
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains
THOMAS R.JONES*†, EMMANUEL J.H.J.WIERTZ‡, LEI SUN*, KENNETH N.FISH§, JAY A.NELSON§, AND HIDDE L.PLOEGH‡
*Department of Molecular Biology, Infectious Diseases Section, Wyeth-Ayerst Research, Pearl River, NY 10965; ‡Center for Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139; and §Department of Molecular Microbiology and Immunology, Oregon Health Sciences University, Portland, OR 97201
ABSTRACT The human cytomegalovirus (HCMV) early glycoprotein products of the US11 and US2 open reading frames cause increased turnover of major histocompatibility complex (MHC) class I heavy chains. Since US2 is homologous to another HCMV gene (US3), we hypothesized that the US3 gene product also may affect MHC class I expression. In cells constitutively expressing the HCMV US3 gene, MHC class I heavy chains formed a stable complex with β2-microglobulin. However, maturation of the N-linked glycan of MHC class I heavy chains was impaired in US3+ cells. The glycoprotein product of US3 (gpUS3) occurs mostly in a high-mannose form and coimmunoprecipitates with β2-microglobulin associated class I heavy chains. Mature class I molecules were detected at steady state on the surface of US3+ cells, as in control cells. Substantial perinuclear accumulation of heavy chains was observed in US3+ cells. The data suggest that gpUS3 impairs egress of MHC class I heavy chains from the endoplasmic reticulum.
Human cytomegalovirus (HCMV) causes serious disease in congenitally infected infants and immunocompromised/ immunosuppressed adults (1). As is common to other members of the family herpesviridae, HCMV causes latent or persistent viral infections. However, this state has not been well characterized for HCMV. Virus derived from such latent or persistent infections can be responsible for the HCMV-associated disease state in immunocompromised or immunosuppressed patients. One requirement for viruses that exhibit a latent or persistent phase is that they encode mechanism(s) that manipulate the host’s immune surveillance system (2).
Viruses encode proteins that target and modulate many different aspects of the host’s immune system (3). A target common to many of these viruses are the major histocompatibility complex (MHC) class I antigens (4, 5). Class I products play a central role in recognition and subsequent lysis by cytotoxic T lymphocytes (6), a key component of the cellular arm of the human immune system. For example, adenoviruses encode proteins that repress the transcription of class I heavy chain genes (7) or that can bind to and retain class I heavy chains in the endoplasmic reticulum (ER) (8, 9). The herpes simplex virus type 1 US 12 gene product, called ICP47, binds to the MHC-encoded TAP peptide transporter and prevents the delivery of cytosolic antigenic peptides to assembling class I molecules in the ER (10–12). Murine cytomegalovirus encodes multiple functions that affect class I expression, either at the level of synthesis or expression at the cell surface (13–15). Epstein-Barr virus affects presentation of certain peptides by MHC class I complexes (16). In that case, a cis-acting Gly-Ala repeat within the viral Epstein-Barr virus-encoded nuclear antigen 1 (EBNA-1) protein is an inhibitory signal that prevents its antigenic processing, thus affecting the yield of EBNA-1-derived peptides to be presented. HCMV causes posttranslational down-regulation of MHC class I heavy chains (17–19). In HCMV-infected cells, class I heavy chains show unusually rapid turnover (t1/2=20 min or less) and are degraded early in the course of their biosynthesis, proceeding no further than the ER (17, 19).
Through the analysis of defined deletion mutants of HCMV, we recently identified two HCMV early genes, US11 and US2, each of whose products was sufficient to cause increased turnover of MHC class I heavy chains (ref. 20; T.R.J. and L.S., unpublished results). US11 encodes a type I membrane glycoprotein that resides in the ER and causes the rapid dislocation of newly synthesized class I heavy chains from the ER to the cytosol, where they are degraded by the proteasome (21). A second gene whose expression results in destabilization of class I heavy chains is US2 (T.R.J. and L.S., unpublished results). US2 is member of a gene family that also contains US3, a gene adjacent to US2 in the HCMV genome (22). Given the homology between the proteins encoded by US2 and US3, it was of interest to determine whether US3 also affected the expression of MHC class I heavy chains. Unlike US2, whose expression begins at early times after infection (T.R.J. and L.S., unpublished results), US3 is transcribed abundantly under immediate-early conditions and at lower levels at early and late times after infection, due to multiple negative regulatory mechanisms (23–26). In contrast to US2, expression of US3 does not destabilize heavy chains, but maturation and transport to the cell surface of MHC class I heavy chains are impaired. We show that a US3 gene product forms a complex with β2-microglobulin-associated class I heavy chains, which accumulate predominantly in the ER of US3+ cells.
MATERIALS AND METHODS
DNA Sequence. The numbering system of Chee et al. (22) was used for the HCMV AD169 DNA sequence (GenBank accession no. X17403).
Virus and Cells. HCMV AD169 was obtained from the American Type Culture Collection and propagated. RV47, a US2 and US3 deletion mutant of HCMV, has been described (27). The origin and growth of human foreskin fibroblast cells
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HCMV, human cytomegalovirus; MHC, major histocompatibility complex; ER, endoplasmic reticulum; GST, glutathione S-transferase.
|
† |
To whom reprint requests should be addressed at: Building 205, Room 276, Wyeth-Ayerst Research, Pearl River, NY, 10965. e-mail: jonest@war.wyeth.com. |
and U373-MG astrocytoma (U373) control cells were as described (20). Both the US2+ and US3+ cell line used in this study, originally designated 55–310 cells and 5–214 cells (respectively) in laboratory records, were derived from U373 cells. To obtain these cell lines, HCMV US2 and US3 genes were cloned into plasmids such that their expression was under the control of the HCMV major immediate-early promoter. The US2 open reading frame was cloned as a 0.776-kb BanII-XhoI DNA fragment (bases 193,779–193,003) in vector pIEsp-puro (20), to yield pIEspUS2-puro. The US3 open reading frame was cloned as a 0.638-kb AvaII-SacII DNA fragment (bases 194,700–194,062) in vector pIE-puro (20), to yield pIEpuro-US3(AS). Stably transfected US2+ and US3+ cell lines were obtained via transfection of these plasmids and selection in puromycin-containing medium as described (20).
Glutathione S-Transferase (GST)-US3 Fusion Protein. A GST-US3 fusion protein was made using a modified vector, pGST-Nco (provided by I.Mohr, Wyeth-Ayerst Research, Pearl River, NY). Using the polymerase chain reaction, the region of the US3 gene representing the region encoding amino acids 33–112 (22) was amplified in a manner that resulted in the insertion of a 5′ NcoI and a 3′ EcoRI sites to facilitate directional cloning between those same sites in pGST-Nco vector. After transfection into Escherichia coli DH5, expression of the GST-US3 fusion protein was induced with isopropyl β-D-thiogalactoside. After sonication of the induced bacteria, the fusion protein was located in the pellet fraction, then solubilized, and electroeluted from SDS/ polyacrylamide gels.
Antibodies. Rabbit polyclonal antisera reactive with HCMV US2 and US11 proteins have been described (ref. 20; T.R.J. and L.S., unpublished results). Polyclonal antisera reactive with HCMV US3 protein was derived from New Zealand White rabbits immunized with the GST-US3 fusion protein (Cocalico Biologicals, Reamstown, PA). Rabbit polyclonal antisera designated anti-HC, which reacts free MHC class I heavy chains, has been described (17). Murine monoclonal antibodies W6/32 (28), specific for a conformation-dependent epitope on the heavy chain of human MHC class I proteins, and Ber-T9, specific for the human transferrin receptor, were purchased from Dako. Murine monoclonal antibody TP25.99 (29), specific for a conformation-independent epitope on MHC class I heavy chains, was obtained from S.Ferrone (New York Medical College, Valhalla, NY). BBM.1 monoclonal antibody recognizes both free and heavy chain-associated human β2-microglobulin (30). Anti-vertebrate actin monoclonal antibody clone C4 was purchased from Boehringer Mannheim.
Metabolic Labeling, Immunoprecipitation, and Immunoblot Analysis. Metabolic labeling, immunoprecipitation, and immunoblot techniques were done as described (20). Where indicated, the mild detergent digitonin (Boehringer Mannheim) was used for cell lysis (1% final concentration) and in all immunoprecipitation steps (0.2% final concentration). Digitonin was dissolved in buffer containing 25 mM Hepes (pH7.2)/10 mM CaCl2. For pulse-chase experiments, 1 ml of complete medium containing 2× unlabeled methionine/ cysteine was added directly to the radioactive pulse medium and incubation was continued until the proper harvest time. Radioiodination of cell surface proteins was done with sodium 125I (New England Nuclear) according to standard protocols (31). Endoglycosidase H and N-glycosidase F digestions were done for 18 hr at 37°C after immunoprecipitation using the recommended conditions (Boehringer Mannheim).
Immunofluorescence Microscopy. In some experiments, expression of MHC class I molecules was detected by immunofluorescence microscopy as described (20), except that the blocking step with human serum was omitted. Fixed cells were treated sequentially with TP25.99 primary antibody and fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Pierce) secondary antibody. Confocal immunofluorescence microscopy was performed as described (32), except that TP25.99 monoclonal antibody was used to detect MHC class I heavy chains. The dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6[3]) (Molecular Probes) was used to stain the ER as described (33). These samples were visualized using a Leica confocal laser scanning microscope. Class I heavy chain fluorescence was quantified using the QUANTIMET 500 fluorescence analysis program (Leica).
RESULTS
Expression and Characterization of US3 Gene Products. The open reading frames of the HCMV US2 and US3 genes
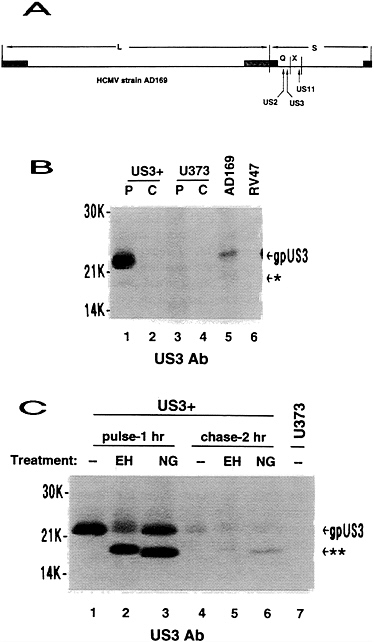
FIG. 1. Genome location and expression from HCMV US3. (A) Relative location of US2, US3, and US11 in the HCMV genome. (B) Uninfected US3+ cells or U373 control cells were metabolically radiolabeled for 2 hr (lanes P) then chased in medium containing excess unlabeled amino acids for 3 hr (lanes C). Human foreskin fibroblasts were infected with either HCMV wild-type AD169 or mutant RV47 at a multiplicity of infection of 3 and radiolabeled from 3 to 7 hr after infection. Immunoprecipitation was done with US3 antibody. (C) US3+ cells were metabolically radiolabeled in pulse-chase fashion as indicated prior to immunoprecipitation, using US3 antibody, and glycosidase treatment. For control purposes, an immunoprecipitation from pulse-labeled U373 control cells is shown. gpUS3, 22-kDa US3 glycoprotein; *, 18-kDa US3 product; **, deglycosylated US3 protein; EH, endoglycosidase H digestion; NG, N-glycosidase F digestion.
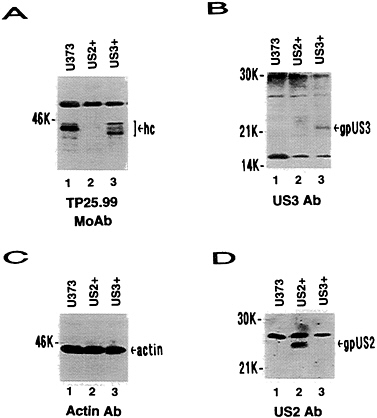
FIG. 2. Western blot analysis of US2+ and US3+ cell proteins probed with TP25.99 monoclonal antibody (A), US3 antibody (B), actin antibody (C), or US2 antibody (D). hc, Class I heavy chains; gpUS3, US3 glycoprotein; gpUS2, US2 glycoprotein.
encode proteins of 200 and 187 amino acids, respectively (22). These proteins have similar hydrophilicity profiles and contain
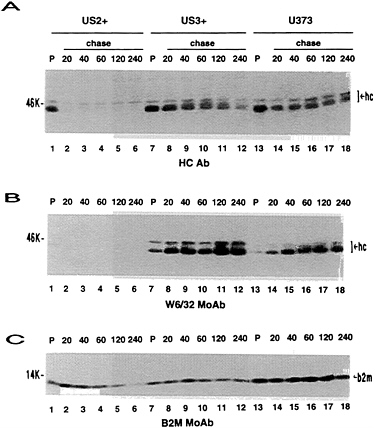
FIG. 3. Stability of MHC class I proteins. US2+ cells, US3+ cells, and U373 control cells were metabolically radiolabeled for a 20-min pulse and then chased for the indicated time (min) in medium containing excess unlabeled amino acids. Immunoprecipitations were done with anti-HC antibody (A), W6/32 monoclonal antibody (B), or BBM.1 monoclonal antibody (C). Lanes 1–12 and 13–18 are from different SDS/PAGE. hc, Class I heavy chains; b2m, β2-microglobulin.
hydrophobic regions near their N and C termini that likely correspond to a signal sequence and transmembrane domain, respectively. Furthermore, the HCMV US3 gene product is homologous to the US2-encoded protein. By Lipman-Pearson alignment, these proteins show 22% identity, or 50% homology by scoring conservative amino acid changes (data not shown). The fact that the US2 product down-regulates expression of MHC class I heavy chains suggests that US3 could also alter class I expression. The strategy employing phenotypic analysis of progressive deletion mutants of HCMV used to identify the role of US11 and US2 in class I heavy chain down-regulation would not have identified a similar phenotype caused by US3, since US3 lies between US2 and US11 (Fig. 1A) (ref. 20; T.R.J. and L.S., unpublished results). Therefore, effects of US3 were directly analyzed in stably transfected U373 cells. These cells are permissive for HCMV replication and have been used to confirm the role of the HCMV US11 and US2 genes in turnover of MHC class I heavy chains (ref. 20; T.R.J. and L.S., unpublished results).
US3 was cloned in a HCMV major immediate-early promoter-based vector so that it would be constitutively expressed in stably transfected cells. Cell lines expressing US3 were identified by Western blot analysis using US3 antisera. Numerous US3+ colonies were obtained, with no indication of detrimental effects of constitutive US3 expression. In radio-labeling-immunoprecipitation experiments, expression of US3 in transfected cells resulted in at least two polypeptides of 22 kDa and 18 kDa (Fig. 1B). These proteins comigrated with proteins detected in HCMV (wild type)-infected cells but were
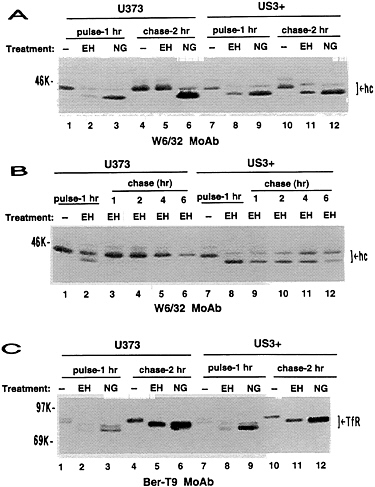
FIG. 4. Impaired maturation of MHC class I heavy chains in US3+ cells. U373 control cells or US3+ cells were metabolically radiolabeled for a 1-hr pulse and then chased for the indicated time (hr) in medium containing excess unlabeled amino acids. (A and B) Immunoprecipitation with W6/32 monoclonal antibody. (C) Immunoprecipitation with Ber-T9 monoclonal antibody. After immunoprecipitation, proteins were either untreated (−), or digested with either endoglycosidase H (EH) or N-glycosidase F (NG). hc, Class I heavy chains; TfR, transferrin receptor.
not observed in cells infected with a HCMV mutant from which US3 had been deleted. However, the relative abundance of the 22-kDa US3 protein was much greater in transfected cells than in infected cells. The reason for this difference has not been investigated but may be related to differential splicing reported in HCMV-infected cells (23). The 22-kDa US3 gene product is an N-linked glycoprotein (gpUS3), since it is sensitive to both N-glycosidase F and endoglycosidase H treatment (Fig. 1C). US3 reveals three consensus N-linked glycosylation sites, but only one is within a hydrophilic domain (residue 59) (22). In the 2-hr chase lanes, about 50% of the remaining gpUS3 remains sensitive to endoglycosidase H, indicative of a slow conversion to a form containing complex glycans. The experiments depicted in Fig. 1, as well as other independent experiments using shorter pulse times (data not shown), demonstrate that gpUS3 has a short half-life (about 1 h), similar to gpUS2 and gpUS11 (ref. 20; T.R.J. and L.S., unpublished results).
US3 and MHC Class I Heavy Chain Expression. Expression of US2- and US11-encoded proteins result in extremely low steady-state levels of MHC class I heavy chains by immunoblot analysis (ref. 20; T.R.J. and L.S., unpublished results). In Fig. 2, US3+ cells were compared with U373 control cells and US2+ cells. In contrast to US2+ cells, near wild-type steady-state levels of MHC class I heavy chains were detected in US3+ cell lines (Fig. 2A). However, the mobility of the major form of class I heavy chain was slightly increased in US3+ cells compared with U373 control cells. In pulse-chase experiments (Fig. 3), a progressive slight reduction in free heavy chains was observed in US3+and U373 control cells, coincident with increasing amounts of β2-microglobulin associated heavy chains. Consistent with previous results (T.R.J. and L.S., unpublished results), free class I heavy chains were rapidly degraded in US2+ cells and only small amounts of β2-microglobulin-associated heavy chains were detected. Unlike US2, expression of US3 has no detectable effect on the stability of MHC class I heavy chains. Furthermore, association of β2-microglobulin with class I heavy chains appeared to proceed normally in US3+ cells.
Impaired Maturation of Class I Heavy Chains. The altered mobility of class I heavy chains in US3+ cells suggested that processing of class I heavy chains was altered by gpUS3. In U373 control cells, β2-microglobulin-associated MHC class I heavy chains were partially endoglycosidase H-sensitive at the end of the 1-hr pulse but were fully endoglycosidase H-resistant after a 2-hr chase (Fig. 4A). In US3+ cells, class I heavy chains were largely endoglycosidase H-sensitive, even after a 2-hr chase. By extending the chase period, we observed that in US3+ cells there was a protracted, but steady, conversion of heavy chains to endoglycosidase H-resistant forms (Fig. 4B). After a 6-hr chase, some 50% of class I heavy chains retained endoglycosidase H sensitivity. Maturation of a control membrane glycoprotein, the transferrin receptor, is unaltered in US3+ cells (Fig. 4C). Thus, maturation of glycoproteins is not generally affected in US3+ cells; the effects of US3 are specific for MHC class I heavy chains.
Localization and State of Class I Heavy Chains. Addition of complex glycans to glycoproteins (and the attendant conversion to endoglycosidase H-resistance) occurs in the medial-and trans-Golgi (34). The slow maturation of class I heavy chains in US3+ cells suggested that ER to Golgi trafficking of class I heavy chains is impaired. Confocal immunofluorescence microscopy revealed intense perinuclear staining for class I heavy chains in US3+ cells but not in U373 control cells (Fig. 5A and C). The fluorescence pattern in US3+ cells was similar to that observed when using the dye DiOC6[3] under conditions that label the ER (33) (Fig. 5B). In a sampling of 100 cells, there was 17-fold more ER-localized fluorescence in US3+ cells, compared with U373 control cells, when probed for class I heavy chains. Thus, the predominant location of the impaired class I heavy chains is the ER in US3+ cells.
Immunofluorescence microscopy of nonpermeabilized cells indicated that substantial amounts of class I heavy chains were on the surface of both US3+ cells and U373 control cells, while cell surface heavy chains were not detected in US2+ cells (Fig. 6). Since cell surface staining appeared similar for U373 control cells and US3+ cells, and given our observation that US3+ cells contain a substantial amount of endoglycosidase

FIG. 5. Confocal immunofluorescence microscopy. (A and C) TP25.99 monoclonal antibody was used to localize MHC class I heavy chains. (B and D) ER staining with the dye DiOC6[3]. The same field is shown in both panels. (A and B) US3+ cells. (C and D) U373 cells.
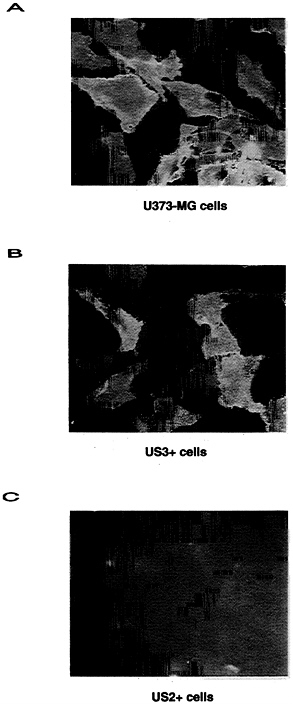
FIG. 6. Immunofluorescence of fixed, but nonpermeabilized, U373 control cells (A), US3+ cells (B), or US2+ cells (C). TP25.99 monoclonal antibody was used to determine the presence of cell surface MHC class I heavy chains.
H-sensitive heavy chains, it was of interest to know the nature of the class I heavy chains that were on the cell surface of US3+ cells. Immunoprecipitation experiments on 125I-surface-labeled cells, revealed that class I heavy chains present on the surface of both US3+ and U373 control cells were entirely endoglycosidase H-resistant and, in US3+ cells, were reduced by about 2-fold (Fig. 7). No 125I-labeled gpUS3 was detected on the cell surface. The difference in mobility of surface-labeled class I molecules observed in US3+ cells, compared with U373 control cells, is consistent with selective escape of certain class I alleles from US3-mediated retention. Preliminary results from isoelectric focusing analysis of such immunoprecipitates confirms this suggestion (data not shown).
Association of Class I Heavy Chains and gpUS3. The cumulative data imply that US3 impairs egress of class I heavy chains from the ER. A possible direct association between gpUS3 and class I heavy chains was investigated in immunoprecipitation experiments (Fig. 8). US3+ and U373 control cells were metabolically labeled and then lysed with either
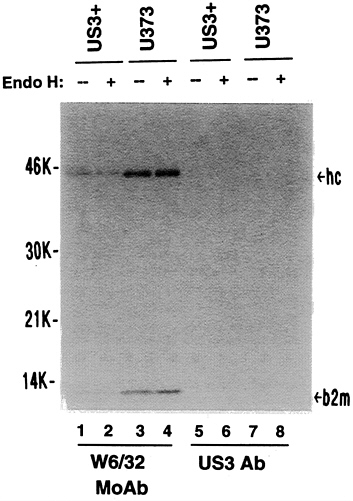
FIG. 7. Maturation state of cell surface MHC class I heavy chains. Cell surface proteins of US3+ or U373 cells were radioiodinated and immunoprecipitated with either W6/32 monoclonal antibody or US3 polyclonal antibody. Subsequently, the extracts were either untreated (−) or digested with endoglycosidase H (+). hc, Class I heavy chains; b2m, β2-microglobulin.
digitonin lysis buffer or the standard lysis buffer containing sodium deoxycholate, sodium dodecyl sulfate, and Nonidet P-40. From the digitonin extracts, class I heavy chains could be recovered for each cell line using either anti-HC antibody or W6/32 antibody but not with the US3 antibody. When using W6/32 antibody, a 22-kDa protein coprecipitated with heavy chains from US3+ cells but not from U373 control cells or from either cell line with anti-HC antibody (Fig. 8, lanes 1, 3, 7, and 9). This 22-kDa protein comigrated with authentic gpUS3 (Fig. 8, lane 12). In the presence of ionic detergents (standard lysis buffer), class I heavy chains and β2-microglobulin, but not the 22-kDa protein, were immunoprecipitated by W6/32 antibody from the US3+ cell extract (data not shown). A reimmunoprecipitation was performed to confirm the identity of the 22-kDa coprecipitating protein. First, W6/32 antibody was used to recover class I heavy chains and associated proteins from digitonin-lysed cells. Then, standard lysis buffer (containing sodium deoxycholate, sodium dodecyl sulfate, and Nonidet P-40) was added to this immunoprecipitate to dissociate heavy chains from the associated 22-kDa protein, followed by immunoprecipitation using US3 antibody (Fig. 8, lanes 4–5 and 10–11). The 22-kDa protein coprecipitating with class I molecules from US3+ cells could be recovered with US3 antibody and comigrated with authentic gpUS3. Thus, this confirms the identity of the coprecipitating protein as gpUS3. We were unable to recover gpUS3 with US3 antibody from the digitonin lysis buffer extracts of US3+ cells, although it readily coprecipitated from that extract with W6/32 antibody (Fig. 8, lanes 8 and 9) and could be recovered from the standard lysis buffer extract with US3 antibody (Fig. 8, lane 12). Therefore, it is possible that there is very little free
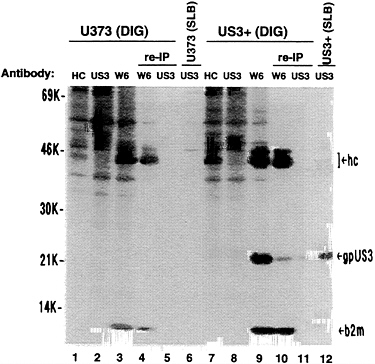
FIG. 8. Coprecipitation of MHC class I heavy chains and gpUS3. U373 cells or US3+ cells were metabolically radiolabeled for 2.5 hr and lysed with either 1% digitonin lysis (DIG) buffer or standard lysis buffer (SLB) as indicated. Proteins were immunoprecipitated with anti-HC antibody (HC) (lanes 1 and 7), US3 antibody (US3) (lanes 2, 6, 8, and 12), or W6/32 monoclonal antibody (W6) (lanes 3, 4, 9, and 10). In some cases, a reimmunoprecipitation from digitonin extracts was done (lanes 4–5 and 10–11). W6/32 monoclonal antibody was used first step, then the immunoprecipitate was exposed to standard lysis buffer (10 min at 37°C), and the released proteins were recovered from the supernatant and immunoprecipitated with US3 antibody (lanes 5 and 11). Proteins immunoprecipitated in the first step but not released to the supernatant during the exposure to standard lysis buffer were electrophoresed also (lanes 4 and 10). hc, Class I heavy chains; gpUS3, US3 glycoprotein; b2m, β2-microglobulin.
gpUS3 in US3+ cells. Its association with class I heavy chains, and possibly with other yet to be identified polypeptides, is rapid and results in immunological shielding of epitopes recognized by US3 antibody.
DISCUSSION
Herein we describe the effects of the product encoded by the HCMV US3 gene, a glycoprotein homologous to that encoded by US2, on expression of MHC class I products. By comparison of stably transfected cell lines that express either US2 or US3, we demonstrated that, in contrast to US2, expression of US3 does not cause rapid turnover of class I heavy chains. Instead, gpUS3 was found associated with β2-microglobulin-complexed class I heavy chains and inhibited their maturation and transport to the cell surface. Confocal immunofluorescence microscopy indicated that class I heavy chains accumulated predominantly in the ER in US3+ cells. Thus, HCMV encodes at least three proteins that posttranslationally down-regulate MHC class I expression and, presumably, function.
The major US3 gene product detected in US3+ cells is a glycoprotein containing endoglycosidase H-sensitive glycans and with a relatively short intracellular half-life (t1/2=1 hr), as was also observed for US2 and US11 (T.R.J. and L.S., unpublished results). The US11 glycoprotein (gpUS11) is an ER-resident protein (21). At present, the intracellular location of gpUS3 has not been determined directly. However, its association with the ER-retained class I heavy chains, and its endoglycosidase H sensitivity, imply that gpUS3 is an ER-resident protein.
HCMV gpUS3 is functionally similar to adenovirus E3–19K gene product, although there is no homology between them. The hydropathy profile of US3 is suggestive of a type I membrane protein, such as E3–19K. US3 and E3–19K are similarly-sized and contain high-mannose-type N-linked glycans. E3–19K is an early gene product that binds and retains class I heavy chains in the ER, thus preventing transport to the cell surface and resulting in decreased recognition and lysis by specific cytotoxic T cells (8, 9, 35–38). Unlike gpUS3, which has a short half-life, the E3–19K glycoprotein is stable (8, 9). This difference may be functionally relevant, adding a dimension of complexity to the dynamic equilibrium between class I molecules and the respective viral gene products. Because at early times after infection US3 acts on heavy chains simultaneous with US2 and US11, the interplay among US2, US3, and US11 is likely to be a complicated affair. In any case, significant down-regulation of class I molecules can be accomplished even in the face of the short half-life these HCMV gene products.
The efficiency of ER retention is influenced by the affinity of the viral protein for the various class I alleles. For example, E3–19K binds to some MHC class I molecules more strongly than to others (39, 40). This also seems to be the case for gpUS3. Over the course of 6 h, maturation of some heavy chains occurs resulting in almost normal cell surface expression of class I at steady state in US3+ cells (Fig. 6 and 7). Our data indicate that heavy chains from the class I alleles expressed in our US3+ cells may not be retained in the ER to the same extent. Evidence for this is 2-fold. (1) Conversion to endoglycosidase H resistance of only the lower but not the upper band of the heavy chain doublet in US3+ cells immunoprecipitated with W6/32 antibody was detected (Fig. 4B). In U373 control cells, conversion to endoglycosidase H resistance of both bands was detected. (2) Surface-labeled heavy chains in US3+ cells have reduced mobility in SDS/PAGE compared with those from U373 control cells (Fig. 7). Combined, these results are consistent with the possibility that not all class I alleles are affected equally by US3.
Multiple genes that act to down-regulate MHC class I expression or function may be common to herpesviruses, which engage in an extended relationship with their host. This is especially true for cytomegaloviruses, with their prolonged replication cycles. Murine cytomegalovirus encodes at least two proteins that affect the expression of class I molecules through posttranslational mechanism(s) (15). One gene has been localized to a 6.8-kb region of the MCMV genome and is expressed at early times after infection. This gene functions in a manner similar to US3, in that it causes class I heavy chains to be retained in the ER (15). A second MCMV gene expressed at later times after infection results in the reduction of cell surface class I molecules by an unknown mechanism (15).
Why should cytomegaloviruses require multiple MHC class I interactive proteins? In vivo, the requirement for multiple class I down-regulatory functions may be reflective of the varied cell types that are infected by HCMV. Alternatively, perhaps all three genes are expressed during in vivo infection, as they are in cultured fibroblasts. In the latter, US2 and US11 are expressed at both early and late times after infection, while US3 is expressed at immediate-early time and at reduced levels at early-late times (refs. 20, 23, and 24; T.R.J. and L.S., unpublished results). Thus, the normal biosynthetic pathway of MHC class I heavy chains is altered throughout infection by HCMV in cultured fibroblasts. Since US3 expression precedes that of US11 and US2, the former may provide an important immunoevasion function to the newly infected cell by preventing effective presentation of immediate-early epitopes and interruption by T cells of the replicative cycle at that point. It was demonstrated that expression from US3 is negatively regulated at early-late times after infection by cellular repressor proteins (26). If abundance or physical state of these
cellular repressor proteins vary among the different cell types infected by HCMV, then expression kinetics of US3 will be altered concominantly, with effects of gpUS3 on class I heavy chains not limited to the first few hours after infection.
One may speculate that class I heavy chains that are retained in the ER by gpUS3 are subsequently attacked by either gpUS11 or gpUS2. Although not formally tested, this may not be the case. gpUS2 and gpUS11 readily exert their effect on free heavy chains (T.R.J. and L.S., unpublished results), and their attack on β2-microglobulin-associated heavy chains is detected shortly after this association (21). Furthermore, gpUS11 has been shown to cause destabilization of newly synthesized heavy chains upon completion of polypeptide synthesis by dislocating them from the ER to the cytosol, probably by reverse transport through the ER translocation complex (21). gpUS2 acts in a similar fashion (E.J.H.J.W., H.L.P., and T.R.J., unpublished results). Therefore, the mechanism of action of these proteins may require class I heavy chains that remain in close association with, or in the proximity of, the translocation complex. In the presence of gpUS3, there are no apparent defects in a very early step of class I biosynthesis, namely, association with β2-microglobulin (Fig. 3). The apparent half-time for this association can vary widely, from less than 5 min to 4 hr for some heavy chain alleles (41, 42). Thus, it is unlikely that heavy chains retained by gpUS3 would still be in the proximity of the translocation complex. Further experiments are required to determine the susceptibility of gpUS3-retained class I heavy chains to gpUS2 or gpUS11 function to properly assess the role of US3 in HCMV infection.
This manuscript is dedicated to the memory of Yakov (Yasha) Gluzman, a mentor and colleague. We thank S.Ferrone for the generous gift of TP25.99 monoclonal antibody. H.L.P. and J.A.N. are supported by funding from the National Institutes of Health (Grants NIH-AI-33456 and NIH-AI-21640, respectively). E.J.H.J.W. is on leave from the National Institute of Public Health and Environmental Protection (Bilthoven, The Netherlands) and further supported by a Talent Stipendium of The Netherlands Organization for Scientific Research and a long-term fellowship from the European Molecular Biology Organization.
1. Alford, C.A. & Britt, W.J. (1990) in Virology, eds. Knipe, D.M. & Fields, B.N. (Raven, New York), 2nd Ed., pp. 1981–2010.
2. Oldstone, M.B.A. (1991) J. Virol. 65, 6381–6386.
3. Gooding, L.R. (1992) Cell 71, 5–7.
4. Maudsley, D.J. & Pound, J.D. (1991) Immunol. Today 12, 429–431.
5. McFadden, G. & Kane, K. (1994) Adv. Cancer Res. 63, 117–209.
6. Townsend, A. & Bodmer, H. (1989) Annu. Rev. Immunol. 7, 601–624.
7. Schrier, P.I., Bernards, R., Vaessen, R.T.M.J., Houweling, A. & Van der Eb, A.J. (1983) Nature (London) 305, 771–775.
8. Andersson, M., Paabo, S., Nilsson, T. & Peterson, P.A. (1985) Cell 43, 215–222.
9. Burgert, H.-G. & Kvist, S. (1985) Cell 41, 987–997.
10. York, I.A., Roop, C., Andrews, D.W., Riddell, S.R., Graham, F.L. & Johnson, D.C. (1994) Cell 77, 525–535.
11. Hill, A., Jugovic, P., York, I., Russ, G., Bennink, J., Yewdell, J., Ploegh, H. & Johnson, D.C. (1995) Nature (London) 375, 411–415.
12. Fruh, K., Ahn, K., Djaballah, H., Sempe, P., van Endert, P.M., Tampe, R., Peterson, P.A. & Yang, Y. (1995) Nature (London) 375, 415–418.
13. Del Val, M., Hengel, H., Hacker, H., Hartlaub, U., Ruppert, T., Lucin, P. & Koszinowski, U.H. (1992) J. Exp. Med. 176, 729–738.
14. Campbell, A.E. & Slater, J.S. (1994) J. Virol. 68, 1805–1811.
15. Thäle, R., Szepan, U., Hengel, H., Geginat, G., Lucin, P. & Koszinowski, U.H. (1995) J. Virol. 69, 6098–6105.
16. Levitskaya, J., Coram, M., Levitsky, V., Imreh, S., Steigerwald-Mullen, P.M., Klein, G., Kurilla, M.G. & Masucci, M.G. (1995) Nature (London) 375, 685–688.
17. Beersma, M.F.C., Bijlmakers, M.J.E. & Ploegh, H.L. (1993) J. Immunol. 151, 4455–4464.
18. Warren, A.P., Ducroq, D.H., Lehner, P.J. & Borysiewicz, L.K. (1994) J. Virol. 68, 2822–2829.
19. Yamashita, Y., Shimokata, K., Saga, S., Mizuno, S., Tsurumi, T. & Nishiyama, Y. (1994) J. Virol. 68, 7933–7943.
20. Jones, T.R., Hanson, L.K., Sun, L., Slater, J.S., Stenberg, R.M. & Campbell, A.E. (1995) J. Virol. 69, 4830–4841.
21. Wiertz, E.J.H.J., Jones, T.R., Sun, L., Bogyo, M., Geuze, H.J. & Ploegh, H.L. (1996) Cell 84, 769–779.
22. Chee, M.S., Bankier, A.T., Beck, S., Bohni, R., Brown, C.M., Cerny, R., Horsnell, T., Hutchinson, C.A., Kouzarides, T., Martignetti, J.A., Preddie, E., Satchwell, S.C., Tomlinson, P., Weston, K. & Barrell, B.G. (1990) Curr. Top. Microbiol. Immunol. 154, 125–169.
23. Weston, K. (1988) Virology 162, 406–416.
24. Tenney, D.J. & Colberg-Poley, A.M. (1991) J. Virol. 65, 6724– 6734.
25. Biegalke, B.J. (1995) J. Virol. 69, 5362–5367.
26. Thrower, A.R., Bullock, G.C., Bissell, J.C. & Stinski, M.F. (1996) J. Virol. 69, 91–100.
27. Jones, T.R. & Muzithras, V.P. (1992) J. Virol. 66, 2541–2546.
28. Barnstable, C.J., Bodmer, W.F., Brown, G., Galfre, G., Milstein, C., Williams, A.F. & Ziegler, A. (1978) Cell 14, 9–20.
29. D’Urso, C.M., Wang, Z., Cao, Y., Tatake, R., Zeff, R.A. & Ferrone, S. (1991) J. Clin. Invest. 87, 284–292.
30. Brodsky, F.M. & Parham, P. (1982) J. Immunol. 128, 129–135.
31. Harlow, E. & Lane, D., eds. (1988) in Antibodies: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
32. Fish, K.N., Britt, W. & Nelson, J.A. (1996) J. Virol. 70, 1855– 1862.
33. Terasaki, M. (1994) in Cell Biology: A Laboratory Handbook, ed. Celis, J.E. (Academic, Orlando, FL), pp. 381–386.
34. Kornfield, R. & Kornfield, S. (1985) Annu. Rev. Biochem. 54, 631–664.
35. Burgert, H.-G., Maryanski, J.L. & Kvist, S. (1987) Proc. Natl. Acad. Sci. USA 84, 1356–1360.
36. Andersson, M., McMichael, A. & Peterson, P.A. (1987) J. Immunol. 138, 3960–3966.
37. Tanaka, Y. & Tevethia, S.S. (1988) Virology 165, 357–366.
38. Hermiston, T.W., Tripp, R.A., Sparer, T., Gooding, L.R. & Wold, W.S.M. (1983) J. Virol. 67, 5289–5298.
39. Severinsson, L., Martens, I. & Peterson, P.A. (1986) J. Immunol. 137, 1003–1009.
40. Beier, D.C., Cox, J.H., Vining, D.R., Cresswell, P. & Engelhard, V.H. (1994) J. Immunol. 152, 3862–3872.
41. Krangel, M.S., Orr, H.T. & Strominger, J.L. (1979) Cell 18, 979–991.
42. Hill, A., Takiguchi, M. & McMichael, A. (1993) Immunogenetics 37, 95–101.







