
This paper was presented at a colloquium, entitled “Genetics and the Origin of Species,” organized by Francisco J.Ayala (Co-chair) and Walter M.Fitch (Co-chair), held January 30–February 1, 1997, at the National Academy of Sciences Beckman Center in Irvine, CA.
DNA variation at the Sod locus of Drosophila melanogaster: An unfolding story of natural selection
RICHARD R.HUDSON, ALBERTO G.SÁEZ, AND FRANCISCO J.AYALA
Department of Ecology and Evolutionary Biology, University of California, Irvine, CA 92697
ABSTRACT Patterns of variation at the Sod locus of Drosophila melanogaster suggest that the protein polymorphism at this locus has very recently arisen. In addition, it appears that a previously rare DNA variant has been recently and rapidly driven to intermediate frequency. From the size of the region (>20 kb) that has been swept along with this rare variant, and patterns of linkage disequilibrium in the region, it is inferred that strength of selection was large (s > 0.01) and that the sweep occurred more than 25,000 generations ago. In addition, there are striking similarities to patterns of variation observed at the Est6 and Est-P loci, which are located approximately 1,000 kb from Sod.
In the late 1960s, protein electrophoresis of soluble enzymes emerged as an important tool of population geneticists. It promised to elucidate the amount and the nature of genetic variation within species and, hence, clarify important aspects of evolution. Electrophoretic surveys of hundreds of populations were carried out. Large amounts of variation were documented. Such studies in some cases were very informative about geographic structure and in some cases revealed unsuspected species or subspecies. But the evolutionary nature of the variation was unclear from the start and remains so. John Gillespie (1) expresses this viewpoint well: “Naturally occurring variation is an enigma…despite the ease of measurement, we remain essentially ignorant of the forces that maintain variation.” The debate has continued for 30 years about whether most protein polymorphisms are the result of mutation and genetic drift of selectively equivalent alleles (the neutralist position) or whether most protein polymorphisms are maintained by some form of balancing selection (the selectionist position). This neutralist-selectionist debate has stimulated a great deal of empirical work (surveys of variation in populations, as well as experimental work on enzyme properties and population “cage” experiments). A large amount of population genetic theory was also developed with the goal of helping to resolve this debate. Yet, it seems safe to say that this controversy was not clearly resolved by the theory and empirical efforts applied to electrophoretic variation. [The empirical and theoretical efforts to understand molecular variation in populations are well reviewed by Kimura (2) and Gillespie (1), who hold very different views on the causes of molecular evolution.]
Approximately 15 years ago, the first studies of variation at the DNA sequence level began to appear. New hope arose that genetic studies at this ultimate level of genetic resolution would help resolve the neutralist-selectionist controversy. For example, new theoretical analysis showed that the existence of a balanced polymorphism (if maintained for long evolutionary periods of time) results in a characteristic peak of variation in the region of DNA surrounding the site where the balancing selection acts (3). Fig. 1 shows the expected levels of variation in a region surrounding a balanced polymorphism and the observed levels of variation (4) at the Adh locus of Drosophila melanogaster. It seemed that a resolution of the controversy was just around the corner. One needed only to survey DNA variation at a large number of loci known to exhibit electrophoretic polymorphisms. Each of those loci at which balancing selection was acting would be expected to show a large peak of variation at linked sites. Hence, the effects of balancing selection would be clearly manifest and the controversy would be over, one way or the other.
Alas, surveying populations for DNA sequence variation is labor intensive and expensive, and progress has been slow. With the advent of PCR methodology the pace has certainly accelerated, but a clear picture has not yet emerged. To advance this data collection effort, we (and our collaborators) have undertaken an in-depth study of variation at the DNA level at the Sod locus in D. melanogaster and related species. The Sod locus was chosen for a number of reasons, including the fact that two electrophoretically distinguishable alleles, designated Fast and Slow, are commonly found segregating at high frequencies in populations around the world and the fact that a number of experiments suggest that Sod variation may be subject to some form of natural selection (5–9). The electrophoretic polymorphism at this locus appeared to be a good candidate for an old balanced polymorphism, for which the signature of a peak of linked variation would be visible. It is also known that the common Slow and Fast alleles differ by a single amino acid (10). We now have DNA sequences of many copies of this gene and regions surrounding the gene.
In the following pages, we will summarize the patterns of variation that have been observed at this locus in D. melanogaster. We will also describe our continuing efforts to understand the evolutionary history and significance of the variation at this locus. In particular, we will address the question: Is the Sod polymorphism an old balanced polymorphism?
The Initial Survey
An initial survey of DNA sequence variation at the Sod locus was carried out several years ago (11). A region 1,410 bp long was sequenced in 41 lines of D. melanogaster from localities in California and Barcelona, and included 19 sequences coding for the Slow form of the enzyme and 22 coding for the Fast allele. This sampling of approximately equal numbers of Slow and Fast alleles was done so that the level of variation within alleles could be effectively compared with the level of divergence between alleles. (The frequency of the Slow allele in the populations sampled is roughly 10%.) A total of 63 nucleotide site polymorphisms and 6 insertion/deletion polymorphisms were observed in the sample. Overall the amount of variation is typical of what has been observed at other loci in D. melanogaster. In addition, Tajima’s test (12) and the test of
© 1997 by The National Academy of Sciences 0027–8424/97/947725–5$2.00/0 PNAS is available online at http://www.pnas.org.
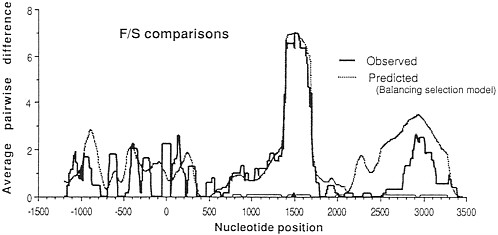
FIG. 1. Observed and predicted levels of divergence between Fast (F) and Slow (S) alleles in a sliding window across the Adh region of D. melanogaster. (The window is 100 silent sites wide.) The site coding for the F/S difference is at position 1500. Note the large peak of variation centered on the F/S site. For details see Kreitman and Hudson (4).
Hudson et al. (13) revealed no significant departures from equilibrium neutral models. There were, however, several surprising and interesting aspects of the sequence variation that suggested the recent action of natural selection. First, among the 22 Fast alleles sequenced, it was found that 9 were identical in sequence for the entire 1410 bp long region examined. This sequence was designated the Fast A haplotype. Second, all 19 Slow alleles were identical to each other and differed from the Fast A haplotype by a single nucleotide, the nucleotide that accounts for the amino acid difference between the Fast and Slow forms of the enzyme. This site will be referred to as the Fast/Slow site. Thus, the Slow/Fast polymorphism is clearly not an old balanced polymorphism; in fact, the Slow allele is obviously very recently derived from the Fast A haplotype. Summarizing, we find that about half of a random sample of sequences at this locus would consist of sequences that are nearly identical to each other, and the other half of the sample would be much more heterogeneous, differing from each other at roughly 20 sites of 1,410. This pattern of variation was demonstrated to be highly incompatible with an equilibrium neutral model (11).
Our working hypothesis is that a rare variant (perhaps a new mutation) has recently and rapidly increased in frequency to around 50%. As it increased in frequency, the haplotype in which it was embedded was pulled up in frequency at the same time. Although selection on the Fast/Slow site might have driven the Slow allele to its present frequency, such selection by itself cannot account for the observed high frequency of the Fast A haplotype. Thus, selection on some other site would appear to be involved. It should be noted that the putative polymorphic site upon which selection acts is not necessarily in the region sequenced, but must be tightly linked to it.
Such a selective event, whereby a rare variant is driven to intermediate frequency, could potentially affect a large region of DNA. (We will refer to such an event as a partial selective sweep.) Calculations of Kaplan et al. (14) suggest that a selection coefficient equal to 0.01 can sweep away variation at sites up to 10,000 bp from the site of selection (assuming rates of recombination that are typically observed in D. melanogaster). Fig. 2 shows the patterns of variation to be expected before, immediately after, and some period after, such a partial selective sweep. Immediately after the rise in frequency of the previously rare variant, all chromosomes bearing the selected variant will be identical across essentially the whole region, which was swept along with the selected variant. As time progresses, the “selected haplotype” (the haplotype in which the selected variant arose), will slowly be broken up by recombination events. Eventually, after much time has passed and further mutations accumulate, if the variation at the selected site is maintained by balancing polymorphism, a peak of linked variation should emerge. This pattern would emerge at a point in time much later than that shown in Fig. 2.
From the Sod data of Hudson et al. (11), we know that the region partially swept of variation is bigger than 1,410 bp. In addition, it appears that some recombination has occurred between the sequences since the putative partial selective sweep.
Sequence Variation at Linked Regions
To further investigate this putative selective history, additional lines were sequenced at the Sod locus and at three tightly linked regions. We were particularly interested in assessing the size of the region that had been swept along with the selected site and to assess the amount of recombination and mutation that has occurred since the partial selective sweep. With this additional information, inferences can be made about the strength of selection and the time since the partial sweep occurred. Details of this survey will appear elsewhere, but we will summarize the preliminary results here.
In this study, 15 lines of D. melanogaster from El Rio vineyard (Lockeford, San Joaquin County, California) and the Canton S strain of D. melanogaster were sequenced at the Sod locus and three neighboring regions. [The Sod locus of six of these lines, designated here 112, 565F, 581F, 255S, 510S, and 438S, were also sequenced in the earlier study (11).] The three neighboring regions, denoted 2021, 6Kbr3r, and 1819, are located approximately 12.7 kb upstream of Sod, 3.7 kb downstream of Sod, and 19.2 kb downstream of Sod, respectively. Fig. 3 shows the locations and sizes of each of these regions.
The polymorphic sites in this sample are indicated in Fig. 4. It is important to note that the Sod locus shows a similar pattern of variation to that observed in the earlier study. The four Slow alleles sequences are not, however, identical in this sample, but consist of two sequences identical to the Slow alleles found earlier and two other sequences that each differ from the other Slow allele sequences at a single site. Of 12 Fast alleles, 5 are the Fast A sequence, 2 more differ by a single site from Fast A, 2 others differ by 3 sites from Fast A, and the 3 others differ considerably from Fast A. Two Fast alleles in
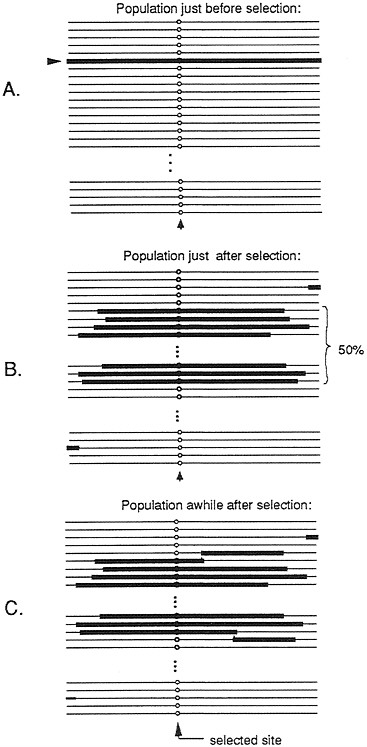
FIG. 2. Representation of the pattern of variation in a population of chromosomes just before (A), just after (B), and some time after (C) a partial selective sweep in which a mutation is rapidly driven to a frequency of 50% by natural selection. The site where selection acts is designated by a circle. The parts of chromosomes highlighted by a thick line indicate segments that are descended from the chromosome on which the original mutant arose.
particular are quite similar to each other and differ from Fast A at roughly 30 sites. Thus, in this new sample, roughly 75% of the Fast alleles are Fast A or a very similar haplotype. Though the sample sizes are very small this new sample suggests that the frequency of the homogeneous class of haplotypes may be somewhat larger than that suggested by the earlier samples.
What patterns of variation are observed at the linked regions? Both the 6Kbr3r, which is 3.7 kb downstream from Sod and the 1819 region, which is roughly 19.2 kb downstream from Sod show patterns of variation that are very similar to the pattern observed at Sod. That is, most of the sequences are very similar to each other, forming a very homogeneous subset, whereas the other sequences (between 2 and 5 of 16 lines) are relatively diverged from the homogeneous subset, and show some divergence among themselves. Thus, the region showing the Sod pattern of variation extends at least 20 kb downstream from Sod. As indicated earlier, if this pattern of variation is due to a partial selective sweep, quite strong selection (selection coefficient on the order of 0.01) is required. A second important feature of the data from these downstream regions is the pattern of recombination. We note that some of the lines, which form part of the homogeneous subset at the Sod locus, are lines that constitute part of the heterogeneous subset at the 6Kbr3r region. For example, the 510S line, which codes for the Slow Sod allele, forms part of the homogeneous class of haplotypes in the Sod locus. But this line is quite diverged from the homogeneous class of haplotypes in the 6Kbr3r region. Conversely, line 498F is quite diverged from the Fast A haplotype in the Sod locus, but is a member of the very homogeneous subset (differing at a single site from the most common haplotype) in the 6Kbr3r region. These patterns suggest that considerable recombination has occurred between the 6Kbr3r region and the Sod locus in the time since the selective event. The lack of complete linkage disequilibrium between Sod and 6Kbr3r suggests that the selective sweep is not extremely recent. To be somewhat more precise, since linkage disequilibrium decays approximately as exp(–rt), where r is the recombination rate per generation and t is the time measured in generations, we can estimate t. Because linkage disequilibria between sites in the Sod locus and sites in the 6Kbr3r region are substantially decayed, rt is unlikely to be less than one. The recombination rate in D. melanogaster females is estimated to be about 2×10-5/kb per generation (15) (except in regions near centromeres and telomeres). Taking into account that recombination does not occur in males, we estimate that the recombination rate between Sod and 6Kbr3r, which are approximately 4 kb apart, is approximately 4×10-5. This suggests that the time since the selective sweep is roughly 25,000 generations (=1.0/{4×10-5}) or longer. (Twenty-five thousand generations correspond to 5,000 years, assuming an average of 5 generations per year.)
The time since the putative selective event can also be inferred from the amount of variation that has accumulated within the relatively homogeneous subsets. We will assume provisionally that the sampled lines that constitute the homogeneous subset are related by a star genealogy (i.e., all lineages of the sampled regions remain distinct back to a time near the time of the selective event). This is a reasonable assumption if the effective population size is large and the selective event recent. The low observed frequency of most variants in the homogeneous set is also consistent with this assumption. In the Sod region the 3 lines, 581F, 498F, and 968F, constitute a heterogeneous and diverged subset, and the other 13 lines can be considered to constitute the homogeneous subset. The number of polymorphic sites among this homogeneous subset is nine. Similarly, in the 6Kbr3r region there are 10 sequences in the homogeneous subset, and there are 2 sites polymorphic, and finally in the 1819 region there are 13 or 14 lines in the homogeneous subset, depending on whether one counts line 521F as being a member of the subset or not. If the homogeneous subset includes 521F, then there are 12 polymorphic sites in the subset in this sequenced region. The sequence in the Sod region is 1,408 bp long, of which 439 bp are protein coding. Since in protein coding sequences about 25% of changes are synonymous, the sequenced Sod region is approximately equivalent to 1,079 (=1408-0.75*439) bp of noncoding sequence. Assuming that the other sequenced regions are noncoding and denoting the neutral mutation rate at noncoding and silent sites by µ [assumed to be 16×10-9 per site per year (16, 17)], we find that the expected number of polymorphic sites in the homogeneous subset is µt (13*1,079+10*764+14*937)= µt*34,785, where t is the time back to the selection event (in
FIG. 3. Schematic representation of the P1 clone, 112, which was derived from a Canton S strain. The boxes indicate the position of the sequenced regions. The length of these sequences and the distance between their borders are shown. The exons of the Sod coding region are shown as solid areas and indicated with the labels 1 and 2.
years). If we set µt*34,785 equal to 23, the observed number of polymorphisms in the homogeneous subset, and solve for t, we find t ˜ 41,000 years. Many of these polymorphisms within the homogeneous subset could be the result of conversion from haplotypes in the heterogeneous subset. In particular, the polymorphisms at sites 124, 142, 394, and 1143 in the Sod region, and sites 140 and 690 in the 1819 region could have resulted from conversion from haplotypes observed in the heterogeneous subset. This leaves only 17 mutations, which leads to an estimate of t of 31,000 years. Other polymorphisms in the homogeneous subset could have resulted from conversion or recombination and, in addition, the neutral mutation rate that we have used is based on substitution rates at silent sites in coding regions and may underestimate the neutral mutation rate in noncoding regions. Hence, our estimate may be biased upward, but is consistent with our conclusion from the pattern of recombination, which is that the selective event is probably older than 5,000 years.
The pattern of variation in the 2021 region, which is 12.7 kb upstream from the Sod locus, is completely different. There is no homogeneous subset of any appreciable size. There is a high level of polymorphism and no hint of the partial selective sweep evident in the other regions. The 2021 region is apparently outside the region of the partial sweep, indicating that the upstream boundary is somewhere between the Sod locus and the 2021 region. It would be desirable to confirm that regions further upstream continue to exhibit the “normal” pattern of variation, and to narrow

FIG. 4. Polymorphic sites found in the lines sampled from El Rio, California and the Canton S strain (designated 112). For each sequenced region the line designations appear on the left and the positions of polymorphic sites are indicated at the top. The lines are in a different order in each of the four regions to put similar haplotypes together in each region.
the position of this upstream boundary by examining additional regions between Sod and 2021.
Discussion and Conclusions
The Slow/Fast polymorphism is clearly not an old balanced polymorphism. On the other hand, the data suggest that natural selection has acted recently and strongly on variation in the neighborhood of Sod. The data appear compatible with a model in which a rare variant has recently risen rapidly in frequency, and is perhaps now subject to some form of balancing selection. The data at this point do not allow us to put an upper bound on the size of the region, which has been partially swept of variation. The 2021 region, which is 12.7 kb upstream from Sod, appears to be outside the swept region. Hence, one boundary of the swept region appears to be between the Sod locus and the 2021 region. Because the 1819 region, roughly 20 kb downstream of Sod appears to be in the swept region, we conclude that the swept region is greater than 20 kb in length. This in turn suggests that surprisingly strong selection acted on the selected site (selection coefficient on the order of 0.01 or higher). The pattern of linkage disequilibrium is like Fig. 2C, from which we infer that the time since the sweep is 25,000 generations or more.
These results force one to consider the possibility that balanced polymorphisms may typically be short-lived, arising and being maintained for a time too short to result in the strong peak of linked variation, such as that observed at Adh. The two best documented cases of long-maintained balanced polymorphisms are in the major histocompatibility complex in mammals (18, 19) and the S-locus (mating incompatibility determining locus) of some plants (20). These two cases involve large numbers of maintained alleles and remarkably old lineages (which presumably result in a peak of variation at linked sites.) These two examples may be the very rare exception. The most celebrated case of a balanced polymorphism is the sickle cell variant at the ß-globin locus of humans. This is a case where strong balancing selection is well documented, and it is also well documented that the currently segregating sickle cell variants are recently arisen (and have arisen independently in different populations.) Perhaps the sickle cell case, and the Sod case, are illustrative of the most common situation for protein polymorphisms, not the ancient stable polymorphisms that result in peaks of variation at linked sites. These short-lived polymorphisms might be compatible with models that incorporate temporal and spatial variability in selection coefficients (1).
On the other hand, it is important to consider alternatives to the partial selective, sweep hypothesis. This is particularly so given some similarities between the pattern of variation seen at Sod and the results of surveys at Est6 (21, 22) and Est-P (23), which are located approximately 1 centimorgan or 1,000 kb from Sod (24). The pattern of variation in the Sod locus and in the 1819 region in our recent study is remarkable for having two similar haplotypes that are highly diverged from the rest of the sample. (Note in Fig. 4 lines 968F and 498F in the Sod locus and lines 968F and 112 in the 1819 region.) This pattern of variation seems surprising and may not be expected under the sweep hypothesis. A very similar pattern was also observed in a recent survey at the Est-P locus (23). In that survey, which used a subset of the same lines used in our study, line 357F is highly diverged from the rest of the sample. Est-P and Sod are too far apart to be affected by the same selected sweep with normal rates of recombination. The presence of a small number of highly diverged haplotypes suggests the possibility of a distinct isolated subpopulation that has recently merged with another population. This could have been a geographically isolated population, but another possibility is that the diverged haplotype is or was part of an inversion. In(3L)P is a common and widespread inversion that contains Est-P and Sod (25), but no third chromosome inversions were found to be segregating in the El Rio population (26). It is not known if the diverged haplotypes could represent sequences that have “escaped” from an inversion, as has been previously suggested (22) for sequences at the Est6 locus, which is very closely linked to Est-P. Parenthetically, we note that two highly divergent lines were found in a survey of variation in the Adh region of D. melanogaster (4). The possibility of an inversion was investigated, and no direct evidence for such an inversion was found.
It is also worth noting that an earlier study of DNA sequence variation at Est6 found patterns that “suggest that allozyme 8 has both arisen and proliferated, relatively recently” (21). Thus, at Est6 as well as at Sod, it appears that certain variants have recently been driven to high frequency. We conclude that the patterns of DNA sequence variation at Est6 and Est-P have intriguing similarities to those at Sod. These may reflect similar independent selection events, but the possibility of some event or process affecting this large segment of chromosome 3 must also be entertained.
We thank Kevin Bailey, Evgeniy S.Balakirev, and Eladio Barrio for contributions to the early stages of this project; Shiliang Qin and John W.Jacobs for use of a laboratory of the Hitachi Chemical Research Center, Inc. at the University of California, Irvine; and Evgeniy S. Balakirev, Jordi Bascompte, and Francisco Rodriguez-Trelles for discussions; and Nelsson Becerra for technical assistance. This work was supported by National Institutes of Health Grant GM-42397 to F.J.A. and by a postdoctoral fellowship to A.G.S. from the Spanish Ministry of Education and Science.
1. Gillespie, J.H. (1991) The Causes of Molecular Evolution (Oxford Univ. Press, New York).
2. Kimura, M. (1983) The Neutral Theory of Molecular Evolution (Cambridge Univ. Press, Cambridge, U.K.).
3. Hudson, R.R. & Kaplan, N.L. (1988) Genetics 120, 831–840.
4. Kreitman, M. & Hudson, R.R. (1991) Genetics 127, 565–582.
5. Lee, Y.M., Misra, H.P. & Ayala, F.J. (1981) Proc. Natl. Acad. Sci. USA 78, 7052–7055.
6. Singh, R.S., Hickey, D.A. & David, J. (1982) Genetics 101, 235–256.
7. Peng, T., Moya, A. & Ayala, F.J. (1986) Proc. Natl. Acad. Sci. USA 83, 684–687.
8. Peng, T.X., Moya, A. & Ayala, F.J. (1991) Genetics 128, 381–391.
9. Tyler, R.H., Brar, H., Singh, M., Latorre, A., Graves, J.L., Mueller, L.D., Rose, M.R. & Ayala, F.J. (1993) Genetica 91, 143–149.
10. Lee, Y.M. & Ayala, F.J. (1985) FEBS Lett. 179, 115–119.
11. Hudson, R.R., Bailey, K., Skarecky, D., Kwiatowski, J. & Ayala, F.J. (1994) Genetics 136, 1329–1340.
12. Tajima, F. (1989) Genetics 123, 585–595.
13. Hudson, R.R., Kreitman, M. & Asuadé, M. (1987) Genetics 116, 153–159.
14. Kaplan, N.L., Hudson, R.R. & Langley, C.H. (1989) Genetics 123, 887–899.
15. Chovnick, A., Gelbart, W. & McCarron, M. (1977) Cell 11, 1–10.
16. Sharp, P.M. & Li, W.-H. (1989) J. Mol. Evol. 28, 398–402.
17. Rowan, R.G. & Hunt, J.A. (1991) Mol. Biol. Evol. 8, 49–70.
18. Klein, J. (1986) Natural History of the Major Histocompatibility Complex (Wiley, New York).
19. Takahata, N. (1993) in Mechanisms of Molecular Evolution, eds. Takahata, N. & Clark, A.G. (Japan Sci. Soc., Tokyo), pp. 1–21.
20. Clark, A.G., (1993) in Mechanisms of Molecular Evolution, eds. Takahata, N. & Clark, A.G. (Japan Sci. Soc., Tokyo), pp. 79–108.
21. Cooke, P.H. & Oakeshott, J.G. (1989) Proc. Natl. Acad. Sci. USA 86, 1426–1430.
22. Odgers, W.A., Healy, M.J. & Oakeshott, J.G. (1995) Genetics 141, 215–222.
23. Balakirev, E.S. & Ayala, F.J. (1996) Genetics 144, 1511–1518.
24. Hartl, D.L. & Lozovskaya, E.R. (1995) The Drosophila Genome Map: A Practical Guide (Landes, Austin, TX).
25. Voelker, R.A., Cockerham, C.C., Johnson, F.M., Schaffer, H.E., Mukai, T. & Mettler, L.E. (1978) Genetics 88, 515–527.
26. Smit-McBride, Z., Moya, A. & Ayala, F.J. (1988) Genetics 120, 1043–1051.





