
This paper was presented at a colloquium entitled “Genetics and the Origin of Species,” organized by Francisco J.Ayala (Co-chair) and Walter M.Fitch (Co-chair), held January 30–February 1, 1997, at the National Academy of Sciences Beckman Center in Irvine, CA.
Phylogenetics and the origin of species
(allelic genealogies/gene trees/lineages/mitochondrial DNA/phylogeography)
JOHN C.AVISE AND KURT WOLLENBERG
Department of Genetics, University of Georgia, Athens, GA 30602
ABSTRACT A recent criticism that the biological species concept (BSC) unduly neglects phylogeny is examined under a novel modification of coalescent theory that considers multiple, sex-defined genealogical pathways through sexual organismal pedigrees. A competing phylogenetic species concept (PSC) also is evaluated from this vantage. Two analytical approaches are employed to capture the composite phylogenetic information contained within the braided assemblages of hereditary pathways of a pedigree: (i) consensus phylogenetic trees across allelic transmission routes and (ii) composite phenograms from quantitative values of organismal coancestry. Outcomes from both approaches demonstrate that the supposed sharp distinction between biological and phylogenetic species concepts is illusory. Historical descent and reproductive ties are related aspects of phylogeny and jointly illuminate biotic discontinuity.
…genetics has so profound a bearing on the problem of the mechanisms of evolution that any evolution theory which disregards the established genetic principles is faulty at its source. —Theodosius Dobzhansky, 1937
Dobzhansky (1) began Genetics and the Origin of Species with “an observational fact more or less familiar to everyone…the discontinuity of the organic variation.” After addressing the sources of genetic variability in sexually reproducing populations and the evolutionary processes configuring this variation, Dobzhansky then culminated his tome with three concluding chapters extending earlier sentiments by Lamarck, Darwin, A. R.Wallace, and others who had identified an important role for reproductive isolation in the origin and maintenance of biotic discontinuity in the living world. Genetics and the Origin of Species provides one of the seminal treatments of what would become known as the “biological species concept” (2), and it remains today among the most eloquent of expositions on the evolutionary significance of speciation as the juncture “at which the once actually or potentially interbreeding array of forms becomes segregated in two or more separate arrays which are physiologically incapable of interbreeding” (1). In Dobzhansky’s judgment, “biological classification is simultaneously a man-made system of pigeonholes devised for the pragmatic purpose of recording observations in a convenient manner and an acknowledgment of the fact of organic discontinuity” (1). Throughout this century, the biological species concept (BSC) unquestionably has provided the primary philosophical framework orienting thought and research on speciation (3).
Thus, a recent development in the field of systematics could hardly be of deeper import to evolutionary biology: the rise in popularity (4) of a competing view that depreciates or, in the extreme, disavows entirely (5, 6) any relevance of reproductive isolation for species concepts. Various formulations of a “cladistic” or “phylogenetic species concept” (PSC) have been advanced (7–16), but all agree that species concepts and definitions should emphasize criteria of phylogenetic relationship (descent) and not reproductive relationship (interbreeding) (14). For example, a phylogenetic species as defined by Cracraft (10) constitutes “the smallest diagnosable cluster of individual organisms within which there is a parental pattern of ancestry and descent.” with diagnosis based strictly on one or more synapomorphic (shared-derived) characters that identify a monophyletic aggregate of individuals (17).
A widespread perception of overt conflict between the BSC and the PSC is underscored by numerous published statements such as the following: “as a working concept, the biological species concept is worse than merely unhelpful and non-operational—it can be misleading” (18); “a focus on the processes involved in breeding systems and barriers is unnecessary for…species recognition” (15); “reproductive isolation should not be a part of species concepts” (5); “a concept consistent with the PSC should replace ‘biological species’ concepts” (6); “evolutionary biologists should abandon the BSC” (17); and “the BSC and all other essentialist definitions should be scrapped once and for all” (19).
In light of these developments, this colloquium, which is timed to celebrate the 60th anniversary of the publication of Dobzhansky’s classic, is an appropriate forum to reflect once again upon the BSC, one of the book’s orienting foundations. The following questions will be addressed: (i) Is an appropriately constructed phylogenetic concept of species truly revisionary and antithetical to the BSC? (In our opinion, no.) (ii) Does the PSC as typically formulated provide an adequate philosophical or operational framework for achieving the stated goal of clarifying biotic relationships according to phylogenetic descent? (No, because it has failed to take into adequate account established population genetic principles.) (iii) Can desirable elements of the BSC and a properly reformulated PSC be reconciled in ways that will contribute to the scientific understanding of biotic discontinuity? (Yes.)
Here we introduce a heuristic approach that is inherently phylogenetic yet applies at the microevolutionary level of recent biological species and their constituent populations. The approach focuses on the individual and collective genealogical transmission pathways available to nuclear alleles in sexual organismal pedigrees. Each allele is defined here as a length of DNA that has been free of recombination within it during the ecological or evolutionary time under consideration, and whose branching pathways of descent therefore describe a nested, nonreticulate transmission history (“allelic
© 1997 by The National Academy of Sciences 0027–8424/97/947748–8$2.00/0 PNAS is available online at http://www.pnas.org.
pathway” or “allelic genealogy”) entirely suitable for phylogenetic or cladistic examination (20, 21).
Genealogical Pathways and Organismal Phytogenies. Phylogeny at the level of populations and species. Ever since the publication of Genetics and the Origin of Species, geography and demography have played key roles in most biological speciation scenarios (22). During the evolutionary sequence of events by which an extended reproductive community of organisms (a field for gene recombination) becomes sundered, a curtailment of population genetic exchange by environmental separation typically is envisioned as a necessary prerequisite for the eventual evolution of the intrinsic (genetic) reproductive isolating barriers (RIBs) that are the hallmark of the BSC (Fig. 1). The initial genomic sundering may involve sister populations distributed across broad areas (species D and E in Fig. 1), small founding populations on the periphery of a species’ range (species A) (23), or, in some cases (24), local syntopic populations separated by microhabit at (species B). In each case, population genomic differentiation facilitated by environmental impediments to interbreeding initiates or eventually may lead to an elaboration of intrinsic reproductive barriers. Biological speciation also can take place suddenly in small populations via reproductive sundering processes such as polyploidization, chromosomal rearrangements, or changes in the mating system (20). Species A and B in Fig. 1 could be interpreted as examples.
Each such geographic-demographic model yields logical predictions about the coarse-focus phytogeny for particular extant populations or biological species (25). For example, from a traditional perspective, taxa D and E (Fig. 1) are sister biological species that comprise a clade. On the other hand, the widely distributed species C that recently spawned a peripheral isolate A, or a syntopic species B, is paraphyletic with respect to each of these latter forms. As emphasized by Patton and Smith (26), most mechanisms of speciation currently advocated by evolutionary biologists “will result in paraphyletic taxa as long as reproductive isolation forms the basis for species definition.” Such statements pertain to the historical subdivisions of gene pools at the levels of species or well-demarcated populations. In reality, intermediate situations also exist in which biotic subdivisions display incomplete phylogenetic separation because of a semipermeability in the extrinsic or intrinsic barriers to genetic exchange.
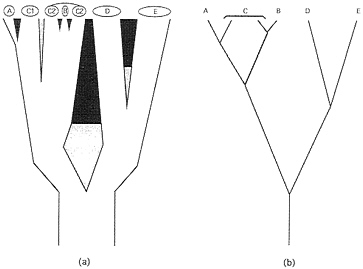
FIG. 1. (a) Phylogeny for live biological species (A-E) and two geographically separated populations (C1 and C2) of C. Branch widths are proportional to the populations’ or species’ sizes and also indicate a geographic orientation. Thus, A is a peripheral isolate from C1, and B arose within the range of C2. The sundering agents are intrinsic RIBs (black areas), extrinsic barriers to gene flow (gray areas), or both in temporal order of appearance (gray then black). (b) Simplified “stick” representation of the phylogeny in a.
Phylogeny at the level of alleles. In principle, any representation of phytogeny for separated populations or species might be examined under finer focus by reference to organismal pedigrees (Fig. 2). Ineluctably, pedigrees define extended pathways of genetic transmission that constitute rivulets in “the stream of heredity (that) makes phytogeny” (27). Consider, for example, the matrilineal pathway of transmission (F → F → F → F…, where F signifies female) for mitochondrial (mt) DNA (Fig. 3 Upper Left). All extant females in taxon E trace genealogically through female ancestors to a shared progenitress at t−5, those in D coalesce at t−9, and those in the D+E assemblage stem to a common ancestor at t−12. The great-great…-great matrilineal grandmother of all extant individuals in the pedigree existed at t−20. With respect to the mainlines in the A-C complex (which coalesce at t−11), C1 is paraphyletic to A, and C2 is paraphyletic to B. All such statements reflect the realities of allelic-level ancestry through heredity, as to be distinguished from any estimates of ancestry in empirical appraisals based on molecular or any other data.
Similarly, other gender-described classes of genealogical pathways can be envisioned. In any pedigree for sexually reproducing organisms, only four such transmission routes are mutually exclusive in every generation: the matrilineal pathway already mentioned; the patrilineal analogue (M → M → M → M…, where M signifies male; the route, for example, of the mammalian Y chromosome); and the generation-to-generation alternating reciprocal pathways “M → F → M → F…” and “F → M → F → M….” As traced through the organismal pedigree under consideration (Fig. 3), a comparison of these “independent” pathways illustrates two fundamental points. First, the coalescent trees for the
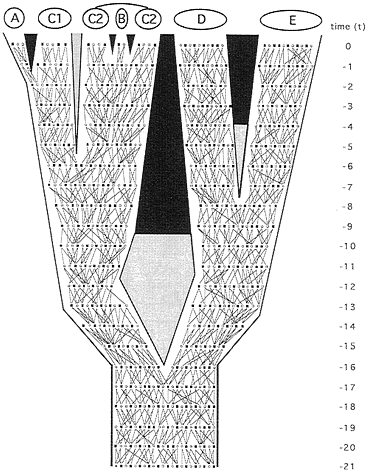
FIG. 2. Same phytogeny as in Fig. 1 but here depicting organismal pedigrees through 21 discrete generations leading to the present. The two lines tracing from each male (■) or female (○) in any generation identify the parents of that individual. They also describe the geographic dispersal of offspring (which is assumed to be distance-limited) and the mating events.
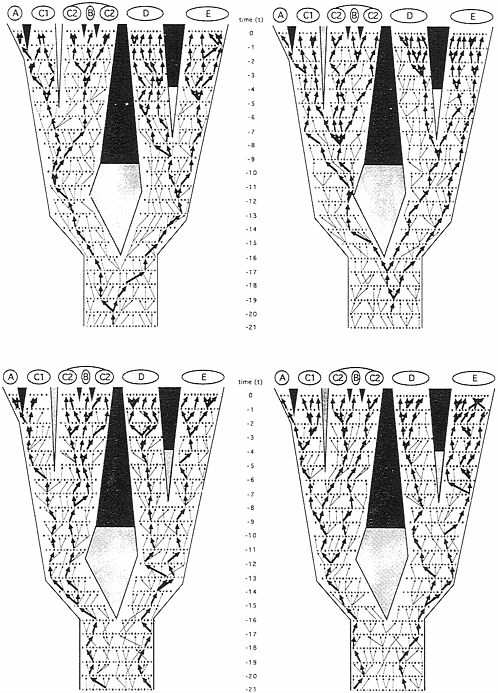
FIG. 3. Identical phylogeny and pedigree to Fig. 2, but here in which the four allelic transmission pathways that are mutually exclusive in every generation have been highlighted by arrows. (Upper Left) Matrilineal pathway reflecting the “F → F→ F → F…” transmission route (e.g., of mtDNA). (Upper Right) Patrilineal pathway reflecting the “M → M → M → M…” transmission route (e.g., of the Y chromosome). (Lower Left) Generation-to-generation pathway through alternating genders “M → F → M → F….” (Lower Right) The reciprocal of the latter, “F → M → F → M….” Heavy arrows mark transmission routes through this pedigree that extend to the current generation; light arrows mark the same respective transmission routes that terminated before reaching the extant generation.
allelic pathways can differ from one another in both depth and pattern. For example, extant individuals in species E share a common ancestor in the patrilineal phylogeny at t− 19 (Fig. 3 Upper Right), whereas they do so in the “M → F → M → F…” tree at t−3 (Fig. 3 Lower Left). Second, such transmission routes can differ from the species- or population-level phylogeny for reasons of lineage sorting, even in the total absence of introgression. With respect to patrilineal genealogy (Fig. 3 Upper Right), for example, the sister species D and E do not display a relationship of reciprocal, mono-
phyly (28), nor do they comprise a sister clade to the A-C group.
The total number of allelic transmission pathways as defined in this manner by gender is 2(G+1), where G (≥1) is the number of generations monitored (29). In the current case, this count is 222=4,194,304. Each such multigeneration pathway describes a unique, gender-defined route of potential allelic transmission through the organismal pedigree. These multitudinous genealogical pathways also differ from the four “independent” pathways pictured in Fig. 3, although the degrees of overlap vary widely. For example, the “F → F → F…→ F → M” pathway is identical to the matrilineal pathway (Fig. 3 Upper Left) except for the most recent generation, where the transmission was to sons. This particular example would describe the history of mtDNA in extant males. The reciprocal pathway (M → M → M…→ M → F) describes the history of family surnames as displayed by unmarried females in many human societies: a patrilineal legacy to living daughters (30). All other 4,194,298 genealogical pathways (e.g., F → F → M → M…→ F → F → M → M, or F → M → F → F…→ F → M → F → F) have been equally available to any piece of autosomal DNA trickling through the organismal pedigree under the rules of Mendelian inheritance. This statement holds regardless of the sex ratio in the population, because every individual has a mother and a father.
This gender-defined conception of allelic pathways differs somewhat from the usual definition of a “gene tree” for nuclear loci, but is introduced here to emphasize analogies to the conventionally understood pathways for mtDNA and the Y chromosome (as well as to clarify and stimulate thought about the potential numbers and patterns of allelic transmission routes). In actuality, extant alleles at any real autosomal gene collectively will have traversed many different genealogical pathways, such that the particular gender-based transmission routes normally remain unspecifiable by locus (31, 32).
This allelic-level heuristic conception of microphylogeny emphasizes that different pieces of DNA can have different genealogical histories both within and between closely related forms. This situation is an inevitable outcome of the quasi-independent transmission histories of alleles within and among loci through the organismal pedigrees of sexual reproducers.
Coalescent theory. A theory of gene coalescence as a function of population demography (33–41) and biological speciation mode (28, 42–46) has developed in response to the novel interpretive challenges provided by molecular genealogical data, particularly that from nonrecombining animal mtDNA (47, 48). As a phylogenetically oriented subdiscipline of population genetics, coalescent theory formally addresses lineage sorting and branching processes through organismal pedigrees, such that demographic and life-history factors, including population size, dispersal, and mating pattern, assume importance. Only cursory background will be noted here.
Imagine a large, idealized population with nonoverlapping generations and a constant number of Nm males and Nf females who contribute to a gametic pool from which the next generation of diploid zygotes is randomly derived. For particular allelic pathways of the sort pictured in Fig. 3, the expected mean time to common ancestry for pairs of alleles or haplotypes, measured in generations, is Nm or Nf, and the mean coalescence time for large numbers of sampled haplotypes is approximately 2Nm or 2Nf (49). These expectations apply to an mtDNA gene tree, a Y-linked gene tree, or to any hypothetical nuclear allelic pathway described by a specified gender-based transmission route when Nm=Nf. For unequal sex ratio, the average time to common ancestry would have to be adjusted to reflect the relative numbers of generations an allele spends in each gender in the particular allelic transmission pathway specified In the real world, a collective genealogy for multiple alleles at a true autosomal locus will not be delineated so clearly, because in each generation the alleles could have been transmitted along any of four possible routes (F → F, M → M, F → M, or M → F). Expected coalescent times at a real autosomal locus are 4-fold greater (50) than those for the idealized allelic pathways described above.
Natural populations depart from this idealized model, but expectations are approximated by substituting effective population sizes of males (Ne(m)) and females (Ne(f)) into the equations. Also, at real loci the theory applies strictly to neutral alleles. For example, coalescent times under balancing selection are extended because allelic extinction is inhibited, whereas coalescent times under selective sweeps are truncated. Recent extensions of coalescent theory have examined the consequences of nonequilibrium population demographies (51–55) and varied selection regimes (56–61) on gene genealogies.
The lineage-sorting processes underlying coalescent theory also apply across the sundering events that generate phylogenetic nodes and branches at the levels of populations or species (28, 42–45). Consider again the sister species D and E in Fig. 3. At the present time (t=0), E is paraphyletic to D in terms of patrilineal genealogy (Fig. 3 Upper Right), but a reasonable expectation under lineage sorting is that this genealogical relationship will soon convert to one of reciprocal monophyly [“exclusivity” in other parlance (62–64)] as one or the other of the two ancient patrilines in E goes extinct. In general, a neutral allelic tree is highly likely to have evolved to a status of exclusivity in two sister populations or species separated for more than about 4Ne generations, where Ne refers to the size of the isolates (28, 65).
A discordance also exists between the patrilineal genealogy (Fig. 3 Upper Right) and the deepest node (distinguishing A-C from D-E) in the species phylogeny. In the future, depending on which of the two deep patrilines currently within E first goes extinct, this discordance will either (i) disappear or (ii) become cemented in place. Outcome (ii) illustrates how a true allelic-tree/species-tree discordance also may characterize species that stem from closely spaced population nodes (relative to effective population size) at distant times in the evolutionary past. The probabilities of such discordance as functions of Ne and internodal numbers of generations are presented by Nei (49).
In summary, coalescent theory as applied across the phylogenetic nodes in populational or species trees has shown that the phylogenetic status of a given pair of biological species with respect to allelic genealogy is itself evolutionarily dynamic, with a usual time course subsequent to biological speciation being poly- or paraphyly → reciprocal monophyly. Furthermore, the demographic and geographic modes of speciation have major impact on the developing phylogenetic status of allelic genealogies in related biotas (28). Thus, at microevolutionary scales, concepts of phylogeny cannot be divorced from those of population genetics and demography. As a sundering agent at the level of populations and species, extrinsic and intrinsic barriers to interbreeding are keynote evolutionary agents motivating genealogical partitions at the level of allelic lineages.
Points of Conflict Between the PSC and the BSC. Phylogenetic complaints against the BSC. Proponents of the PSC have leveled several criticisms against the BSC. One widespread grievance is that appeals to reproductive isolation in species recognition necessitate unjustifiable and untestable judgments about the future; namely, whether contemporary barriers to interbreeding are permanent or temporary.
However, analogous prospective judgments are no less inherent in species concepts based on criteria of perceived phylogenetic separation (66). Thus, whether or not this criticism of the BSC is fully justified, it cannot be used to argue in favor of the PSC.
We will briefly address the three other perceived flaws of the BSC that were emphasized in a recent review (6).
(i) Reproductive compatibility among populations is a shared primitive (rather than derived) feature, so it provides no criterion for identifying monophyletic units or clades. In other words, “a serious potential problem of the BSC is the occurrence of paraphyletic, or nonhistorical, groups” (6). We appreciate the premise of this sentiment, but fail to see why it is so anathematical to admit paraphyly in species concepts (apart from the fact that this practice violates an operational ethic of cladistic analysis). As illustrated above, many modes of biological speciation initially entail paraphyly both at the levels of population trees and allelic trees. The philosophical stance that we favor acknowledges the reality of paraphyly for many biological species but then capitalizes upon such lineage information in particular instances to recover the historical population demographies that accompanied biological speciation. By tapping the genealogical archives of extant organisms, the bygone demographies of ancestral populations and speciation events may be inferred (67). In this important sense, paraphyly can hardly be equated with “nonhistory.”
(ii) A focus on reproductive compatibilities and patterns of interbreeding can cause a “misrepresentation of the significance of hybridization among differentiated taxa” (6). This criticism of the BSC stems from the fact that widely varying levels of hybridization and introgression have been employed by tax-onomists as justifications for naming biological species. However, such problems are implementational more than epistemological. The Linnaean binomial system of nomenclature lends itself poorly to the summary of situations with intermediate levels of introgression. Under the genealogical perspective promoted in this paper, the mosaic phylogenetic histories of allelic pathways within an organismal pedigree (including introgressed lineages) are of greater empirical content and conceptual import than are the necessarily simplified taxonomic summaries.
(iii) “A long-recognized drawback of the BSC is its difficulty in ranking allopatric populations…. Because reproductive isolation is an epiphenomenon (or emergent property) of divergence, it is not easily related to descriptions of how characters vary geographically” (6). This criticism validly notes an inherent implementational difficulty for species-level taxonomy under the BSC. However, even in this restricted context, alternative concepts such as the PSC may fare no better. If individual synapomorphies are to be used to define species as under the conventional PSC, then with sufficient empirical effort nearly every local population will be distinguishable from nearly every other by some character, and the challenge remains as to how to rank the differences. Such ranking might be accomplished more appropriately by considerations of multicharacter genealogies (see below), but these too are emergent properties of population-level divergence through extended pedigrees. Furthermore, because demographic factors such as gene flow and effective population size exert overriding influence on phylogeographic patterns, strict application of the monophyly criterion for species definition will strongly bias toward formal taxonomic recognition of smaller as opposed to larger populations (the latter more often will be paraphyletic with respect to close relatives). Thus, the conventional PSC itself can fail to capture important aspects of organismal history across geography.
Diagnostic complaints against the PSC. The overriding difficulty with the traditionally formulated PSC concerns the diagnosability criteria by which clades in sexually reproducing organisms are to be recognized at microevolutionary scales. In the light of coalescent theory, any approach that promulgates clade diagnosis on the basis of synapomorphs at only one or a few genes makes little sense. Consider again Fig. 3. Many phylogenetic species, each comprising a “diagnosable cluster of individual organisms within which there is a parental pattern of ancestry and descent” (10), could be identified depending on which allelic genealogy was scrutinized. Many more such “clades” could be identified by various other of the 4.2 million gender-defined transmission pathways in this same pedigree (Figs. 4 and 5). Such diagnostic possibilities are not merely hypothetical. With the resolving power already available in molecular assays of rapidly evolving genes such as mtDNA or autosomal microsatellites, many local populations, family units, and even individual organisms can readily be distinguished by recently derived mutations (20, 68). What conceptual or utilitarian rationale exists for defining each such diagnosable biological unit as a distinct species?
The “clades” identified by the synapomorphies in different allelic trees almost inevitably group sexually interbreeding individuals into overlapping arrays, such that the phylogenetic units recognized by different pieces of DNA are neither mutually exclusive nor nested. For example, all extant C1 males form a clade in the allelic tree displayed in the lower right of Fig. 3, whereas they are variously allied to A or C2 and B in the patrilineal genealogy (Upper Right). Only after reproductive ties have been severed for times considerably longer than effective population sizes do deeper topologies in multiple allelic genealogies tend to come into congruence with one another, and with the coarse-focus topologies of the population-level phylogenies that they comprise. From this perspective, speciation under phylogenetic precepts should be viewed more properly as the evolutionary process by which patterns of predominant nonconcordance among shallow allelic genealogies are converted to patterns of predominant concordance in deeper allelic trees.
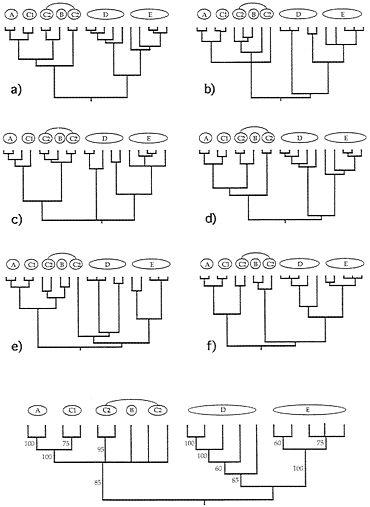
FIG. 4. Examples of six (a-f) additional gender-defined allelic transmission pathways (analogous to those in Fig. 3) through the organismal pedigree (Fig. 2). Each genealogy terminated in extant females and, hence, is a “female-tip” gene tree. Lineages that did not coalesce at t-21 were assumed to do so at t-22. At the bottom of the figure is a consensus tree for 20 such randomly chosen female-tip genealogies. Numbers on branches indicate the percentage of allelic trees in which that “clade” was found.
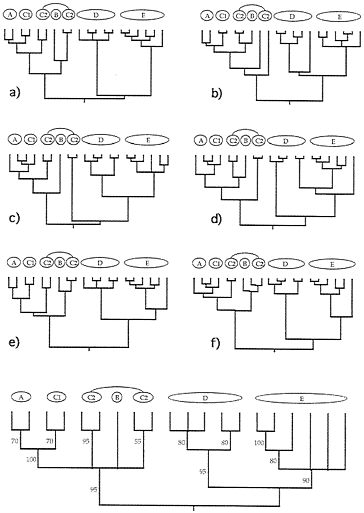
FIG. 5. Examples of six (a-f) additional “male-tip” allelic pathways through the organismal pedigree of Fig. 2 and a consensus tree for 20 such randomly chosen male-tip genealogies (see legend to Fig. 4).
In summary, no species concept that results in an overly simplified caricature of organismal phylogeny can hope to capture the rich and varied fabrics of genealogical histories in the multiple pieces of DNA that make up those “composite” phylogenies. As phrased by Maddison (21), “the species phylogeny is more like a statistical distribution, being composed of various trees (the gene trees), each of which may indicate different relationships.” The challenge then becomes to describe these statistical distributions and to properly interpret the demographic and evolutionary processes that have shaped them.
Reconciling a Multilocus PSC with the BSC. Most early definitions of phylogenetic species failed to accommodate the realities of microevolutionary genealogy in the pedigrees of sexually reproducing organisms. Avise and Ball (69) therefore concluded that if a broader framework of the PSC was to contribute to a significant advance in systematic practice, a shift from issues of diagnosability to considerations of multilocus magnitudes and patterns of genealogical differentiation would be required. Recent verbal reformulations of PSC-like concepts according to principles of multilocus genealogical concordance (63, 64, 69, 70) have begun to heed this call, although much remains to be developed in a formal multilocus coalescent theory of speciation.
To develop conceptual connections between the phylogenetic histories of particular genes and the composite histories of populations and species, we now introduce a modification of traditional coalescent theory that jointly examines multiple allelic genealogies through an organismal pedigree. Two general approaches are employed that, in effect, acknowledge and summarize the genealogical heterogeneity across the multitudinous transmission pathways available to nuclear (and cytoplasmic) alleles. These approaches bear some analogy to the philosophies of “separate” (71) versus “combined” (72) phylogenetic treatments of multiple, potentially conflicting data sets (see ref. 73).
Qualitative concordance. The first of these heuristic approaches compares topologies across multiple allelic genealogies (e.g., through use of consensus trees). For example, three of the four allelic genealogies in Fig. 3 portray a status of reciprocal monophyly (exclusivity) for D and E and also suggest that these species form a clade distinct from the A-C assemblage. The fourth allelic pathway (Upper Right) contradicts these patterns, but would be overruled in a majority consensus representation.
Extending this approach, we have examined many additional genealogical transmission pathways through the organismal pedigree in Fig. 2. Such gender-defined allelic pathways terminate either in extant females (“female-tip” trees) or males (“male-tip” trees). Six random examples from among the millions of pathways definable for each gender-tipped class are shown in Figs. 4 and 5, together with consensus-tree summaries of additional such representative allelic genealogies. Notice both the heterogeneity of detailed branching pattern across the different allelic genealogies and the fact that the consensus trees nonetheless properly capture the major features in the overall organismal pedigree and phylogeny (compare with Fig. 2).
Quantitative coancestry. The second heuristic approach considers composite indices of genetic relatedness between organisms and groups individuals into genealogical assemblages accordingly. Here, the computer program SAS (74) was applied to the known pedigree in Fig. 2 to calculate true coefficients of coancestry between all pairs of extant individuals. A coancestry (or kinship) coefficient is defined as the chance that an allele randomly drawn from one individual is identical by descent (autozygous) within the pedigree to an allele drawn from another individual (75), and is equivalent in value to the inbreeding coefficient for these individuals’ hypothesized offspring. Such probabilities are positive functions of the number of genealogical pathways connecting a pair of individuals to all ancestors in the pedigree and inverse functions of the lengths of those pathways. The coancestry matrix for extant individuals in Fig. 2 then was clustered by UPGMA (76), with results presented in Fig. 6.
Although the composite genealogical “tree” in Fig. 6 is artificially branched and mostly dichotomous, it again captures the major features of the organismal pedigree (Fig. 2) upon which (ultimately) it was based. Thus, D and E are sister groups separated by a relatively deep node; C1 appears paraphyletic to A; C2 appears paraphyletic to B; and the A-C assemblage joins the D-E group at the oldest node in the phenogram
Readers may object that these qualitative consensus and quantitative coancestry approaches at attempted recovery of a known pedigree, being based as they are on the allelic-tree properties of that pedigree, involve circularities of reasoning. We agree. In a genealogical sense, a composite organismal pedigree cannot be fundamentally distinct from a statistical compilation of the multitudinous transmission pathways within it.
A BSC-PSC reconciliation. Both of these multilocus genealogical approaches demonstrate that reproductive barriers (the hallmark of the BSC) are important, even within a strictly phylogenetic species framework, because they generate through time increased genealogical depth and concordance across allelic pathways. This point has seemed rather obvious to us, yet it has not been appreciated fully by most calls in the literature for a replacement of the BSC by the PSC. It has been the intent of this paper to illustrate, using a novel but simple conceptual construct based on considerations of nonanastomatic allelic pathways, how reproductive and phylogenetic aspects of biological differentiation are related intimately.
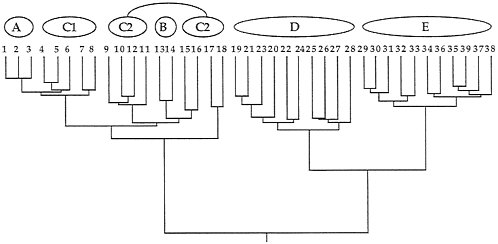
FIG. 6. Phenogram based on a cluster analysis of the matrix of coancestry coefficients for the 39 extant individuals in the pedigree of Fig. 2. Note the close resemblance of this representation to that of the original pedigree.
Thus, reproductive barriers tend to demarcate and distinguish deep biotic discontinuities from those that are “trivial” in the sense of being recent and/or idiosyncratic to small fractions of the genome. It is no mere coincidence, for example, that biological species D and E as defined by reproductive criteria (Figs. 1–3) constitute recognizable assemblages of individuals genealogically (Figs. 4–6). Conversely, genealogical considerations can be seen as important under the philosophical framework of the BSC because they force explicit attention on historical and demographic aspects of speciation. For example, the genealogical paraphyly of C to A (Figs. 4–6), and the high coefficients of coancestry between these two arrays of individuals (Fig. 6), jointly imply a recent and perhaps bottlenecked separation of A from C, as indeed was the case (Figs. 1–3).
As applied to taxonomy, Avise and Ball (69) suggested that the BSC be retained as a philosophical orientation for species recognition (notwithstanding the operational difficulties acknowledged above), whereas “significant” phylogeographic partitions, as registered by relatively deep and concordant genealogical separations in multiple lineage pathways, provide a justifiable philosophical and empirical basis for the recognition of additional historically important “subspecific” units. For at least two reasons, such suggestions probably will have minor impact on either the procedures or outcomes of traditional systematics (although this “boon” alone should not be interpreted as a primary justification for genealogical concordance concepts). First, systematists at their best always have sought concordant evidence from multiple characters before making firm taxonomic judgments (77). Second, although the recovery of nuclear gene genealogies has become technically feasible in recent years (e.g., refs. 78 and 79), such molecular appraisals remain laborious and challenging. Thus, in practice, conventional classes of information from multiple character state distributions (molecular or otherwise) no doubt will continue to provide the surrogate phylogenetic information to be included in appraisals of genealogical concordance and the biotic discontinuities thereby registered.
Concluding Thoughts. Biological speciation lies at a pivotal boundary where the partially braided collection of allelic pathways of interbreeding individuals bifurcates into two such collections (32). Hennig (80) characterized this boundary as the dividing line between the realms of “tokogenetic” associations (genetic relationships between individuals) and “phylogenetic” associations (genetic relationships between species), or the border between reticulate and divergent relationships. This boundary also demarcates the areas of inquiry traditionally associated with two of the major disciplines within evolutionary biology: phylogenetic biology (macroevolution) and population genetics (microevolution). The PSC has roots in the field of systematics but, as applied at microevolutionary levels, has ignored principles of Mendelian and population genetics (at its peril). Conversely, the BSC has roots in population genetics but now might profit from an infusion of appropriate phylogenetic considerations to illuminate previously underemphasized elements of genealogical history, both of alleles and of populations over microevolutionary scales.
Two approaches can be taken to accommodate the distinct world views of phylogenetic biology and population genetics. The first is to claim that phylogenetic concepts are devoid of jurisdiction and meaning at intraspecific levels (80). However, as evidenced by considerations of allelic genealogies, this perception is incorrect. Thus, a more fruitful endeavor is to attempt affirmative rapprochements between phylogenetic biology and population genetics by drawing conceptual and empirical connections between these disciplines (47). Considerations of multilocus allelic coalescent processes, perhaps as suggested in this paper, provide an interesting avenue for the further exploration of such possibilities.
Biological taxonomy and classification can be viable disciplines without any recognition of evolution, just as rocks and minerals can be named and classified into groups. Indeed, Darwin’s (81) elucidation of evolutionary processes had virtually no impact on the day-to-day practice of naming and grouping species. Today, many phylogenetic biologists argue that the recognition of pattern in phylogeny should not be confused with nor unduly influenced by whatever hypothesized processes (e.g., reproductive isolation) might have contributed to the historical configurations (9, 15, 16, 82). The “pattern cladists” make valid cautionary points about objectivity in scientific explanation, and in practice “species” in the natural world can be identified and pigeonholed under appropriate phylogenetic procedures without consideration of evolutionary-shaping processes. However, to cleanse from species concepts all references to reproductive isolation would be to leave an unduly sterile epistemological foundation for the origin and maintenance of the biotic discontinuities so evident to Dobzhansky 60 years ago. If concepts resembling the BSC had not existed throughout this century, in the light of modern coalescent theory and associated multilocus genealogical concordance principles, they surely now would demand invention.
O’Hara (66) has likened the challenge of phylogenetic summary in biology to that of cartographic representation in geography. Phytogenies and maps alike are simplifications of reality, generalized representations with events selectively deleted according to the level and nature of detail required. An
interstate road map of the United States may be helpful in driving cross-country but is of no use in navigating the Freedom Trail in Boston for which a fine-grained local map provides the appropriate resolution. Similarly, phylogenetic summaries capture varying degrees of generalization about the streams and watersheds of heredity that make phylogeny, and a given depiction should be matched to the problem at hand. It has been the thesis of this paper that the “species problem” cannot be properly addressed from a phylogenetic perspective without reference to the fine-focus details of pedigrees and of lineage sorting processes at microevolutionary scales, and that an incorporation of such perspectives can resolve many of the apparent conflicts previously emphasized between the PSC and the BSC. To paraphrase and adapt the quotation from Dobzhansky (1) that opened this paper: population genetics has so profound a bearing on the problem of the mechanisms of speciation that any speciation theory that disregards established population genetic principles is faulty at its source.
We thank the National Science Foundation for continued support of the Avise laboratory.
1. Dobzhansky, T. (1937) Genetics and the Origin of Species (Columbia Univ. Press, New York).
2. Mayr, E. (1940) Am. Nat. 74, 249–278.
3. Otte, D. & Endler, J.A., eds. (1989) Speciation and Its Consequences (Sinauer, Sunderland, MA).
4. Martin, G. (1996) Nature (London) 380, 666–667.
5. McKitrick, M.C. & Zink, R.M. (1988) Condor 90, 1–14.
6. Zink, R.M. & McKitrick, M.C. (1995) Auk 112, 701–719.
7. Rosen, D.E. (1979) Bull. Am. Mus. Nat. Hist. 162, 267–376.
8. Eldredge, N. & Cracraft, J. (1980) Phylogenetic Patterns and the Evolutionary Process. (Columbia Univ. Press, New York).
9. Nelson, K.C. & Platnick, N.I. (1981) Systematics and Biogeog-raphy (Columbia Univ. Press, New York).
10. Cracraft, J. (1983) Curr. Ornithol. 1, 159–187.
11. Cracraft, J. (1987) Biol. Philos. 2, 329–346.
12. Donoghue, M.J. (1985) Bryologist 88, 172–181.
13. Mishler, B.D. & Brandon, R.N. (1987) Biol. Philos. 2, 397–414.
14. de Queiroz, K. & Donoghue, M.J. (1988) Cladistics 4, 317–338.
15. Wheeler, Q.D. & Nixon, K.C. (1990) Cladistics 6, 77–81.
16. Nixon, K.C. & Wheeler, Q.D. (1990) Cladistics 6, 211–223.
17. Cracraft, J. (1989) in Speciation and Its Consequences, eds. Otte, D. & Endler, J.A. (Sinauer, Sunderland, MA), pp. 28–59.
18. Frost, D.R. & Hillis, D.M. (1990) Herpetologica 46, 87–104.
19. Mallet, J. (1995) Trends Ecol. Evol. 10, 490–491.
20. Avise, J.C. (1994) Molecular Markers, Natural History and Evolution (Chapman & Hall, New York).
21. Maddison, W. (1995) in Experimental and Molecular Approaches to Plant Biosystematics, eds. Hoch, P.C. & Stephenson, A.G. (Missouri Botanical Garden, St. Louis), pp. 273–287.
22. Mayr, E. (1963) Animal Species and Evolution (Harvard Univ. Press, Cambridge, MA).
23. Giddings, L.V., Kaneshiro, K.Y. & Anderson, W.W. (1989) Genetics, Speciation and the Founder Principle (Oxford Univ. Press, New York).
24. Bush, G.L. (1975) Annu. Rev. Ecol. Syst. 6, 339–364.
25. Harrison, R.G. (1991) Annu. Rev. Ecol. Syst. 22, 281–308.
26. Patton, J.L. & Smith, M.F. (1989) in Speciation and Its Consequences, eds. Otte, D. & Endler, J.A. (Sinauer, Sunderland, MA), pp. 284–304.
27. Simpson, G.G. (1945) Bull. Am. Mus. Nat. Hist. 85, 1–350.
28. Neigel, J.E. & Avise, J.C. (1986) in Evolutionary Processes and Theory, eds. Nevo, E. & Karlin, S. (Academic, New York), pp. 515–534.
29. Avise, J.C. (1995) Conserv. Biol. 9, 686–690.
30. Avise, J.C. (1989) Nat. Hist. 3, 24–27.
31. Doyle, J.J. (1995) Syst. Botany 20, 574–588.
32. Brower, A.V.Z., DeSalle, R. & Vogler, A. (1996) Annu. Rev. Ecol. Syst. 27, 423–450.
33. Griffiths, R.C. (1980) Theor. Popul. Biol. 17, 370–50.
34. Kingman, J.F.C. (1982) Stochastic Processes Appl. 13, 235–248.
35. Tajima, F. (1983) Genetics 105, 437–460.
36. Avise, J.C., Neigel, J. & Arnold, J. (1984) J. Mol. Evol. 20, 99–105.
37. Avise, J.C., Ball, R.M., Jr., & Arnold, J. (1988) Mol. Biol. Evol. 5, 331–334.
38. Tavaré, S. (1984) Theor. Popul. Biol. 26, 119–164.
39. Takahata, N. & Nei, M. (1985) Genetics 110, 325–344.
40. Hudson, R.R. (1990) Oxford Surv. Evol. Biol. 7, 1–44.
41. Donnelly, P. & Tavaré, S. (1995) Annu. Rev. Genet. 29, 401–421.
42. Pamilo, P. & Nei, M. (1988) Mol. Biol. Evol. 5, 568–583.
43. Takahata, N. (1989) Genetics 122, 957–966.
44. Wu, C.-I. (1991) Genetics 127, 429–435.
45. Hudson, R.R. (1983) Evolution 37, 203–217.
46. Hey, J. (1994) in Molecular Ecology and Evolution: Approaches and Applications, eds. Schierwater, B., Streit, B., Wagner, G.P. & DeSalle, R. (Birkhaeuser, Basel), pp. 435–449.
47. Avise, J.C., Arnold, J., Ball, R.M., Jr., Bermingham, E., Lamb, T., Neigel, J.E., Reeb, C.A. & Saunders, N.C. (1987) Annu. Rev. Ecol. Syst. 18, 489–522.
48. Avise, J.C. (1989) Evolution 43, 1192–1208.
49. Nei, M. (1987) Molecular Evolutionary Genetics (Columbia Univ. Press, New York).
50. Birky, C.W., Jr., Maruyama, T. & Fuerst, P. (1983) Genetics 103, 513–527.
51. Slatkin, M. & Hudson, R.R. (1991) Genetics 129, 555–562.
52. Takahata, N. (1991) Genetics 129, 585–595.
53. Rogers, A.R. & Harpending, H. (1992) Mol. Biol. Evol. 9, 552–569.
54. Marjoram, P. & Donnelly, P. (1994) Genetics 136, 673–683.
55. Rogers, A.R. (1995) Evolution 49, 608–615.
56. Kaplan, N.L., Darden, T. & Hudson, R.R. (1988) Genetics 120, 819–829.
57. Kaplan, N.L., Hudson, R.R. & Langley, C.H. (1989) Genetics 123, 887–899.
58. Hudson, R.R. & Kaplan, N.L. (1995) Genetics 141, 1605–1617.
59. Takahata, N. (1990) Proc. Natl. Acad. Sci. USA 87, 2419–2423.
60. Takahata, N. (1993) Mol. Biol. Evol. 10, 2–22.
61. Takahata, N. & Nei, M. (1990) Genetics 124, 967–978.
62. de Queiroz, K. & Donoghue, M.J. (1990) Cladistics 6, 61–75.
63. Graybeal, A. (1995) Syst. Biol. 44, 237–250.
64. Baum, D.A. & Shaw, K.L. (1995) in Experimental and Molecular Approaches to Plant Biosystematics, eds. Hoch, P.C. & Stephenson, A.G. (Missouri Botanical Garden, St. Louis), pp. 289–303.
65. Moore, W.S. (1995) Evolution 49, 718–726.
66. O’Hara, R.J. (1993) Syst. Biol. 42, 231–246.
67. Ayala, F.J. & Escalante, A.A. (1996) Mol. Phylogenet. Evol. 5, 188–201.
68. Avise, J.- C. & Hamrick, J.L., eds. (1996) Conservation Genetics: Case Histories from Nature (Chapman & Hall New York).
69. Avise, J.C. & Ball, R.M., Jr. (1990) Oxford Surv. Evol. Biol. 7, 45–67.
70. Mallet, J. (1995) Trends Ecol. Evol. 10, 294–299.
71. Miyamoto, M.M. & Fitch, W.M. (1995) Syst. Biol. 44, 64–76.
72. Kluge, A.G. (1989) Syst. Zool. 38, 7–25.
73. Hillis, D.M., Mable, B.K. & Moritz, C. (1996) in Molecular Systematics, eds. Hillis, D.M., Moritz, C. & Mable, B.K. (Sinauer, Sunderland, MA), pp. 515–543.
74. SAS Institute (1988) SAS/STAT User’s Guide (SAS Institute, Cary, NC), Release 6.09.
75. Hartl, D.L. & Clark, A.G. (1989) Principles of Population Genetics (Sinauer, Sunderland, MA), 2nd Ed.
76. Sneath, P.H.A. & Sokal, R.R. (1973) Numerical Taxonomy (Freeman, San Francisco).
77. Wilson, E.O. & Brown, W.L., Jr. (1953) Syst. Zool. 2, 97–111.
78. Palumbi, S.R. & Baker, C.S. (1994) Mol. Biol. Evol. 11, 426–435.
79. Slade, R.W., Moritz, C. & Heideman, A. (1994) Mol. Biol. Evol. 11, 341–356.
80. Hennig, W. (1966) Phylogenetic Systematics (Univ. Illinois Press, Urbana).
81. Darwin, C. (1859) On the Origin of Species (John Murray, London).
82. Brady, R.H. (1985) Cladistics 1, 113–126.








