
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Use of virion DNA as a cloning vector for the construction of mutant and recombinant herpesviruses
(γ-2 herpesvirus/gene transfer/viral oncogenesis/T lymphocyte)
S. MONROE DUBOISE, JIE GUO, RONALD C. DESROSIERS, AND JAE U. JUNG *
Department of Microbiology and Molecular Genetics, New England Regional Primate Research Center, Harvard Medical School, Southborough, MA 01772–9102
ABSTRACT We have developed improved procedures for the isolation of deletion mutant, point mutant, and recombinant herpesvirus saimiri. These procedures take advantage of the absence of NotI and AscI restriction enzyme sites within the viral genome and use reporter genes for the identification of recombinant viruses. Genes for secreted engineered alkaline phosphatase and green fluorescent protein were placed under simian virus 40 early promoter control and flanked by NotI and AscI restriction sites. When permissive cells were cotransfected with herpesvirus saimiri virion DNA and one of the engineered reporter genes cloned within herpesvirus saimiri sequences, recombinant viruses were readily identified and purified on the basis of expression of the reporter gene. Digestion of recombinant virion DNA with NotI or AscI was used to delete the reporter gene from the recombinant herpesvirus saimiri. Replacement of the reporter gene can be achieved by NotI or AscI digestion of virion DNA and ligation with a terminally matched fragment or, alternatively, by homologous recombination in cotransfected cells. Any gene can, in theory, be cloned directly into the virion DNA when flanked by the appropriate NotI or AscI sites. These procedures should be widely applicable in their general form to most or all herpesviruses that replicate permissively in cultured cells.
Large DNA viruses, such as the orthopoxviruses and herpesviruses, are useful as gene transfer vectors, because they can easily accommodate substantial amounts of additional DNA in their genomes (1–3). Accessory genes unnecessary for replication of these viruses can be eliminated to modulate the virus’ properties and to help ensure their safety. In the case of herpesviruses, persistence of the viral genetic information may be useful for some applications where continued foreign gene expression is desirable. We have been manipulating the genetic information of herpesvirus saimiri (HVS) to obtain a better understanding of its natural life cycle and to use it as a gene transfer vector for experimental vaccine and therapeutic strategies.
HVS infection is endemic and apparently apathogenic in squirrel monkeys (Saimiri sciureus; refs. 4–6). The virus is extremely oncogenic, however, in other nonhuman primates, producing fulminant T-cell lymphoproliferative diseases (7, 8). HVS is the prototypic and best characterized γ-2 herpesvirus (Rhadinovirus; refs. 9 and 10). The only known human γ-2 herpesvirus, the recently discovered human herpesvirus 8, or Kaposi sarcoma associated herpesvirus, shows greatest homology with HVS and shares a similar genomic organization (11, 12). Studies on the mechanisms of HVS oncogenicity are expected to contribute to the understanding of Kaposi sarcoma and possibly other disorders believed to be associated with human herpesvirus 8 infection (13–15).
Previous characterization of HVS oncogenesis has demonstrated that sequences near the left end of the viral genome are unnecessary for viral replication but essential for oncogenesis and in vitro transformation of common marmoset lymphocytes (16–19). This region contains an oncogene designated as saimiri transforming protein (STP; refs. 20 and 21). The complete nucleotide sequence of HVS subgroup A strain 11 has revealed other open reading frames that possibly contribute to the transforming capacity of the virus (22). Among these are genes encoding a viral cyclin related to cyclin D1 (23, 24), a homolog of the superantigen of mouse mammary tumor virus (25), a CD-59 homolog (26), a G protein-coupled receptor homologous to the cellular interleukin 8 receptor (27), a Bcl-2 homolog (28), and an interleukin 17 homolog (29, 30). Striking sequence differences have been noted between viruses in subgroup A and the more oncogenic subgroup C in the region of the genome containing the STP oncogene (20, 21). Highly oncogenic subgroup C isolates of HVS immortalize human and rhesus monkey T lymphocytes in vitro (31, 32), and they contain a divergent STP gene (20, 21) and a unique gene encoding tyrosine kinase interacting protein (Tip), which interacts with the major T-cell tyrosine kinase Lck (33–35). Construction of replication-competent deletion mutant viruses allows in vitro and in vivo assessments of the contribution of these genes to viral induced transformation. Nononcogenic deletion mutants of HVS may also provide a basis for development of γ-2 herpesvirus gene transfer vectors capable of persistently infecting lymphoid cells.
In contrast to other γ herpesviruses such as Epstein-Barr virus that fail to grow permissively in cell culture systems, HVS is capable of productive lytic infection of cultured New World primate monolayer cells (36). This greatly facilitates the generation of mutant and recombinant viruses. Progress has recently been reported in cell culture production of human herpesvirus 8 (37), but procedures for serial propagation, production in large quantities, and for genetic manipulation of human herpesvirus 8 have not been defined. In the work reported here, we describe methods that have significantly facilitated selection of mutant and recombinant HVS strains with the potential for use as nononcogenic HVS gene transfer vectors. We also report direct cloning of a replaceable foreign gene expression cassette into HVS. The methods described
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HVS, herpesvirus saimiri; STP, saimiri transforming protein; Tip, tyrosine kinase interacting protein; SEAP, secreted engineered alkaline phosphatase; GFP, green fluorescent protein; SV40, simian virus 40.
|
* |
To whom reprint requests should be addressed at: New England Regional Primate Research Center, Harvard Medical School, P.O. Box 9102, Southborough, MA 01772–9102. e-mail: jjung@warren.med.harvard.edu. |
should have general utility for genetic manipulation of other herpesviruses.
MATERIALS AND METHODS
Cell Culture, Virus, and Virion DNA. Herpesvirus saimiri strain C-488 was propagated in low passage (<30 passages) owl monkey kidney cells (OMK 637) grown in minimal essential medium (MEM) supplemented with penicillin, streptomycin, L-glutamine, and 10% (vol/vol) heat-inactivated fetal bovine serum (GIBCO/BRL). Tests of reporter plasmid expression were conducted in COS-1 cells cultivated in Dulbecco’s modified Eagle’s medium with high glucose supplemented with 10% fetal bovine serum, penicillin, streptomycin, and L-glutamine.
Virion DNA was prepared by removing cell debris from supernatants of infected OMK cells by low-speed centrifugation, pelleting the virus by centrifugation at 40,000×g for 2 hr in an SS-34 rotor, disrupting the virus at 60°C for 2 hr in lysis buffer (10 mM Tris, pH 8.5/1 mM EDTA/1% sarkosyl/0.1 mg of proteinase K per ml), and then extracting the aqueous solution first with an equal volume of phenol and then twice with chloroform. All pipetting was done with sterile tips that were cut to facilitate manipulation of intact viral genomes without significant shearing. Virion DNA prepared in this manner was sufficiently pure and intact for use in transfection of OMK cells for selection of desired recombinant viruses.
Reporter Plasmid Construction. Reporter gene expression cassettes contained either secreted engineered alkaline phosphatase (SEAP) or green fluorescent protein (GFP) from Aequorea victoria under the control of the simian virus 40 (SV40) early promoter and enhancer. Polymerase chain reaction (PCR) using Vent polymerase (New England Biolabs) was employed to amplify components of the expression cassettes from commercially available plasmids and to add selected restriction enzyme sites. SV40 early promoter, enhancer, and polyadenylylation elements with a NotI site available for insertion of a reporter gene were amplified from a modified pSVβ (CLONTECH) from which the LacZ reporter had been eliminated by NotI digestion and ligation to produce a reporterless vector. The 5′ primer, CGCGGTACCGATATCGCCGGCGCGCCGGTACAGCTTGTGGAATGTGTGTCA, added KpnI, EcoRV, SgrAI, and AscI restriction sites while the 3′ primer, CGCTCTAGAGCTCACGTGGCGCGCCGGCGGATAAAAACCTCCCACACCT, added XbaI, SacI, PmlI, AscI, and SgrAI sites. Digestion of the PCR product with KpnI and XbaI allowed cloning of the control elements into corresponding sites in pNEB193 (New England Biolabs).
The SEAP reporter was amplified from pBC12/PL/SEAP (Tropix, Bedford, MA) using primers that added flanking NotI sites. Primers used were AGAGAATTCGCGGCCGCATATCGTCGACAAGCTTCTGC and CAGTCTAGAGCGGCCGCGGGTTAACCCGGGTGCGCGGCG. NotI-digested PCR product was cloned into the NotI site of the vector containing SV40 control elements to produce the plasmid pSV40/SEAP. The similar GFP expression cassette was constructed by deleting the SEAP gene from the expression cassette with SalI and SmaI and inserting GFP from pGFP (CLONTECH) digested with SalI and StuI.
Reporter function was tested by DEAE-dextran-mediated transfection into COS-1 cells (38), followed by assay of expression at 48–72 hr posttransfection. SEAP production and secretion was detected by liquid scintillation counter measurement of chemiluminescence produced in assays of cell culture medium using Phospha-Light reagents (Tropix, Bedford, MA) according to the manufacturer’s recommendations. GFP production was detected visually by observation of bright green fluorescence produced at an excitation wavelength of 495 nm using an Olympus IMT-2 fluorescence microscope.
Gene Deletion Plasmid Construction. A 3.6-kb plasmid clone (p488PX) of a PstI/XbaI fragment derived from the left end of the herpesvirus saimiri group C strain 488 genome and containing the STP-C488 oncogene has been described (21). This PstI/XbaI fragment was transferred into pNEB193. A 508-bp deletion, including 273 bp of STP-C488, was introduced by replacement of a SpeI/EcoRV fragment (nucleotides 1318– 1825 of HVS-C488) with an XbaI/EcoRV fragment from pSV40/SEAP containing the SEAP expression cassette. Similarly, a deletion in the Tip gene (nucleotides 1226–454 of HVS-C488) was made by replacement of a StuI/HpaI fragment (nucleotides 879–438) with the SV40/SEAP cassette in a PmlI/EcoRV fragment.
Transfections and Isolation of Recombinant HVS. Production of recombinant HVS by mixed transfection of infectious virion DNA and linearized plasmid containing specific mutations has been described (18, 19). In this study, mutant virus containing a deletion in STP-C488 was produced by homologous recombination in subconfluent monolayers of OMK cells cotransfected by Ca2+ coprecipitation of virion DNA and deletion plasmid at a molar ratio of ≈1:200. Plasmid DNA for transfection was linearized with FspI, which was then heat-inactivated at 65°C for 20 min. Transfected cells were incubated in MEM with 10% fetal bovine serum at 37°C until the cell monolayer was completely destroyed by the cytopathic effect of virus replication at 10–12 days after transfection.
Serial 10-fold dilutions in MEM of virus produced from transfections were added to individual wells (0.3 ml per well) of 48-well tissue culture plates (Corning). At 10–12 days after infection, wells showing cytopathic effect were identified microscopically to be tested further for evidence of reporter gene expression from recombinant virus. Individual wells containing virus were assayed for SEAP production using the Phospha-Light chemiluminescent assay (Tropix) performed in opaque 96-well microtiter plates read in a MicroBeta scintillation counter (Wallac, Gaithersburg, MD). SEAP expressing HVS recombinant virus was purified by selection of SEAP-positive wells at high dilution during repeated limiting dilution passages of the virus. To produce virus containing the STP-C488 deletion, but with the SEAP reporter removed, recombinant virion DNA was digested with either NotI or AscI. After restriction enzymes were heat-inactivated at 65°C for 30 min, virion DNA was ligated overnight with T4 ligase (Takara Shuzo, Kyoto) and then transfected into OMK cells. The resultant SEAP negative virus was purified by limiting dilution as described above.
To demonstrate rescue of wild-type phenotype, DNA from virions of the STP-C488 deletion virus was used for cotransfection with linearized wild-type p488PX plasmid, followed by limiting dilution purification of SEAP negative virus with STP-C488 restored. By OMK cell cotransfection of virion DNA from SV40/SEAP expressing Tip deletion virus along with linearized pSV40/GFP, replacement of the SEAP expression cassette with the GFP cassette via homologous recombination was performed. GFP expressing Tip deletion virus was isolated by limiting dilution and microscopic screening for GFP fluorescence.
Direct Cloning into STP-Deleted Herpesvirus Saimiri. To demonstrate use of virion DNA directly as a cloning vector, virion DNA prepared from recombinant HVSΔSTP/SV40-SEAP was digested with AscI to remove the SEAP expression cassette. The plasmid pSV40/GFP was similarly digested with AscI. AscI was subsequently heat-inactivated at 65°C for 30 min. The digested viral DNA was further treated with heat-killed phosphatase (Epicentre Technologies, Madison, WI) to dephosphorylate the viral vector DNA ends. Digested and dephosphorylated viral DNA was incubated overnight with ≈100-fold molar excess of SV40/GFP insert DNA and T4 DNA ligase. As a control, an identical mixture except without ligase added was incubated in parallel. The ligation mixture
and the control mixture were then transfected into subconfluent OMK cells, which were observed beginning at 6 days by fluorescence microscopy for the appearance of cells producing GFP. Virus from the transfections was serially diluted in MEM and placed in individual wells of 48-well plates (Corning) for isolation of the GFP-expressing virus. Individual wells containing virus from each transfection were assessed microscopically for GFP expression and by chemiluminescent assay for SEAP expression.
RESULTS
Isolation of a HVS Recombinant Using a SEAP Reporter as a Selection Marker. An SV40 early promoter-driven SEAP expression cassette was designed for insertion into cloned HVS genes considered likely to be nonessential for virus replication. One such gene, the STP-C488 oncogene, was targeted for deletion by inserting the SV40/SEAP reporter in place of a 508-bp SpeI/EcoRV fragment (including most of the STP gene) in a 3.6-kb plasmid clone. The selection method for the recombinant virus is illustrated in Fig. 1. As indicated, cotransfection of OMK cells with infectious virion DNA together with linearized plasmid containing the SV40/SEAP expression cassette in the STP-C488-deleted region resulted in homologous recombination producing STP-C488 deleted virus expressing the reporter gene. To assure purification of recombinant virus, limiting dilution plating onto OMK monolayers was serially repeated 7 times. In the first passage at a dilution of 10−7, ≈10% of wells contained SEAP-positive virus. Enrichment for SEAP positive recombinant virus at each step resulted in only SEAP-positive virus being detected by passage 3.
Removal of the Exogenous Expression Cassette from STP-C488-Deleted HVS. The SV40/SEAP expression cassette was designed to contain restriction enzyme recognition sites not present in the HVS genome. AscI sites flanking the complete expression cassette and NotI sites flanking the SEAP gene were included to allow removal of either the entire expression cassette or the reporter gene only. The ability to remove foreign control elements and genes conveniently is useful for a variety of applications. Ectopic expression of a reporter gene or presence of a foreign promoter could inadvertently result in an altered phenotype. Overnight digestion of virion DNA containing SV40/SEAP at the STP-C488 locus with either AscI or NotI followed by heat inactivation of the enzyme and overnight ligation of the viral DNA resulted in production of
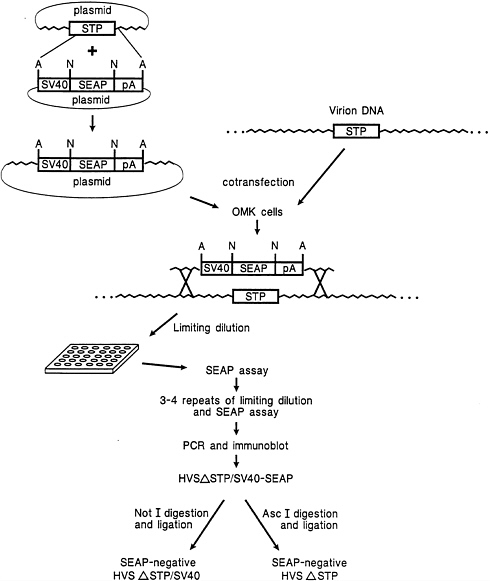
FIG. 1. Method for STP-C488 deletion mutant isolation by homologous recombination. A and N indicate AscI and NotI recognition sites, respectively.
uniformly SEAP-negative virus (see Fig. 1 for schematic). The success of this manipulation suggested that the AscI and NotI restriction sites may also be useful for direct cloning approaches for construction of HVS gene transfer vectors.
Replacement of the SV40/SEAP Expression Cassette in Recombinant HVS Using Homologous Recombination or Direct Cloning Strategies. Fig. 2 illustrates alternative strategies for the use of nononcogenic HVS deletion mutant viruses containing SV40/SEAP for construction of gene transfer vectors. Homologous recombination can clearly be used to replace the SEAP expression cassette with another expression cassette essentially by the same procedure used for selection of the STP-C488-deleted recombinant virus (Fig. 1). In this case, however, loss of SEAP expression serves as the selection marker. To demonstrate the general applicability of this procedure for gene replacement, we employed an additional deletion mutant virus, HVSΔTip/SV40-SEAP.
Replacement of the SV40/SEAP reporter in the HVSΔTip/ SV40-SEAP virus with the comparable SV40/GFP cassette by homologous recombination was performed as outlined in Fig. 2A. Approximately 2% of the wells plated in the first limiting dilution passage after the mixed transfection showed strong green fluorescence. Also, this homologous recombination method was similarly used to restore the wild-type STP-C488 gene to the STP-deleted recombinant virus (marker rescue).
Insertion of a foreign gene (GFP) into the HVSΔSTP/ SV40-SEAP virus was achieved by direct cloning into the AscI sites of virion DNA as described in Materials and Methods and illustrated in Fig. 2B. An SV40/GFP expression cassette was directly inserted as an AscI fragment into virion DNA and transfected into OMK cells. Virus produced by transfection of the ligation mixture was diluted and plated in multiwell plates such that there was less than one infectious particle per well. SEAP and GFP expression was assessed for each infected well (Table 1). SEAP activity was completely eliminated by AscI digestion. From a total of 40 virus infected wells that we observed, cells from 36 wells fluoresced brightly green (Fig. 3 and Table 1). Ligase activity endogenous in the OMK cells was also apparently capable of mediating insertion of SV40/GFP at reduced efficiency under the conditions of high molar excess
Table 1. Reporter assays at passage 1 of transfectants following restriction enzyme digestion and ligation manipulations of recombinant HVS/ΔSTP-C488/SV40/SEAP virion DNA
of insert to virion DNA that was provided; in the assessment of the control (ligase omitted) transfection, GFP-producing virus was detected in 7 (21%) of the 33 wells assessed, while the remaining 26 wells (79%) were SEAP-negative and GFP-negative, indicating that the virus in these wells was derived from self-ligation of the AscI-digested virus (Table 1). The results clearly indicate that the SEAP reporter gene alone or the entire SV40/SEAP expression cassette can be removed directly from virion DNA and that it is possible to directly clone expression cassettes into the AscI sites of the recombinant virus.
DISCUSSION
Identification and isolation of recombinant herpesviruses can be greatly facilitated by use of an appropriate selection method. Advantageous properties of SEAP and GFP reporters
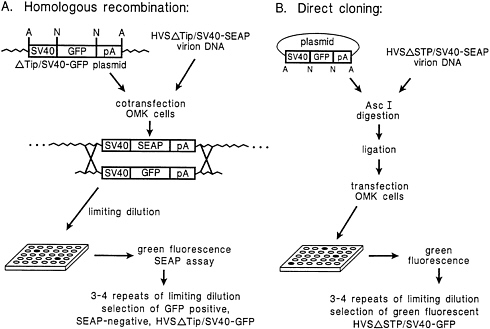
FIG. 2. Two methods for introduction of foreign genes into HVS deletion mutants: (A) homologous recombination and (B) direct cloning. A and N indicate AscI and NotI restriction sites, respectively.

FIG. 3. Expression of GFP cloned directly into STP-C488-deleted HVS digested with AscI. OMK cells infected with HVSΔSTP/SV40-GFP are shown illuminated with (A) visible light, (B) visible and 495-nm light, and (C) 495-nm light only.
have been previously reported (39–41). Application of each of these selection markers to the process of identifying recombinant HVS is a significant advance compared with methods previously used to isolate deletion mutants of HVS or other herpesviruses (1, 18, 19). Assay for SEAP requires only a minute sample of culture medium for a rapid and sensitive test. Assay of cell-free supernatant for SEAP may also be useful for detecting recombinant viruses with minimal lytic activity. Use of GFP requires no sampling and no additional reagents. The level of GFP expression was sufficient in the HVS recombinants to produce a fluorescent signal that was readily detected microscopically. While the wild-type Aequorea GFP used was more than adequate in intensity to detect fluorescence, variants are now available that are brighter, have different excitation and/or emission spectra, and are optimized in codon usage for maximum expression in mammalian cells (42–45). Both SEAP and GFP provide the opportunity for reduction of the time required for recombinant virus identification and isolation. Early detection of recombinants is possible before extensive cytopathic effect is observed and does not disturb the continued growth of the virus to high titers.
Positive selection of SEAP or GFP aids in isolation of initial recombinant HVS and then provides in the purified recombinant the equally useful negative selection tool, loss of the reporter, for isolation of marker-rescued virus or other recombinants as desired. We are currently using these procedures to isolate HVS strains with point mutations in STP and Tip and to isolate HVS recombinants capable of expressing antigens of other organisms for the purpose of vaccination.
Inclusion of unique restriction sites flanking the reporter gene in recombinant HVS greatly facilitates subsequent genetic manipulation and allows use of virion DNA directly as a cloning vector. Similar direct cloning into a large DNA virus, a baculovirus, has been reported (46). Simple removal of the reporter gene may be desirable for some in vivo applications. The NotI and AscI sites were intentionally positioned to allow removal of the reporter gene open reading frame alone, or in combination with the promoter and poly(A) elements. Any gene with or without a promoter can in theory be cloned directly into the virion DNA when flanked by the appropriate NotI or AscI sites. To our knowledge, this is the first report of direct cloning into a herpesvirus. The procedures outlined here are expected to be particularly flexible for a variety of applications.
We thank J.Newton, T.Connors, and A.Hampson for manuscript preparation. This work was supported by Public Health Service Grants CA31363 and AI38131 and Grant RR00168 from the Division of Research Resources.
1. Glorioso, J.C., DeLuca, N.A. & Fink, D.J. (1995) Annu. Rev. Microbiol. 49, 675–710.
2. Ward, P.L. & Roizman, B. (1994) Trends Genet. 10, 267–274.
3. Smith, G.L. & Moss, B. (1983) Gene 25, 21–28.
4. Melendez, L.V., Daniel, M.D., Hunt, R.D. & Garcia, F.G. (1968) Lab. Anim. Care 18, 374–381.
5. Falk, L.A., Wolfe, L.G. & Deinhardt, F. (1972) J. Natl. Cancer Inst. 48, 1499–1505.
6. Desrosiers, R.C. & Falk, L.A. (1982) J. Virol. 43, 352- 356.
7. Melendez, L.V., Hunt, R.D., Daniel, M.D., Fraser, C.E.O., Barahona, H.H., Garcia, F.G. & King, N.W. (1972) in Oncogenesis and Herpesviruses, eds. Biggs, P.M., de The, G. & Payne, L.N. (IARC, Lyons, France), pp. 451–461.
8. Fleckenstein, B. (1982) Biochim. Biophys. Acta 560, 301–342.
9. Fleckenstein, B. & Desrosiers, R.C. (1982) in The Herpesviruses, ed. Roizman, B. (Plenum, New York), pp. 253–332.
10. Jung, J.U. & Desrosiers, R.C. (1994) in Encyclopedia of Virology, eds. Webster, R. & Granoff, A. (Saunders, Philadelphia), pp. 614–622.
11. Chang, Y., Cesarman, E., Pessin, M.S., Lee, F., Culpepper, J., Knowles, D.M. & Moore, P.S. (1994) Science 266, 1865–1869.
12. Moore, P.S., Gao, S.J., Dominguez, G., Cesarman, E., Lungu, O., Knowles, D.M., Garber, R., Pellett, P.E., Mcgeoch, D.J. & Chang, Y. (1996) J. Virol. 70, 549–558.
13. Cesarman, E., Chang, Y., Moore, P.S., Said, J.W. & Knowles, D.M. (1995) N. Engl. J. Med. 332, 1186–1191.
14. Soulier, J., Grollet, L., Oksenhendler, E., Cacoub, P., Cazals-Hatem, D., Babinet, P., d’Agay, M.-F., Clauvel, J.-P., Raphael, M., Degos, L. & Sigaux, F. (1995) Blood 86, 1275–1280.
15. Said, J.W., Chien, K., Takeuchi, S., Tasaka, T., Asou, H., Cho, S.K., de Vos, S., Cesarman, E., Knowles, D.M. & Koeffler, H.P. (1996) Blood 87, 4937–4943.
16. Desrosiers, R.C., Silva, D.P., Waldron, L.M. & Letvin, N.L. (1986) J. Virol. 57, 701–705.
17. Desrosiers, R.C., Bakker, A., Kamine, J., Falk, L.A., Hunt, R.D. & King, N.W. (1985) Science 228, 184–187.
18. Desrosiers, R.C., Burghoff, R.L., Bakker, A. & Kamine, J. (1984) J. Virol. 49, 343–348.
19. Murthy, S.C.S., Trimble, J.J. & Desrosiers, R.C. (1989) J. Virol. 63, 3307–3314.
20. Biesinger, B., Trimble, J.J., Desrosiers, R.C. & Fleckenstein, B. (1990) Virology 176, 505–514.
21. Jung, J.U., Trimble, J.J., King, N.W., Biesinger, B., Fleckenstein, B.W. & Desrosiers, R.C. (1991) Proc. Natl. Acad. Sci. USA 88, 7051–7055.
22. Albrecht, J.-C., Nicholas, J., Biller, D., Cameron, K.R., Biesinger, B., Newman, C., Wittmann, S., Craxton, M.A., Coleman, H., Fleckenstein, B. & Honess, R.W. (1992) J. Virol. 66, 5047–5058.
23. Nicholas, J., Cameron, K.R. & Honess, R.W. (1992) Nature (London) 355, 362–365.
24. Jung, J.U., Stäger, M. & Desrosiers, R.C. (1994) Mol. Cell. Biol. 14, 7235–7244.
25. Yao, Z., Maraskovsky, E., Spriggs, M.K., Cohen, J.I., Armitage, R.J. & Alderson, M.R. (1996) J. Immunol. 156, 3260–3266.
26. Rother, R.P., Rollins, S.A., Fodor, W.L., Albrecht, J.-C., Setter, E., Fleckenstein, B. & Squinto, S.P. (1994) J. Virol. 68, 730–737.
27. Ahuja, S.K. & Murphy, P.M. (1993) J. Biol. Chem. 268, 20691– 29694.
28. Smith, C.A. (1995) Trends Cell Biol. 5, 344.
29. Yao, Z., Painter, S.L., Fanslow, W.C., Ulrich, D., Macduff, B.M., Spriggs, M.K. & Armitage, R.J. (1995) J. Immunol. 155, 5483–5486.
30. Yao, Z., Fanslow, W.C., Seldin, M.F., Rousseau, A.-M., Painter, S.L., Comeau, M.R., Cohen, J.I. & Spriggs, M.K. (1995) Immunity 3, 811–821.
31. Biesinger, B., Müller-Fleckenstein, I., Simmer, B., Lang, G., Wittman, S., Platzer, E., Desrosiers, R.C. & Fleckenstein, B. (1992) Proc. Natl. Acad. Sci. USA 89, 3116–3119.
32. Mittrucker, H.-W., Müller-Fleckenstein, I., Fleckenstein, B. & Fleischer, B. (1992) J. Exp. Med. 176, 909–913.
33. Biesinger, B., Tsygankov, A.Y., Fickenscher, H., Emmrich, F., Fleckenstein, B., Bolen, J.B. & Broker, B.M. (1995) J. Biol. Chem. 270, 4729–4734.
34. Jung, J.U., Lang, S.M., Friedrich, U., Jun, T., Roberts, T.M., Desrosiers, R.C. & Biesinger, B. (1995) J. Biol. Chem. 270, 20660–20667.
35. Jung, J.U., Lang, S.M., Jun, T., Roberts, T.M., Veillette, A. & Desrosiers, R.C. (1995) J. Virol. 69, 7814–7822.
36. Daniel, M.D., Silva, D. & Ma, N. (1976) In Vitro 12, 290–294.
37. Renne, R., Zhong, W.D., Herndier, B., Mcgrath, M., Abbey, N., Kedes, D. & Ganem, D. (1996) Nat. Med. 2, 342–346.
38. Cullen, B.R. (1987) Methods Enzymol. 152, 684–704.
39. Berger, J., Hauber, J., Hauber, R., Geiger, R. & Cullen, B.R. (1988) Gene 66, 1–10.
40. Cullen, B.R. & Malim, M.H. (1992) Methods Enzymol. 216, 362–368.
41. Chalfie, M., Tu, Y., Euskirchen, G., Ward, W.W. & Prasher, D.C. (1994) Science 263, 802–805.
42. Cubitt, A.B., Heim, R., Adams, S.R., Boyd, A.E., Gross, L.A. & Tsien, R.Y. (1995) Trends Biochem. Sci. 20, 448–455.
43. Heim, R. & Tsien, R.Y. (1996) Curr. Biol. 6, 178–182.
44. Rizzuto, R., Brini, M., De Giorgi, F., Rossi, R., Heim, R., Tsien, R.Y. & Pozzan, T. (1996) Curr. Biol. 6, 183–188.
45. Crameri, A., Whitehorn, E.A., Tate, E. & Stemmer, W.P.C. (1996) Nat. Biotechnol. 14, 315–319.
46. Ernst, W.J., Grabherr, R.M. & Katinger, H.W.D. (1994) Nucleic Acids Res. 22, 2855–2866.






