digests hybridized with pON303G and the ie1 exon 4-specific probe, pON2307 (34), respectively.
The three other pools.harvested following cotransfection of cosmids and pON303ΔAcc each contained wt virus that showed pON303 and exon 4 hybridization patterns similar to Towne/Tol11 1.1 (data not shown). The ie1 cDNA resident in ihfie1.3 cells did not appear to recombine into mutant virus based on the failure to detect the novel, intronless exon 4-specific DNA species that would have been generated. Furthermore, recombination between homologous regions of the virus and cDNA resident in the ihfie1.3 cells would have disrupted the differential splicing necessary to produce to IE2579aa, which we assume would result in a replication defective virus. The presence of wt in some pON303ΔAcc-containing transfection pools was most likely to have resulted from a 430-bp overlap (170,521–170,951) between pON303ΔAcc and Tol11 (Fig. 1). This background of wt virus suggests that efficient homologous recombination can occur with as little as 400 nucleotide overlap between input DNA fragments.
RC303ΔAcc Growth on Complementing and Noncomplementing Cells. To determine whether mutant virus was capable of productive infection of cells lacking the ie1 gene, serial 10-fold dilutions of the progeny from ihfie1.3 cells were used to infect complementing and noncomplementing cells in parallel. Mutant virus failed to grow in any of four replicate experiments when 106 normal HF cells were exposed to mois <0.01 (as determined by plaque assay on ihfie1.3 cells) but retained the ability to form plaques on 106 ihfie1.3 cells at mois as low as 0.000001 (data not shown). At an moi of 3 or greater, growth of four different plaque isolates of the mutant was remarkably similar in both cell types, producing progeny at levels comparable to those of wt virus at 96 hours postinfection (hpi) (Fig. 3). When wt Towne growth was compared on both cell types in a separate experiment, there were no differences observed at 24, 48, or 72 hpi (data not shown). At mois between 0.01 and 1.0, in noncomplementing cells mutant virus exhibited a sharp increase in number of plaques and these required approximately three times longer (21 days versus 7 days in complementing cells) to appear. Comparison of titers of four independent RC303ΔAcc plaques from one transfection pool as determined on complementing cells and noncomplementing cells is shown in Table 1. Although mutant virus exhibited a linear relationship between dilution and ability to form plaques on ihfie1.3 cells, plaque formation on noncomplementing HFs appeared to be nonlinear with respect to dilution, with a 10-fold dilution of stock producing an ≈100-fold decrease in plaque formation. Consistent with the slow growth and nonlinear relationship between input dose and plaque formation, mutant virus exhibited a plaquing efficiency on HF that was much lower (5×10−4 to 3×10−3
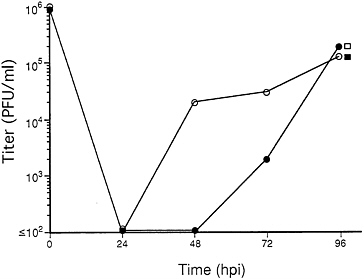
FIG. 3. Growth of RC303ΔAcc and Towne/Tol11 1.1 in complementing ihfie1.3 (open symbols) and noncomplementing HF (solid symbols) cells. Cell cultures were exposed to RC303ΔAcc (circles) or Towne/Tol11 1–1 (squares) at an moi of 3. Cells infected with the ie1-deficient mutant were harvested at 24, 48, 72, and 96 hpi and cells infected with wt were harvested at 96 hpi. Titers of progeny in all cultures were determined by plaque assay on ihfie1.3 cells.
Table 1. RC303ΔAcc plaque assay on complementing and noncomplementing cells
|
RC303ΔAcc |
ihfie 1.3* |
HF† |
Ratio§ |
|
Plaque 1 |
5.0×107‡ |
1.8×105 |
278 |
|
Plaque 2 |
2.2×107 |
1.0×104 |
2200 |
|
Plaque 3 |
2.3×107 |
2.0×104 |
1150 |
|
Plaque 4 |
3.6×107 |
1.1×105 |
327 |
|
*Stained at 7 days postinfection, samples exhibited a linear dilution of infectivity. †Stained at 21 days postinfection, samples exhibited a nonlinear dilution suggesting multiple hit kinetics (data not shown). ‡Titer (plaque-forming units per ml). §Ratio (HF/ihfie 1.3) of input needed to produce 10–200 plaques on 106 cells. |
|||
relative to ihfie1.3 cells) such that only 10–200 plaques formed at an moi of 0.02–0.05 and these took nearly 3 weeks to grow. This behavior suggested that IE1491aa was necessary under low moi conditions and that in the absence of this protein multiple independent virus particles were required to initiate infection. Thus, virion structural proteins might have compensated for the absence of IE1491aa at higher mois. This growth pattern also raised the possibility that IE1491aa made in the complementing cells might have been carried over to noncomplementing cells in the inoculum or that the ie1 cDNA might have been transduced by mutant virus during propagation on ihfie1.3 cells.
To determine whether the growth of-mutant virus in noncomplementing cells was a result of IE1491aa carry-over in the inoculum or a result of reacquisition of the ie1 gene by the virus, two sequential passages of RC303ΔAcc were carried out on HF cells at an moi of 0.5, and progeny viral DNA was evaluated by blot hybridization. IE1491aa contamination in the inoculum would have quickly been eliminated by successive passages on HF cells. Despite the fact that progeny accumulated more slowly in noncomplementing cells and took approximately three times as long to reach peak levels, the total amount of viral DNA that could be recovered from noncomplementing cells was comparable to that recovered from complementing cells (Fig. 2B). Analysis of BamHI and EcoRI digests (data not shown) further demonstrated an absence of alterations in genome structure or of DNA fragments that would suggest an overgrowth of virus that acquired ie1 cDNA from complementing cells. Given the ability of mutant virus to propagate through successive rounds on HF cells, contamination with IE1491aa could not account for the ability of mutant to grow on noncomplementing cells.
Expression of IE2579aaby RC303ΔAcc Is Dependent upon moi. To directly evaluate the stage of mutant virus replication that was disrupted at low mois, expression of IE1491aa and IE2579aa was compared following infection of HFs at mois of 5 and 0.5 using either RC303ΔAcc or wt Towne virus. Towne exhibits growth properties similar to Towne/Tol11 recombinants on HF cells (31). We used a murine monoclonal antibody CH160 that recognizes an epitope within the common amino terminus of these proteins (35). Due to the deletion of exon 4 sequences, mutant virus failed to produce any IE1491aa, whereas wt virus produced readily detectable levels of this protein very early during infection. At an moi (5 plaque-forming units per cell) that resulted in a replication pattern similar to wt, mutant virus produced a pattern of IE2579aa expression similar to wt (Fig. 4A); however, at an moi (0.5 plaque-forming units per cell) that results in inefficient plaque formation and slow growth, expression of IE2579aa was not detected in RC303ΔAcc-infected HF cells (Fig. 4B). Thus, inefficient growth correlated with failure to produce detectable levels of IE2579aa following infection. The identity of the IE1491aa species was confirmed by analysis with exon 4 specific antibody (data not shown). Interestingly, the relative proportion of IE1491aa and IE2579aa accumulating during infection
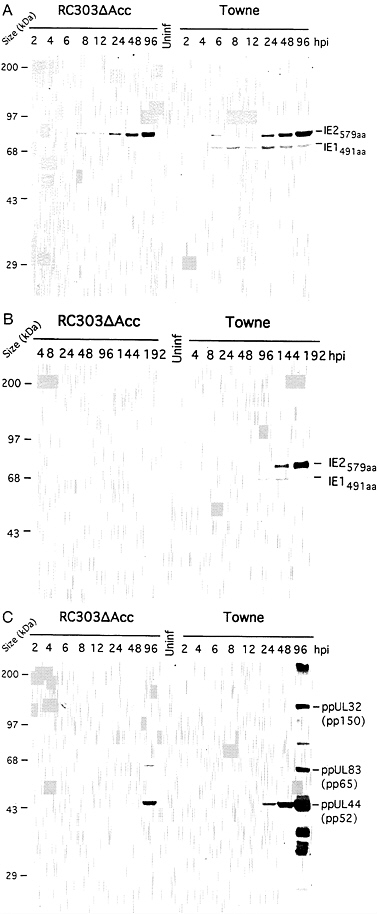
FIG. 4. Immunoblot analysis of proteins expressed by RC303ΔAcc (Left) and Towne (Right). (A) Detection of IE1491aa and IE2579aa with exon 2-specific monoclonal antibody CH160 at 2, 4, 6, 8, 12, 24, 48, and 96 hpi with an moi of 5. (B) Detection of IE1491aa and IE2579aa with CH160 at 4, 8, 24, 48, 96, 144, and 192 hpi with an moi of 0.5. (C) Detection of γ proteins reactive with pooled human CMV-seropositive sera at 2, 4, 6, 8, 12, 24, 48, and 96 hpi with an moi of 5.
with wt virus was also influenced by the moi, with both appearing to accumulate together at high moi, but with IE1491aa appearing much earlier than IE2579aa at the lower moi. When mutant virus expression of β and γ proteins (ppUL44,
ppUL83, ppUL57, and ppUL32) was evaluated at an moi of 0.5, all failed to accumulate in HF cell lysates examined up to 192 hpi (data not shown), a pattern that was consistent with inefficient growth and slow spread of this virus. Even at high moi, accumulation of γ proteins, ppUL32, ppUL83, and ppUL44 were delayed in RC303ΔAcc-infected HF cells compared with a Towne control (Fig. 4C). Thus, RC303ΔAcc exhibited a very strong moi dependence for accumulation of IE2579aa, a regulatory protein that is likely to be essential for productive replication. This very early block to expression of IE2579aa is consistent with our prediction that autoregulation of the ie1/ie2 promoter-enhancer via the cellular transcription factor NF-κB is an important IE1491aa function (16, 17). Other activities of IE1491aa may also be disrupted in RC303ΔAcc, but not manifest as strong an impact on viral replication as this autoregulatory function.
DISCUSSION
RC303ΔAcc is a severely replication defective CMV mutant whose phenotype reveals a role for IE1491aa in productive replication as was predicted from transient assays (16, 17). In culture, IE1491aa is required for successful replication at mois of <0.01, but this requirement may be overcome when sufficient numbers of viral particles are used to initiate infection. The moi-dependent expression of IE2579aa by RC303ΔAcc provides further evidence of the importance of IE1491aa in regulating levels of ie2 expression and replication under conditions where virion transactivators are likely to be limiting. The behavior of ie1-deficient CMV parallels the behavior of herpes simplex virus ICP0 (Vmw110) mutants (36) and predicts a very important role for IE1491aa in the naturally infected human host (37). It is formally possible that IE1491aa fulfills a function analogous to ICP0 (38). These predictions can be tested by inoculation of human tissue implants in SCID-hu mice (39) or by evaluating latency and reactivation properties in granulocyte-macrophage progenitors (40).
Many studies (26, 27, 32, 41) consistently suggested that CMV, like other herpesviruses, carries virion tegument functions responsible for transactivation of α genes. IE1491aa is not a virion protein (unpublished data) and cannot account for the ability of inactivated viral particles to transactivate the ie1/ie2 promoter-enhancer (32). The moi-dependent growth properties of IE1491aa-deficient virus support a role for two tegument proteins ppUL82 (pp71) (27) and ppUL69 (26), in transactivation of the ie1/ie2 promoter-enhancer, although experiments testing the ability of these gene products or UV-inactivated virions will be needed to address this issue more directly. It appears that CMV may encode two different classes of proteins capable of transactivating α gene expression: (i) one packaged into the virion and (ii) one, IE1491aa, expressed immediately upon entering cells. In the absence of IE1491aa, virion transactivators alone may not be able to initiate replication at low mois. Whether IE1491aa can compensate for virion transactivators remains to be determined when mutants disrupting UL82 or UL69 become available. In transient assays, a putative minor ie2 protein, IE2425aa (42), may transactivate expression of the ie1/ie2 promoter-enhancer, but this would not account for the properties of RC303ΔAcc described here. A range of factors, particularly those found in serum (43), that increase levels of cellular transcription factors also stimulate expression from the ie1/ie2 promoter-enhancer during cell growth or differentiation. All of our experiments were carried out at high serum concentrations in actively growing HF cells that may have reduced the requirement for IE1491aa. Studies using resting, serum-starved HF cells are under way. Our comparison of mutant viral growth on complementing and noncomplementing cells (Fig. 3) showed that complementing cells provided a slight advantage to replication even at high mois and suggested that IE1491aa may play additional roles subsequent to α gene activation. Finally, both IE1491aa and IE2579aa are able to block adenovirus E1a-induced apoptosis (25). We did not observe any dramatic alteration in the phenotype of cells infected with RC303ΔAcc that would be consistent with any increased tendency toward apoptosis. An appreciation of how a balance is struck between all of these factors will require considerable additional work in different cell types as well as in different growth and differentiation states.
While the highly passaged, candidate vaccine strain, Towne, is attenuated and fails to reactivate in the immunosuppressed transplant recipient (44), strain Toledo, a virus that has undergone a limited number of passages in cell culture, maintains a level of virulence that can be observed after inoculation of susceptible individuals (45). These virulence differences may be attributed to the loss of a large genomic segment in highly passaged strains (2). The strategy reported here would allow the derivation of avirulent, replication defective mutant virus from any strain of CMV. The mutant virus we have described places 13 kbp of unique sequence from strain Toledo into a Towne background. Although replication defective, this recombinant contains a large array of viral genes that are not present in Towne. As a vaccine, this would be expected to induce a broader ranging immune response but be completely safe even in immunocompromised individuals.
Accumulating evidence suggests that hematopoietic progenitors in bone marrow are important sites of viral latency (40, 46). Cultured granulocyte-macrophage progenitors can support latent infection with either the Towne or Toledo strains and infection is characterized by restricted, latency-specific gene expression (34, 47). Productive viral replication can be induced when these cells are cocultivated with permissive fibroblasts for an extended period of time (40), suggesting that cellular growth or differentiation state may play an important role in the balance between latency and reactivation. By removing ie1 exon 4, RC303ΔAcc disrupts the antisense CMV-latency specific transcript expressed in granulocyte-macrophage progenitors. We are currently evaluating the ability of the ie1 mutant to latently infect this cell type and to reactivate following cocultivation with ihfie1.3 cells. Recombinant CMV lacking IE1491aa may be an appropriate avirulent vector to introduce genes into hematopoietic progenitors without a risk of reactivation or dissemination.
We thank Kirsten Lofgren and Tai-An Cha for providing sequence information on the cosmid ends, Danushka Formankova for immunoblot analysis, and Maria Kirichenko for performing plaque assays. We particularly appreciate the contribution of Jiake Xu who purified the glutathione S-transferase fusion protein from pON2307 and raised ie1 exon-4 specific rabbit antiserum. We acknowledge Mark Penfold, Mark Prichard, Barry Slobedman, Dirk Dittmer, Jiake Xu, and Cynthia Bolovan for helpful comments on the manuscript. Some of the experiments reported here were carried out by E.S.M. at Aviron, a company to which he serves as a scientific advisor. This work was supported by U.S. Public Health Service Grant R01 AI33852.
1. Chee, M.S., Bankier, A.T., Beck, S., Bohni, R., Brown, C.M., Cerny, R., Horsnell, T., Hutchison, C.A.I., Kouzarides, T., Martignetti, J.A., Preddie, E., Satchwell, S.C., Tomlinson, P., Weston, K.M. & Barrell, B.G. (1990) Curr. Top. Microbiol. Immunol. 154, 125–170.
2. Cha, T.A., Tom, E., Kemble, G.W., Duke, G.M., Mocarski, E.S. & Spaete, R.R. (1996) J. Virol. 70, 78–83.
3. Alford, C.A. & Britt, W.J. (1995) in Fields Virology, eds. Fields, B.N., Knipe, D.M. & Howley, P.M. (Lippincott-Raven, New York), pp. 2493–2534.
4. Stenberg, R.M., Witte, P.R. & Stinski, M.F. (1985) J. Virol. 56, 665–675.
5. Stenberg, R.M., Thomsen, D.R. & Stinski, M.F. (1984) J. Virol. 49, 190–199.
6. Stenberg, R.M., Depto, A.S., Fortney, J. & Nelson, J.A. (1989) J. Virol. 63, 2699–2708.
7. Thomsen, D.R., Stenberg, R.M., Goins, W.F. & Stinski, M.F. (1984) Proc. Natl. Acad. Sci. USA 81, 659–663.
8. Boshart, M., Weber, F., Jahn, G., Dorsch-Hasler, K., Fleckenstein, B. & Schaffner, W. (1985) Cell 41, 521–530.
9. Mocarski, E.S. (1995) in Fields Virology, eds. Fields, B.N., Knipe, D.M. & Howley, P.M. (Lippincott-Raven, New York), pp. 2447–2492.
10. Malone, C.L., Vesole, D.H. & Stinski, M.F. (1990) J. Virol. 64, 1498–1506.
11. Stenberg, R.M., Fortney, J., Barlow, S.W., Magrane, B.P., Nelson, J.A. & Ghazal, P. (1990) J. Virol. 64, 1556–1565.
12. Pizzorno, M.C., O’Hare, P., Sha, L., LaFemina, R.L. & Hayward, G.S. (1988) J. Virol. 62, 1167–1179.
13. Lang, D. & Stamminger, T. (1993) J. Virol. 67, 323–331.
14. Macias, M.P. & Stinski, M.F. (1993) Proc. Natl. Acad. Sci. USA 90, 707–711.
15. Stinski, M.F. (1978) J. Virol. 26, 686–701.
16. Cherrington, J.M. & Mocarski, E.S. (1989) J. Virol. 63, 1435– 1440.
17. Sambucetti, L.C., Cherrington, J.M., Wilkinson, G.W.G. & Mocarski, E.S. (1989) EMBO J. 8, 4251–4258.
18. Margolis, M.J., Pajovic, S., Wong, E.L., Wade, M., Jupp, R., Nelson, J.A. & Azizkhan, J.C. (1995) J. Virol. 69, 7759–7767.
19. Hunninghake, G.W., Monks, B.G., Geist, L.J., Monick, M.M., Monroy, M.A., Stinski, M.F., Webb, A.C., Dayer, J.M., Auron, P.E. & Fenton, M.J. (1992) Mol. Cell. Biol. 12, 3439–3448.
20. Hayhurst, G.P., Bryant, L.A., Caswell, R.C., Walker, S.M. & Sinclair, J.H. (1995) J. Virol. 69, 182–188.
21. Hagemeier, C., Walker, S.M., Sissons, P.J. & Sinclair, J.H. (1992) J. Gen. Virol. 73, 2385–2393.
22. Walker, S., Hagemeier, C., Sissons, J.G. & Sinclair, J.H. (1992) J. Virol. 66, 1543–1550.
23. Lukac, D.M., Manuppello, J.R. & Alwine, J.C. (1994) J. Virol. 68, 5184–5193.
24. LaFemina, R.L., Pizzorno, M.C., Mosca, J.D. & Hayward, G.S. (1989) Virology 172, 584–600.
25. Zhu, H., Shen, Y. & Shenk, T. (1995) J. Virol. 69, 7960–7970.
26. Winkler, M., Schmolke, S., Plachter, B. & Stamminger, T. (1995) Scand. J. Infect. Dis. Suppl. 99, 8–9.
27. Liu, B. & Stinski, M.F. (1992) J. Virol. 66, 4434–4444.
28. Greaves, R.F., Brown, J.M., Vieira, J. & Mocarski, E.S. (1995) J. Gen. Virol. 76, 2151–2160.
29. Roy, N. (1993) Ph.D. thesis (Cornell Univ. School of Med., New York).
30. Halbert, C.L., Demers, G.W. & Galloway, D.A. (1991) J. Virol. 65, 473–478.
31. Kemble, G., Duke, G., Winter, R. & Spaete, R. (1996) J. Virol. 70, 2044–2048.
32. Spaete, R.R. & Mocarski, E.S. (1985) J. Virol. 56, 135–143.
33. Spaete, R.R. & Frenkel, N. (1982) Cell 30, 295–304.
34. Kondo, K., Xu, J. & Mocarski, E.S. (1996) Proc. Natl. Acad. Sci. USA 93, 11137–11142.
35. Plachter, B., Britt, W., Vornhagen, R., Stamminger, T. & Jahn, G. (1993) Virology 193, 642–652.
36. Stow, N.D. & Stow, E.C. (1986) J. Gen. Virol. 67, 2571–2585.
37. Cai, W., Astor, T.L., Liptak, L.M., Cho, C., Coen, D.M. & Schaffer, P.A. (1993) J. Virol. 67, 7501–7512.
38. Stow, E.C. & Stow, N.D. (1989) J. Gen. Virol. 70, 695–704.
39. Brown, J.M., Kaneshima, H. & Mocarski, E.S. (1995) J. Infect. Dis. 171, 1599–1603.
40. Kondo, K., Kaneshima, H. & Mocarski, E.S. (1994) Proc. Natl. Acad. Sci. USA 91, 11879–11883.
41. Stinski, M.F. & Roehr, T.J. (1985) J. Virol. 55, 431–441.
42. Baracchini, E., Glezer, E., Fish, K., Stenberg, R.M., Nelson, J.A. & Ghazal, P. (1992) Virology 188, 518–529.
43. Stamminger, T., Fickenscher, H. & Fleckenstein, B. (1990) J. Gen. Virol. 71, 105–113.
44. Plotkin, S.A., Smiley, M.L., Friedman, H.M., Starr, S.E., Fleisher, G.R., Wlodaver, C., Dafoe, D.C., Friedman, A.D., Grossman, R.A. & Barker, C.F. (1984) Lancet i, 528–530.
45. Plotkin, S.A., Starr, S.E., Friedman, H.M., Gonczol, E. & Weibel, R.E. (1989) J. Infect. Dis. 159, 860–865.
46. Minton, E.J., Tysoe, C., Sinclair, J.H. & Sissons, J.G. (1994) J. Virol. 68, 4017–4021.
47. Kondo, K. & Mocarski, E.S. (1995) Scand. J. Infect. Dis. Suppl. 99, 63–67.






