
Biologic Imaging: From Mouse Genome to Human Disease
Michael Phelps
Department of Molecular and Medical Pharmacology
University of California at Los Angeles School of Medicine
Los Angeles, California
A partnership of Congress and the Atomic Energy Commission in the Atoms for Peace program created a peaceful and important use for radioactivity and created the field of nuclear medicine. Nuclear medicine now provides about 13 million clinical procedures per year and is an important part of the biologic research programs of not only the United States but the world. My task is going to be to focus on molecular nuclear medicine and help to bring nuclear medicine into a partnership with modem biology and genetics.
My colleagues and I are going to focus on using the power of imaging to look at the biologic basis of organ function in living organisms. Imaging has enormous power. As scientists, we seek to assemble pieces of a puzzle and to put together enough pieces and lines of evidence to show the whole picture of a process being studied. Imaging seeks to create a picture from the very beginning.
The Department of Energy (DOE) is playing a critical role in developing a segment of imaging technologies called biologic imaging. Biologic imaging covers many domains. It combines biology, modem genetics, and the principles of imaging to enable us to look at a whole system—a molecule, a cell, or a whole organism.
Now, why take an image? Medically, we take an image because we want to find out what is causing a problem. Another reason is to see what happens to a drag after a patient swallows it or we intravenously inject it. Imaging lets us watch a drug be distributed throughout a body and understand both how it interacts with its target and how it interacts with the rest of the body.
How do we get images? We use a trace amount of unusual radionuclides—positron-emitting radionuclides of carbon, nitrogen, oxygen, and fluorine (as a substitute for hydrogen or hydroxyl groups). We can use a simple molecule, glucose, and a particular form of it, 2-dioxyglucose, in which a fluorine substitutes for a hydroxyl group. This molecule was created at Brookhaven National Laboratory, developed as a drag at Washington University and then as a tracer for autoradiography.
In our technique of PET, the presence of the positron-emitting radionuclide fluorine-18 on the molecule will tell us where the molecule is after we administer it to a patient. The positron combines with an electron, and then the massive electron and the positron are annihilated, producing back-to-back photons that are easily transmitted through the body and detected by a camera, which is called a PET scanner. The PET scanner consists of a circumferential array of detectors that can detect the photons. This technology was developed with DOE support; when the original system was developed at Washington University, there were 2 sources of financial support: DOE and the Women's Advertising Association of St. Louis.
I am going to talk about 2 studies that used different PET techniques. The first study used glucose metabolism to discern changes in the energy requirements or work of the brain when normal people performed various tasks.
We asked them to open their eyes and look at a scene, which activated the visual cortex; then, we asked them to listen to the mystery story, ''The Shadow.'' They also performed a simple motor task: they touched their thumb to their finger in a forward and rear direction. Some years ago, Jennifer Jones and I appeared before Congress and asked for $8.7 billion. At the end of her presentation, I showed a slide to remind Congress that we now have technologies to determine whether they were watching what Jennifer showed them and listening to what she said, whether they thought about it, whether they remembered it, and whether they would do anything about it. They did give the money.
The other study, at Washington University, used a different approach. We required a subject to perform the task for extended periods—20 or 30 min. A subject could perform a measurement and the task in 30 s. We could map in a single person all the things that we had mapped in multiple subjects. People looked at words, and activated the visual cortex. Then they spoke the words—a motor task that activated the motor cortex. They thought about the words, funnel cortex. This all led to what is called brain mapping, with not only PET, but MRI. We can actually watch a person perform tasks and watch the brain deal with them. We can watch the brain anticipate a task, receive information, sort through it and understand it, and execute a response.
A study of Huntington's disease started an unusual relationship. It was initiated by me and Dave Kuhl at UCLA. Huntington's disease is the textbook genetic disorder. It appears to have originated from one person, a sailor, and is a dominant hereditary disease of total penetration expressed in midlife. We began to look at people who were symptomatic for Huntington's disease with PET. Then we started to look at their children to see whether we could identify who carried the gene before having any symptoms. At the same time, Jim Gazella at Harvard began to use markers to try to find the gene. Gene hunters were looking for the gene, and we were using imaging to wander through the body to look for where the gene was expressed and what it did to organ systems.
We showed in asymptomatic children that we could identify metabolic abnormalities. The Huntington's patient has a severe metabolic deficiency. We could identify changes 7 yr before people ever had symptoms. Gazella finally identified the marker: the Huntington's gene was on the short arm of chromosome 4, and it was called the G8 probe (for Gazella's eighth try). He was lucky the first time. It took a lot of time after that to identify the gene, which has now been done.
Strangely, when we initially did this, about 30% of the patients were discordant; that is, our answer differed from Gazella's and the marker. People said that the metabolic mapping with PET must be wrong. Fortunately or unfortunately, we agreed with the geneticist Mendel. Our prediction agreed with Mendel, and Gazelle's did not. As it turned out, the initial marker was not very accurate. As its accuracy improved, there was complete concordance between PET and the genetic marker.
The next entry was in Alzheimer's disease. We began a project with Duke University in familial Alzheimer's. In Alzheimer's disease, the traditional imaging studies all produce normal results. But there was a hallmark of Alzheimer's disease with PET: a bilateral metabolic deficiency occurred in the parietal cortex and spread throughout the cortex, and we could watch it develop over time. We began a program in which people were examined to determine whether they carried the APOE4 protein or the gene that expressed that protean. These were people who had Alzheimer's in their families, but no symptoms, that is, no short-term memory deficits or cognitive decline at the time of the study. Patients who carried APOE4, which increases the risk of Alzheimer's disease, had the metabolic abnormality characteristic of Alzheimer's. People who did not carry APOE4 did not have the abnormality. We can probably see the expression of the gene developing in Alzheimer's patients 5 yr before they become symptomatic.
How can such things happen? Our organ systems have compensatory actions and reactions to disease. As a gene defect or any disease defect begins, the body begins to compensate. Parkinson's disease patients lose 70-90% of their dopamine before even the most minor symptoms can be identified. A biologic marker can identify a developing disease not only in the earliest symptomatic phases, but long before the disease produces alterations in organ function or loss of regulated function. For example, it is thought that breast cancer begins about 9 yr before we pick it up.
Discussant
Nora D. Volkow
Medical and Chemistry Departments
Brookhaven National Laboratory
Upton, NY
and
Department of Psychiatry
State University of New York
Stony Brook, NY
Coauthors
Joanna S. Fowler, Gene-Jack Wang,
S. John Gatley, and Yu-Shin Din
Medical and Chemistry Departments
Brookhaven National Laboratory
Upton, NY
Positron emission tomography (PET) allows the measurement of regional concentrations of positron-emitting isotopes. There are positron emitters for the natural elements of life (table 1), so compounds can be labeled without affecting their pharmacologic behavior.1 The noninvasiveness of PET provides a unique tool for drug investigations in humans. Until recently, the distribution of psychoactive drugs in the brain, their binding properties, and their effects on neurotransmitters have been investigated in laboratory animals or postmortem in humans. Pharmacologic studies in living humans have been limited to the assessment of a drag's behavioral effects or to the assessment of drug metabolism, concentration, and clearance in body fluids. With PET, it has become feasible to investigate the temporal course of the behavior of the drug (pharmacokinetics) and the mechanisms of action of the drag (pharmacodynamics) directly in the human brain. The short half-life of the positron emitters used with PET (table 1) enables repeated studies, to assess multiple biochemical characteristics affected by a drag, or longitudinal studies in a given subject. Various experimental strategies can be used with PET for the investigation of the effects of drags, including labeling of the drag itself, neurochemical measurements, and functional measurements. This paper illustrates the power of PET in the investigation of drags of abuse. Similar strategies can be applied to investigate therapeutic drugs.
LABELED DRUGS TO VISUALIZE DRUG DISTRIBUTION AND PHARMACOKINETICS
Several drugs of abuse have been labeled with positron emitters, usually carbon-11, and investigated in the human brain (table 2). That has enabled quantification of the (percentage) uptake of a drag in the brain, its regional distribution, its binding profile, and its pharmacokinetics. PET can also be used to assess the binding and pharmacokinetics of drugs in other organs.
The use of labeled drugs and PET in drug research is illustrated by the case of cocaine, which is considered to be among the most reinforcing of the drugs of abuse.2 The reinforcing effects of cocaine have been associated with its ability to block the dopamine transporter (DAT).3 Blockade of DAT by cocaine results in accumulation of dopamine (DA) in the synapse, which enhances the magnitude of the DA signal (figure 1). One unresolved question regarding cocaine like drugs is why there are compounds that block DAT with similar or higher affinity for DAT but are not reinforcing, as is the case for mazindol. 4,5 Another of these drugs, methylphenidate (Ritalin), has an affinity similar to that of cocaine for DAT. Although methylphenidate has been shown to have reinforcing
TABLE 1 Positron Emitters Most Commonly Used for PET Studies
|
Radioisotope |
Half-Life, min |
Decay |
|
Carbon-11 |
20.4 |
ß+ (0.960 MeV) |
|
Fluorine-18 |
110 |
ß+ (0.635 MeV) |
|
Oxygen-15 |
2 |
ß+ (1.73 MeV) |
|
Nitrogen-13 |
10 |
ß+ (1.19 MeV) |
TABLE 2 Drugs of Abuse Labeled with Positron Emitters and Imaged with PET
|
Drug class |
Labeled drug |
|
Psychostimulant |
|
|
Sedative hypnotic |
[11C]Flumazenil57 |
|
Opiate |
[11C]Morphine, [11C]heroin, [11C]codeine,58 [11C]buprenorphine59 |
|
Nicotine |
[11C]Nicotine60 |
effects when administered intravenously6 it is abused much less frequently than is cocaine.7 One question that arises is whether methylphenidate, the most frequently prescribed psychoactive drug in children in the United States, where it is used for the treatment of attention-deficit disorder,8 is as potentially addictive as cocaine or whether variables other than affinity for DAT influence the reinforcing effects. Of particular interest is whether these 2 drags differ in their pharmacokinetics, inasmuch as previous studies showed that the shorter the interval between intake and perceived effects of a drag, the greater its reinforcing effects.9,10
To address that issue, we compared the distribution and pharmacokinetics of cocaine and methylphenidate in the human brain by labeling cocaine11 and methylphenidate12 with 11C and then measuring their concentrations in the brain with PET.13 [11C]Cocaine and [11C]methylphenidate were almost identical in this distribution in the brain; both concentrated in the basal ganglia, the brain region with the highest DAT concentration. Furthermore, by performing pharmacologic experiments in baboons, we determined that both drags bound to DAT14,15 and that they bound either to the same DAT site or to an overlapping site, in that pretreatment with a pharmacologic dose of either drag was able to block the binding.
PET allows the measurement of not only the distribution of a drug in the brain but also the temporal course of the concentration of the drag in various brain regions, so it was possible to compare the pharmacokinetics of cocaine and methylphenidate directly. The time course for the distribution of [11C]cocaine and of [11C]methylphenidate at the level of the basal ganglia is shown in figure 2. Cocaine goes into the brain very rapidly, achieving peak concentration in 4-8 min, and it also clears from the brain rapidly; after 30 min, there is almost no activity. Methylphenidate, like cocaine, goes into the brain very rapidly, achieving peak concentration in 8-10 min, but its clearance from the brain is much slower than is that of cocaine; at 90 min, there is still a substantial amount of methylphenidate in the brain.
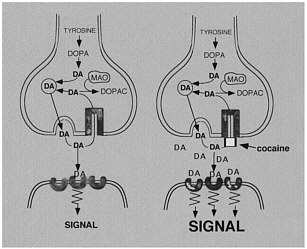
FIGURE 1 Effects of cocaine on DA synapse.
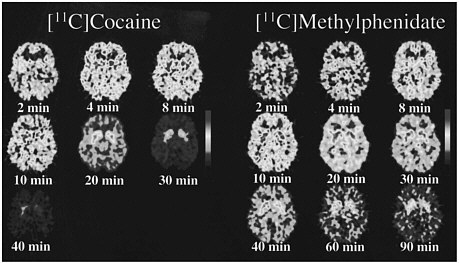
FIGURE 2 PET images obtained with [11C]cocaine and with [11C]methylphenidate shown for 1 plane centered at basal ganglia at different times after administration of radiotracer. Note faster clearance of [11C]cocaine than of [11C]methylphenidate.
Because PET-imaging studies can be done in human subjects, one can simultaneously measure the pharmacokinetics of the drag in the brain and the temporal course for "self reports" of drug effects. That permits assessment of the relationship between the 2 measures. Figure 3 shows the relationship between the kinetics of [11C]cocaine and of [11C]methylphenidate in the basal ganglia and the time course for self-reports of a "high," which is among
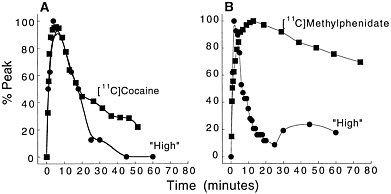
Figure 3 (A) Time-activity curve for [11C]cocaine in basal ganglia and temporal course for self-reported "high" induced by intravenous cocaine (0.6 mg/kg iv). (B) Time-activity curve for [11C]methylphenidate in basal ganglia and temporal course for "high" induced by intravenous methylphenidate (0.5 mg/kg iv). Adapted with permission from Ref. 13.
the best predictors for drug self-administration in humans.16 In the case of cocaine, both the fast uptake of the drug in the brain and the fast clearance parallel the time course of the short-lasting "high." In the case of methylphenidate, the initial perception of the "high" induced by its intravenous administration follows the fast uptake of [11C]methylphenidate in the brain. As for cocaine, the methylphenidate-induced "high" is very short lasting, and it returns to values close to baseline 30 min after administration, despite the drug's remaining in the brain for a much longer period. Thus, the ''high" induced by the intravenous administration of cocaine or of methylphenidate appears to be related to the fast uptake of the drug in the brain but not to its continuous presence there.
We postulate that the potential for a drug to induce addiction is due not only to its reinforcing effects but also to its ability to facilitate repeated administration. In the case of cocaine, fast uptake and clearance from brain, which correspond well with the short duration of the "high," promote repeated administration, which is characteristic of the binge behavior observed in cocaine addicts, who repeat the administration of cocaine every 30-40 min.17 In the case of methylphenidate, we postulate that, although fast uptake evokes the response of a "high" similar to cocaine's, slow clearance does not promote frequent repeated administration. If methylphenidate were taken repeatedly at the time that the "high" returns to baseline, it would rapidly lead to DAT saturation. On the basis of these imaging studies, it appears that it is not sufficient for a drug to block DAT rapidly, but that to enable repeated administration it has to clear from the brain rapidly. Therefore, one can postulate that one reason why cocaine is more reinforcing and more addictive than other drugs that block DAT is its fast pharmacokinetics.
NEUROCHEMICAL MEASUREMENTS
PET can be used to assess the effects of a drug on a neurotransmitter system, including its efficacy at its molecular target and its effects on neurotransmitter concentration.
Drug Efficacy
PET has been used to measure the efficacy of drugs of abuse in producing a specific biochemical effect. For example, in the case of cocaine, studies were conducted to quantify its ability to block DAT in the human brain and to determine the extent of blockade required for cocaine to be perceived as reinforcing.18 The studies used [11C]cocaine as a radiotracer for DAT. Subjects were repeatedly scanned after administration of a placebo and of different pharmacologic doses of intravenous cocaine. Pharmacologic doses of cocaine compete with [11C]cocaine for binding to DAT in proportion to the number of DATs that they occupy. By comparing the difference between [11C]cocaine binding obtained after administration of the placebo and that obtained after administration of pharmacologic doses of cocaine, one can calculate DAT occupancies with appropriate mathematical models. Figure 4 shows images obtained with [11C]cocaine after administration of a placebo and after administration of cocaine at 0.1 mg/kg and 0.3 mg/kg. Even the 0.1-mg/kg dose, which several of the cocaine abusers did not recognize as different from the placebo, substantially inhibited [11C]cocaine binding in striatum. Figure 5 shows the extent of DAT occupancy for 4 doses of intravenous cocaine—2 (0.05 and 0.1 mg/kg) that are not perceived as reinforcing and 2 (0.3 and 0.6 mg/kg) that are often used by cocaine abusers. For cocaine to be perceived as reinforcing, about 60% of DAT needed to be blocked. These results showed that cocaine is highly effective in inducing DAT blockade. In this study, it was also shown that the intensity of cocaine-induced "high" was significantly correlated with the extent of DAT blockade and that a substantial proportion of DAT had to be blocked for cocaine to induce a "high" (figure 6).
Chronic Effects
To understand the actions of drugs, it is important to recognize that the responses to drags can vary among individuals presumably as a result of differences in biochemical characteristics of their brains. That is relevant in the case of addiction because the chronic administration of drags of abuse can lead to biochemical changes that affect later responses to the drags. The response of the brain of an addicted subject to the drag causing the addiction is likely to be different from the response of the brain of a nonaddicted subject. It is relevant to
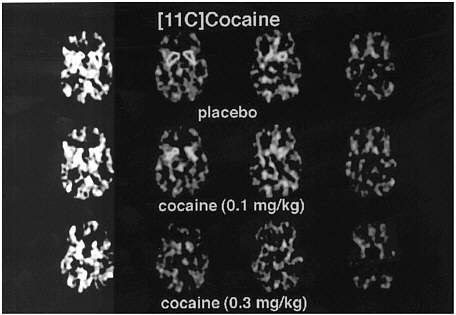
FIGURE 4 Images obtained with [11C]cocaine for 4 consecutive planes at basal gangliaafter administration of placebo or intravenous cocaine at doses of 0.1 mg/kg and 0.3 mg/kg. Notice marked reduction in binding of [11C]cocaine when pharmacologic doses of cocaine are coadministered with radiotracer.
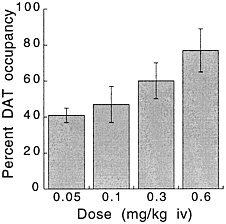
FIGURE 5 Extent of DAT occupancy induced by different doses of intravenous cocaine. Values are means and standard deviations.
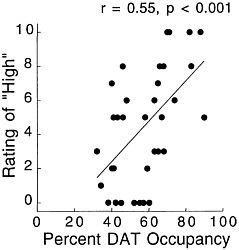
FIGURE 6 Correlation between DAT occupancy induced by cocaine and subjects' self-reported "high." Adapted with permission from Ref. 18.
investigate the biochemical changes induced by the chronic administration of drugs of abuse, because these changes are going to affect responses to the drug and might help one to understand the biochemical changes that underlie the process of addiction. A main neurotransmitter target in addiction is the DA system; increase in extracellular DA in the nucleus accumbens, the brain structure most associated with drag reinforcement, is crucial to the reinforcing effects of most drags of abuse.19,20 With PET, one can assess various components of the DA system—receptors, transporters, and synthetic and catabolic enzymes.21 Studies evaluating the effects of drags of abuse on the DA system have shown that chronic use of cocaine leads to long-lasting decreases in DA D2 receptors.22,23 Similar decrements in DA D2 receptors have also been documented in heroin addicts24 and in alcoholics.25 Studies of the effects of chronic cocaine use on DAT have been less consistent: Some studies report increases;26 others show no changes.27
Studies measuring the effects of drags of abuse on enzymes have focused mainly on enzymes responsible for DA synthesis (dopa decarboxylase)28 and dopamine metabolism (monoamine oxidase [MAO] A and B29 and catecholo-methyltransferase).30 Studies using [18F] to measure the rate of synthesis of DA have shown significant reductions in cocaine abusers compared with controls.31 Studies using [11C]deprenyl-D2 and [11C]clorgyline as radioligands to measure the concentration of MAO A and B have been conducted only in cigarette smokers.32,33 These studies showed that the concentration of MAO A and B was significantly lower in the brains of cigarette smokers than in the brains of nonsmoking subjects (see figure 7 for MAO B). Because subjects in these studies were asked to refrain from smoking for at least 4 h before being examined, the inhibition of MAO B cannot be accounted for by the presence of the drag itself. Figure 8 summarizes the data on brain concentration of MAO B in 8 nonsmokers (as controls), in 8 active smokers, and in 4 former smokers. Smokers had MAO A and B concentrations which averaged 30% and 40% lower than those of nonsmokers and former smokers. The effects are not due to a genetic difference between smokers and nonsmokers, inasmuch as they reverse once smoking is terminated. Inhibition of 1 main enzyme that metabolizes DA in cigarette smokers is likely to result in a higher concentration of DA at the DA terminal. Thus, one could postulate that the inhibition of MAO in cigarette smokers would raise the increases in DA concentration induced by drags, including nicotine. That, in turn, could increase the reinforcing effects of drugs of abuse, in that it is the increase in DA concentration in the nucleus accumbens that is associated with the reinforcing effects of most drugs of abuse.20 Those findings could explain why people addicted to other types of drags almost universally smoke.34
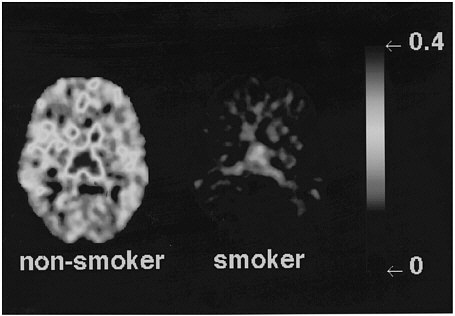
FIGURE 7 Images obtained with [11C]deprenyl-D2, tracer used to image concentration of MAO B, for 1 plane at basal ganglia in a healthy subject and in an active cigarette smoker.
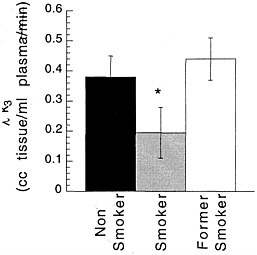
FIGURE 8 Brain concentration of MAO B, expressed as model parameter λk3, in 8 nonsmokers (as controls), 8 active cigarette smokers, and 4 former cigarette smokers.* p < 0.0001.
Neurotransmitter Release
It is possible to assess changes in intrasynaptic DA with PET by using DA D2 receptor ligands, such as [11C]raclopride, which have a relatively low affinity for DA D2 receptors, so that they compete with endogenous DA for binding to the receptor site.35,36 Drugs that increase DA concentration decrease the specific binding of raclopride, whereas drugs that deplete DA increase its binding.36,37,38 Furthermore, with a striatal-slice preparation, a very good correlation had been found between the frequency of electric stimulation and the displacement of [3H]raclopride from its binding sites; these effects are prevented by DA depletion with pharmacologic agents.39 The competition of endogenous DA with raclopride presented the opportunity to measure changes in synaptic DA in vivo with PET by observing the degree of reduction in the binding of [11C]raclopride by pharmacologic agents.40,41,42 In fact, the magnitude of the changes in DA concentrations assessed with microdialysis in the nonhuman-primate brain has been shown to correlate well with changes in [11C]raclopride binding simultaneously recorded in the same animals with PET.43 PET studies in humans have been done with [11C]raclopride to assess the effects of reinforcing drugs on synaptic DA concentration.44,45 For those studies, subjects were scanned twice—at baseline and after administration of the drugs that are known to increase DA synaptic concentration directly or indirectly (figure 9). Studies in normal controls have shown that the magnitude of the change in [11C]raclopride binding elicited by methylphenidate decreases as a function of age.44 Similar studies in cocaine abusers showed that the magnitude of methylphenidate-induced reduction in [11C]raclopride binding was half that observed in age-matched controls.46 Cocaine addicts also showed a blunted response to the methylphenidate-induced "high" compared with nonaddicted subjects. The blunting is likely to reflect decreased DA brain function in cocaine addicts. We cannot rule out the possibility that the blunting preceded the drug use in the cocaine addicts, but the findings illustrate how drug history can affect a person's response to a psychoactive drug.
FUNCTIONAL MEASUREMENTS
Regional brain function can be monitored with PET by measuring energy metabolism or by measuring cerebral blood flow (CBF). Brain metabolic rates for glucose and for oxygen are measured with 2-deoxy-2[18F]fluoro-d-glucose (FDG) and with oxygen-15-labeled oxygen, respectively. The most frequently used PET radiotracer for CBF measurements is 15O-labeled water.47 Because under normal conditions energy metabolism and CBF are tightly coupled with brain activity, the above tracers can be used to measure regional brain function.48 An advantage of FDG is that it reflects glucose metabolism, which is the main source of energy for the brain; a disadvantage is that it reflects metabolic activity over a 30-min period, so rapid processes might not necessarily be recorded. The main advantage of 15O-water is the feasibility of performing repeated studies in the same person within a relatively short period because of the short half-life of 15O (120 s). A disadvantage of measuring CBF is that many psychoactive drugs have vasoactive properties in addition to their direct effects on nervous tissue; this makes it difficult to separate the direct effects of the drug on the brain from its vasoactive effects.
Functional tracers can be used to investigate the effects of both acute and chronic drug administration and so enable a determination of which brain regions are the most sensitive to the effects of a given drug. With that information, one can predict which neurotransmitters are involved in producing the effects of the drug. Also, because the drug is given to subjects who are alert at the time of the study, associations can be made between the behavioral effects of the drug and its regional brain-metabolic consequences. Although the use of functional tracers to assess the effects of drugs might not be as precise in localizing the site of action of a drug as is the use of more-specific tracers, it nonetheless provides a measure that reflects the final consequences of the effects of the drug in the human brain. That is important because, even though the effects of a drug might involve a specific interaction with a receptor site, the secondary consequences of that interaction might be the ones relevant for its pharmacologic effects.
Brain Metabolism
A PET study of the effects of 2 sequential doses of methylphenidate on brain glucose metabolism, measured with FDG in normal controls, exemplifies the strategy. Two intravenous doses of methylphenidate were given 120
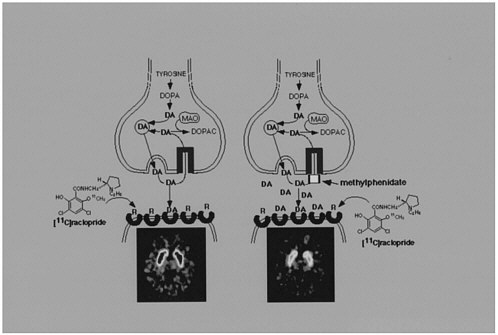
FIGURE 9 Use of PET to measure relative changes in DA concentration induced by methylphenidate with [11C]raclopride. Methylphenidate increases synaptic concentration of dopamine and occupies DA D2 receptors; this results in reduction in [11C]raclopride binding.
min apart to assess the effects of increases in DA concentration on regional brain metabolism. Parallel measurements were obtained of DA D2 receptors with [11C]raclopride to evaluate the extent to which the changes in regional brain glucose metabolism induced by methylphenidate could be related to the concentration of DA D2 receptors in a given subject. It was shown that methylphenidate's effects on global brain metabolism varied among subjects, increasing it in 6, decreasing it in 2, and having no effect in 7 subjects but consistently increasing cerebellar metabolism in all subjects. As a result, none of the regional changes was significant (except in the cerebellum). The metabolic changes induced by methylphenidate in the cerebellum and in the frontal and temporal cortices were significantly correlated with D2 availability. Frontal metabolism and temporal metabolism were increased in subjects with high D2 receptor density and decreased in subjects with fewer D2 receptors. The significant association between D2 receptors and metabolic changes in frontal and temporal cortices and in the cerebellum suggests that methylphenidate's metabolic effects in these brain regions are due in part to DA changes and that the difference in D2 receptors is one mechanism accounting for the variability in response to methylphenidate. The results also show that changes in brain DA concentration, as induced by methylphenidate, can lead either to increases or to decreases in cortical and subcortical metabolism. That corresponds well to the recognized role of DA as a neurotransmitter that modulates the activity of brain regions, enabling both excitatory and inhibitory signals.49 The significant correlation between methylphenidate-induced changes in metabolism and D2 receptors suggests that the effects of DA on brain activity depend in part on the state of the DA system, as had been shown in laboratory animals.50,51 The dependence of the response to methylphenidate on the state of the DA system could help to explain differences in vulnerability to psychostimulant administration.
Metabolic studies have also been done to assess the effects of chronic administration of drugs—cocaine, alcohol, and marijuana—on the human brain. The studies have shown different regional patterns of brain metabolic abnormalities, depending on the substance abused, and have shown different levels of recovery from the metabolic abnormalities on detoxification. For example, chronic cocaine administration and alcohol administration are associated with a marked reduction in frontal metabolic activity. In cocaine addicts, hypofrontality
persisted even after protracted withdrawal;52 in alcoholics, there was substantial recovery or detoxification.53 For cocaine abusers, the reduction in frontal metabolism has been shown to be accounted for in part by the decrease in DA brain function.53
Cerebral Blood Flow
Studies with CBF have been particularly useful in documenting cerebrovascular toxicity associated with substance abuse, as was initially documented with PET in by studies that showed widespread decrements in CBF in cocaine abusers.54 The imaging studies corroborated the clinical reports of cerebral strokes and hemorrhages in cocaine abusers and highlighted this toxic action of cocaine.55 Because repeated studies can be done on the same subjects at different times, they can be used to evaluate the response to therapeutic intervention. For example, imaging studies have documented that the decrements in CBF in cocaine abusers improve with the administration of buprenorphine.56 This exemplifies the value of imaging as a tool for assessing the efficacy of potential treatments for drag addiction.
CONCLUSION
Studies using PET have started to document the mechanisms of reinforcement of addictive substances and to delineate neurochemical changes in the brains of addicted subjects. The findings are still preliminary, but they indicate the potential of PET in substance-abuse research, including
-
The direct assessment of the behavior of drugs of abuse in the human brain. This is relevant because pharmacokinetics and pharmacodynamics can vary among animal species. It also enables the assessment of drug behavior directly in drag-addicted subjects.
-
The association of behavioral and neurochemical characteristics. Because PET studies are done in subjects while they are awake, they permit determination of the relationship between behavior and alterations in local neurotransmitter concentrations, metabolism, and flow. Studies can also be done to assess the relationship between the pharmacokinetics of a given drag and the time course of its pharmacologic effects.
-
The comparison of biochemical characteristics and drag responses between addicted and nonaddicted subjects.
-
Application in the development of therapeutic interventions for drag addiction.
ACKNOWLEDGMENTS
This research was supported in part by the U.S. Department of Energy Office of Health and Environmental Research under contract DE-ACO2-76CH00016 and by the National Institute on Drug Abuse under grants DA06891, DA09490-01, and DA06278.
REFERENCES
1. Fowler JS, Wolf AP, Volkow ND. New directions in positron emission tomography-Part II. In: Bristol JA, editor. Annual Reports in Medicinal Chemistry. San Diego: Academic Press; 1990. p 261-269.
2. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988; 242:715-723.
3. Ritz MC, Lamb RJ, Goldberg SR, Kuhar MS. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987; 237:1219-1223.
4. Chait LD, Uhlenhuth EH, Johanson CE. Reinforcing and subjective effects of several anorectics in normal human volunteers. J. Pharmacol. Exp. Ther. 1987; 242:777-783.
5. Chait LD, Uhlenhuth EH, Johanson CE. The discriminative stimulus and subjective effects of phenylpropanolamine, mazindol and D-amphetamine in humans. Pharmacol. Biochem. Behav. 1986; 24:1665-1672.
6. Parran TV, Jasinski DR. Intravenous methylphenidate abuse: prototype for prescription drug abuse. Arch. Intern. Med. 1991; 151:781-783.
7. National Institute on Drug Abuse, Community Epidemiology Work Group (CEWG). In: Epidemiologic Trends in Drug Abuse. Pub. No. (NIH). Washington DC: Department of Health and Human Services; 1995. p 95-3988.
8. Carrey NJ, Wiggins DM, Milin RP. Pharmacological treatment of psychiatric disorders in children and adolescents: focus on guidelines for the primary care practitioner. Drugs. 1996; 51:750-759.
9. Oldendorf WH. Some relationships between addiction and drug delivery to the brain. In: Frankenheim J, Brown RM, editors. Bioavailability of Drugs to the Brain and the Blood Brain Barrier. Research Monograph 120. Rockville, MD: National Institute on Drug Abuse; 1992. p. 13-25.
10. Balster RL, Schuster CR. Fixed-interval schedule of cocaine reinforcement: effects of dose and infusion duration. J. Exp. Animal Behav. 1973; 20:119-129.
11. Fowler JS, Volkow ND, Wolf AP, Arnett CD, Dewey SL, Schlyer DJ, MacGregor RR, Hitzeman R, Logan J, Bendriem B, Christman D. Mapping cociane binding sites in human and baboon brain in vivo. Synapse. 1989; 4: 371-377.
12. Ding Y-S, Sugano Y, Fowler JS, Salata C. Synthesis of the racemate and individual enantiomers of [11C]methylphenidate for studying presynaptic dopaminergic neurons with positron emission tomography. J. Labelled Cmpnd and Radiopharm. 1994; 34: 989-997.
13. Volkow ND, Ding Y-S, Fowler JS, Wang GJ, Logan J, Gatley JS, Dewey SL, Ashby C, Lieberman J, Hitzemann R, Wolf AP. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in human brain. Arch. Gen. Psychiatry. 1995; 52: 456-463.
14. Volkow ND, Fowler JS, Logan J, Gatley JS, Dewey SL, MacGregor RR, Schlyer DJ, Pappas N, King P, Wolf AP. Carbon-11-cocaine binding compared at sub-pharmacological and pharmacological doses: A PET study . J. Nucl. Med. 1995; 36:1289-1297.
15. Ding Y-S, Fowler JS, Volkow ND, Gatley SJ, Logan J, Dewey S, Alexoff D, Wolf AP. Pharmacokinetics and in vivo specificity of [11C]dl-threo-methylphenidate for the presynaptic dopaminergic neuron Synapse. 1994; 18:152-160.
16. Fischman MW, Foltin RW. Utility of subjective-effects measurements in assessing abuse liability of drugs in humans. British J. Addiction. 1991; 861:563-1570.
17. Johanson CE, Fischman MW. The pharmacology of cocaine related to its abuse. Pharm. Rev. 1989; 41:3-52.
18. Volkow ND, Wang G-J, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley S J, Pappas N, Hitzemann R, Shea K. Relationship between subjective effects of cocaine and dopamine transporter occupancy . Nature. 1997; 386:827-830.
19. Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996; 382:255-259
20. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:5274—5278.
21. Volkow ND, Fowler JS, Gatley JS, Logan J, Wang G-J, Ding Y-S, Dewey, SL. Evaluation of the human brain dopamine system with PET. J. Nucl. Med. 1996; 37:1242-1256.
22. Volkow ND, Fowler JS, Wolf AP, Shlyer D, Shiue Ch-Y, Albert R, Dewey SL, Logan J, Bendriem B, Christman D, Hitzemann R, Henn F. Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am. J. Psychiatry. 1990; 147:719-724.
23. Volkow ND, Fowler JS, Wang G-J, Hitzemann R, Logan J, Schlyer D, Dewey S, Wolf AP. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993; 14:169-177.
24. Wang G-J, Volkow ND, Fowler JS, Logan J, Abumrad NN, Hitzemann R J, Pappas NS, Pascani K. Dopamine D2 receptor availability in opiate-dependent subjects before and after naloxone precipitated withdrawal. Neuropsychopharmacology. 1997; 16:174-182.
25. Volkow ND, Wang G-J, Fowler JS, Logan J, Hitzemann RJ, Ding Y-S, Pappas NS, Shea C, Pascani K. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcoholism Clinical Experimental Research. 1996; 20:1594-1598.
26. Malison RT, Best SE, Zoghbi SS, Zea-Ponce Y, Baldwin RM, Charney DS, Hoffer PB, Seibyl JS, Price LH, Kosten TR, Innis RB. SPECT imaging of dopamine transporters during sustained cocaine abstinence with [123I]ß-CIT. J. Nucl. Med. 1996; 37:44P.
27. Volkow ND, Wang G-J, Fowler JS, Logan J, Hitzemann R, Gatley SJ, MacGregor RR, Wolf AP. Cocaine binding is decreased in the brain of detoxified cocaine abusers. J. Neuropsychopharmacology. 1996; 14:159-168.
28. Gjedde A, Reith J, Dyve A, Leger GC, Guttman M, Diksic M, Evans A, Kuwabara H. Dopa decarboxylase activity in the living human brain. Proc. Natl. Acad. Sci. U.S.A. 1991; 88: 2721-2725.
29. Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H, Aquilonius SM, Burjling P, Halldin C, Hartwig P, Leenders KL, Lundquist H, Oreland L, Stalnacke C-G, Langstrom B. Mapping human brain monoamine oxidase A and B with 11C-suicide inactivatoirs and positron emission tomography. Science. 1987; 235: 481-485.
30. Ding YS, Gatley SJ, Fowler JS, Chen R, Volkow ND, Logan J, Shea CE, Sugano Y, Koomen J. Mapping catechol-o-methyltransferase in vivo: initial studies with [18F]Ro41-0960. Life. Sci. 1995; 58: 195-208.
31. Baxter LR, Schwarts JM, Phelps M, Mazziotta JC, Barrio J, Rawson RA, Engel J, Guze BH, Selin C, Sumida R. Localization of neurochemical effects of cocaine and other stimulants in the human brain. J. Clin. Psychiatry . 1988; 49:23-26.
32. Fowler JS, Volkow ND, Wang G-J, Pappas N, Logan J, MacGregor RR, Alexoff D, Shea C, Wolf AP, Warner D, Zezulkova I, Cilento R. Neuropharmacological actions of cigarette smoke: brain MAO B inhibition. Nature. 1996; 379:733-738.
33. Fowler JS, Volkow ND, Wang G-J, Pappas N, Logan J, Shea C, Alexoff D, MacGregor RR, Schlyer DJ, Zezulkova I, Wolf AP. Brain monoamine oxidase A inhibition in cigarette smokers. Proc. Natl. Acad. Sci. U.S.A. 1996; 93: 14065-14069.
34. Henningfield JE, Clayton R, Pollen W. Involvement of tobacco in alcoholism and illicit drug use. Br. J. Addiction. 1990; 85:279-292.
35. Seeman P, Guan C, Niznik HB. Endogenous dopamine lowers the dopamine D2 receptor density as measured by 3H-raclopride: implications for positron emission tomography of the human brain. Synapse. 1989; 3:96-97.
36. Ross SB, Jackson DM. Kinetic properties of the accumulation of 3H raclopride in the mouse in vivo. Naunyn-Schmied Arch. Pharmacol. 1989; 340:6-12.
37. Young TL, Wong DF, Goldman S, Minkin E, Chen C, Matsumara K, Scheffel U, Wagner HN. Effects of Endogenous dopamine on kinetics of 3H-N-methylspiperone and 3H-raclopride binding in the rat brain. Synapse 1991;7:188-94.
38. Inoue O, Kobayashi K, Tsukada H, Itoh T, Langstrom B. Difference in in vivo receptor binding between 3H-N-methylspiperone and 3H-raclopride in reserpine-treated mouse brain. J Neural Transm 1989;85:1-10.
39. Gifford AN, Gatley S J, Ashby CR. Endogenous released dopamine inhibits the binding of dopaminergic PET and SPECT ligands in superfused rat striatal slices . Synapse. 1996; 22:232-238.
40. Dewey SL, Smith GW, Logan J, Brodie JD, Wei Y-D, Ferrieri RA, King P, MacGregor R, Martin PT, Wolf AP, Volkow ND, Fowler JS. GABAergic inhibition of endogeneous dopamine release measured in vivo with 11C-raclopride and positron emission tomography. J Neurosci 1992;12:3773-80.
41. Dewey SL, Smith GS, Logan J, Brodie JD, Fowler JS, Wolf AP. Striatal binding of the PET ligand 11
42. Hume SP, Myers R, Bloomfield PM, Opacka-Juddry J, Cremer JE, Ahier RG, Luthra SK, Brooks DJ, Lammertsma AA. Quantitation of carbon-11-labeled raclopride in rat striatum using positron emission tomography, Synapse 1992; 12:47-54.
43. Breier A, Su T-P, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentration. Proc. Natl. Acad. Sci. 1997; 94:2569-2574.
44. Volkow ND, Wang G-J, Fowler JS, Logan J, Schlyer D, Hitzemann R, Libermann J, Angrist B, Pappas N, MacGregor RR, Burr G, Cooper T, Wolf AP. Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse. 1994; 16:255-262.
45. Smith GS, Dewey SL, Logan J, MacGregor RR, Brimecombe J, Kasten M, King P, Pappas ND, Volkow N, Fowler J, Wolf AP. Opiate modulation of striatal dopamine release measured with Positron Emission Tomography and 11C-raclopride. Soc. Neurosciences Abstracts 23rd Annual Meeting 1993; 128.9, p 302.
46. Volkow ND, Wang G-J, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Pappas N. Decreased striatal dopaminergic responsivity in detoxified cocaine abusers. Nature. 1997; 386:830-833.
47. Raichle ME, Martin WRW, Herscovitch P, Mintun MA, Markham J. Brain blood flow measured with intravenous H215O. II. Implementation and validation. J. Nucl. Med. 1983; 24:790-798.
48. Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M: The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory procedure and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977; 28:897-916.
49. Cohen JC, Servan-Schreiber D. Context, cortex and dopamine: a connectionist approach to behavior and biology in schizophrenia. Psychological Reviews. 1992; 99:45-77.
50. Deminiere IM, Piazza PV, Le Moal M, Simon H: Experimental approach to individual vulnerability to psychostimulant addiction. Neurosci. Biobehav. Rev. 1989; 13:141-147.
51. Hooks MS, Jones GH, Smith AD, Neill DB, Justice JB: Response to novelty predicts the locomotor and nucleus accumbens dopamine response to cocaine. Synapse. 1991; 9:121-128.
52. Volkow ND, Hitzemann R, Wang G-J, Fowler JS, Wolf AP, Dewey SL. Long-term frontal brain metabolic changes in cocaine abusers. Synapse. 1992; 11:184-190.
53. Volkow ND, Wang G-J, Hitzemann R, Fowler JS, Overall JE, Burr G, Wolf AP. Recovery of brain glucose metabolism in detoxified alcoholics. Am. J. Psychiatry. 1994; 151:178-183.
54. Volkow ND, Mullani N, Gould L, Krajewski K, Adler S. Cerebral blood flow in chronic cocaine users. Brit. J. Psychiatry. 1988; 152: 641-648.
55. Levine SR, Welch KM. Cocaine and stroke. Stroke 1988; 19:779-783.
56. Levin JM, Mendelson JH, Holman LB, Teoh SK, Garada B, Schwartz RB, Mello NK. Improved regional cerebral blood flow in chronic cocaine polydrug users treated with buprenorphine. J. Nucl. Med. 1995; 36:1211-1215.
57. Pappata S, Samson Y, Chavoix C, Prenant C, Maziere M, Baron JC. Regional specific binding of [11C]RO 15 1788 to central type benzodiazepine receptors in human brain: quantitative evaluation by PET. J. Cereb. Blood Flow Metab. 1988; 8:304-313.
58. Hartvig P, Bergström K, Lindberg B, Lundberg PO, Lundqvist H, Långström B, Svärd H, Rane A. Kinetics of 11C-labeled opiates in the brain of rhesus monkeys. J. Pharmacol. Exp. Ther. 1984; 230:250-255.
59. Galynker I, Schlyer DJ, Dewey SL, Fowler JS, Logan J, Gatley SJ, MacGregor RR, Ferrieri RR, Holland MJ, Brodie J, Simon E, Wolf AP. Opioid receptor imaging and displacement studies with [6-O-[11C]methyl]-buprenorphine in baboon brain. Nucl. Med. Biol. 1996; 23: 325-331.
60. Bergstrom M, Nordberg A, Lunell E, Antoni G, Langstrom B. Regional deposition of inhaled 11C-nicotine vapor in the human airway as visualized by positron emission tomography. Clin. Pharmacol. Ther. 1995; 57:309-317.
Discussant
R. Edward Coleman
Professor of Radiology
Duke University Medical Center
Durham, North Carolina
Metabolic Characterization of Oncologic Disease
Positron emission tomography (PET) uses a specific device, the tomograph, for imaging the distribution of a positron-emitting radiopharmaceutical substance that has been administered to a patient. The substance includes a radionuclide that is produced by a cyclotron or generator and incorporated into a substrate, which results in its localization. The substance most widely used for this purpose is fluorine-18 fluorodeoxyglucose (FDG). The 110-min half-life of 18F permits its distribution from the site of production. A single regional site can supply FDG to multiple PET centers. PET has been demonstrated to have important applications in evaluating cardiac and neurologic diseases, but its major clinical application is in oncology, and its use in characterizing malignant diseases and the effect of therapy on them is increasing. In clinical ontology, FDG is used with PET to characterize glucose metabolism of tumors.
Instrumentation for PET has improved dramatically since its development by Michael Phelps and colleagues at the Mallinckrodt Institute of Radiology at Washington University School of Medicine in the early 1970s. Commercially available instruments have an intrinsic spatial resolution of 5 mm and simultaneously produce 30—45 contiguous slices with an axial span of about 15 cm. Imaging of an entire organ, such as the brain or heart, is now feasible from 1 bed position. Sequential longitudinal stepping of the data-acquisition software has made it possible to obtain PET scans of the whole body, which are particularly useful for oncologic applications. Recent developments in technology have resulted in dual-headed gamma cameras that can operate in a coincidence-detection mode that permits PET imaging. These instruments can also be used for routine nuclear-medicine studies. As this new technology is developed further, the number of institutions that can offer PET imaging will increase dramatically.
ONCOLOGIC APPLICATION
FDG imaging of tumors is performed when the patient is fasting to minimize competitive inhibition of FDG uptake by serum glucose. The FDG emission scans are made about 45 rain after the intravenous administration of FDG. A whole-body scan can be acquired in 30-45 min.
Intravenously administered FDG circulates in the blood and is transported across capillary membranes by a carrier-mediated transport process. FDG and glucose are phosphorylated by hexokinase, but FDG-6-phosphate cannot be metabolized through the glycolytic cycle and is accumulated intracellularly. Glucose-6-phosphate undergoes further metabolism through the glycolytic and Krebs cycles to CO2 and water. L. Sokoloff and colleagues1 have developed a mathematical model for relating the net transport of 14C deoxyglucose (DG) and accumulation of DG-6-phosphate in tissue to glucose transport and metabolism. This model, developed for rats and autoradiography, has been extended to humans and FDG PET.
Visual interpretation of the images can be complemented by semiquantitative analysis in clinical studies with FDG PET. These techniques provide an index of glucose metabolism because the amount of 18F in tissue at any time is monotonically related to glucose use. The standardized uptake ratio is a semiquantitative index that is obtained by normalizing the accumulation of FDG in the region of interest to the injected dose and patient body weight. This index of glucose metabolism can also be corrected for lean body weight (instead of total body weight) and serum glucose content, but these corrections have not been found necessary for evaluating some tumors, such as lung cancer.2
The first clinical applications of FDG PET were in the brain. G. DiChiro and colleagues3 demonstrated that the amount of accumulation of FDG in brain tumors distinguished histologically aggressive and less-aggressive tumors and could distinguish viable tumor and necrotic tissue after treatment. Many other tumors have been
studied with FDG PET. Although the largest number of clinical studies with FDG PET have been reported in lung cancer, a large body of information is available on several other malignancies. FDG PET has been demonstrated to be accurate for evaluating head and neck cancer, colorectal cancer, melanoma, lymphoma, breast cancer, hepatic neoplasms, and pancreatic cancer.
APPLICATION OF FDG PET IN LUNG CANCER
Background
The frequency of lung cancer is increasing worldwide. It is the most-common fatal cancer in men and women in the United States, where it claims 145,000 lives each year. The diagnosis and treatment of patients with lung cancer are fraught with problems that benefit from the use of PET. PET is particularly useful in a patient who has a solitary pulmonary nodule (for determining whether it is benign or malignant), in a patient who has documented lung cancer (for determining whether it has metastasized), and in a patient who has had lung cancer and therapy and has a residual mass (for determining whether the mass is cancer or a posttherapy scar).
Clinical Results
FDG PET has been demonstrated to be very accurate for evaluating pulmonary nodules. Most pulmonary nodules are detected with chest radiography, and patients with such nodules generally are referred for computed tomography (CT) for further evaluation. The nodules are generally indeterminate for malignancy on the CT scan. Some characteristics might lead to a diagnosis of a benign or malignant nodule, but most nodules are indeterminate. In an initial study from our institution, we demonstrated 100% sensitivity and 89% specificity of FDG PET for evaluating indeterminate nodules.4 A recently performed multi-institutional study has validated the high sensitivity and specificity of FDG PET for characterizing indeterminate nodules. Before the use of FDG PET, patients with these nodules went to biopsy or thoracotomy for a definitive diagnosis. In surgical series, 30-50% of the nodules that were removed at surgery were benign. The cost effectiveness of PET for evaluating solitary nodules has demonstrated a large savings nationally because its use precludes unnecessary surgery.
Lung cancer spreads first to the lymph nodes in the hilar areas and mediastinum. Autopsy studies have demonstrated a relationship between the size of lymph nodes and the likelihood of malignancy. However, small lymph nodes can contain cancer, and not all large lymph nodes are cancerous. A radiology-diagnostic-oncology group study compared CT and magnetic resonance imaging (MRI) for staging the mediastinum. These procedures were found to have accuracies in the 60-70% range. Several studies have demonstrated an 80-90% accuracy of FDG PET for staging the mediastinum, and these studies have demonstrated that PET is more accurate than is CT or MRI for staging the mediastinum. 5-7
Whole-body imaging with FDG PET provides information that cannot be obtained with any other imaging technique. It has been shown to be very effective in detecting distant disease that is not identified otherwise and in clarifying whether a tumor is present when other techniques yield abnormal results. We have recently shown that FDG PET is very accurate in identifying metastases to the adrenal glands, which are common sites for the spread of lung cancer.8 Other studies have shown that management changes occur in 30-40% of patients who undergo whole-body scans.6,9 The whole-body PET study is replacing multiple-imaging studies for staging malignancies (figure 1).
CT and MRI cannot adequately characterize posttreatment pulmonary abnormalities as persistent tumor, scarring, or necrosis. A tissue biopsy that is negative for tumor cannot be accepted as definitive, because of limitations in the accurate sampling of regions of tumor in the midst of scarring. Patients who have chest radiographic findings that indicate tumor recurrence can be accurately characterized with FDG PET10. Not only can PET be used to evaluate tumor survival, but it can also be used to evaluate distant recurrence through whole-body scanning.
A recent study has documented the cost effectiveness of whole-body PET in the evaluation of patients who have lung cancer.11 The use of FDG PET with CT results in large savings in health-care expenditures because of the avoidance of unnecessary surgery. These savings are realized without any effect on life span.
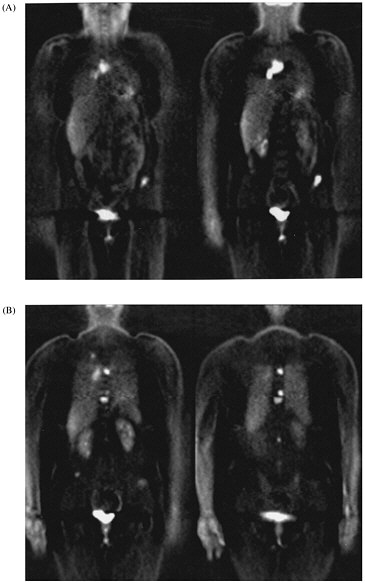
FIGURE 1. Selected coronal images from whole-body FDG PET scan of a 45-yr-old woman who presented with solitary pulmonary nodule and no other evidence of cancer or metastatic disease. (A) Images through anterior portion of body reveal metastases to lymph nodes in chest and to pelvis. (B) Images through posterior portion of body reveal small lung cancer in right lung and multiple vertebral body metastases.
SUMMARY
FDG PET is being used widely for characterizing neurologic and oncologic disease. Its use continues to increase as more data become available. Although initially used for characterizing the degree of malignancy of brain tumors and the effect of therapy on brain tumors, FDG PET is now being used to characterize most malignancies of the body and to follow the effects of therapy. Whole-body FDG PET is an excellent, cost-effective way to detect and stage malignancy.
REFERENCES
1. Sokoloff L, Reivich M, Kennedy C, et al. The C-14 deoxyglucose method for the measurement of local cerebral glucose utilization theory, procedure, and normal values in the conscious and anesthetized albino rat. Neurochemistry 1977; 28:897-899.
2. Lowe VJ, Duhaylongsod FG, Patz EF, Delong DM, Hoffman JM, Wolfe WG, Coleman RE: Pulmonary abnormalities and PET data analysis: A retrospective study. Radiology 1997; 202:435-439.
3. DiChiro G. Positron emission tomography using F(F-18)-fluorodeoxyglucose in brain tumors: a powerful diagnostic and prognostic tool. Invest Radiol 1986; 2:360.
4. Patz EF, Lowe V, Hoffman JM, Paine S, Burrows P, Coleman RE, Goodman PC: Evaluation of focal pulmonary abnormalities with 18F-fluoro-2-deoxyglucose and positron emission tomography. Radiology 188:487-490, 1993.
5. Wahl RL, Quint LE, Greenough RI, et al. Staging of mediastinal non-small cell lung cancer with FDG PET, CT and fusion images: Preliminary prospective evaluation. Radiology 1994; 191:371.
6. Valk PE, Pounds TR, Hopkins DM, and others. Staging non-small cell lung cancer by whole-body positron emission tomographic imaging. Ann Thorac Surg 1995; 60 (6):1573-1577.
7. Patz EF, Lowe VJ, Goodman PC, Herndon J. Thoracic nodal staging with PET imaging with 18FDG in patients with bronchogenic carcinoma. Chest 1995; 108:1617-1619.
8. Erasmus JJ, Patz EF, McAdams HP, Murray JG, Herndon J, Coleman RE, Goodman PC: Evaluation of adrenal masses in patients with bronchogenic carcinoma using 18F-fluorodeoxyglucose positron emission tomography. AJR 1997; 168:1357-1360.
9. Lewis P, Griffin S, Marsden P, et al: Whole-body18F-fluorodeoxyglucose positron emission tomography in preoperative evaluation of lung cancer. Lancet 1994; 1265-1266.
10. Patz EF, Lowe VJ, Hoffman JM, et al. Persistent or recurrent bronchogenic carcinoma: Detection with PET and 2-[18F]-2-Deoxy-D-glucose. Radiology 1994, 191:379-382.
11. Gambhir SS, Hoh CK, Phelps ME, Madar I, Maddahi J. Decision tree sensitivity analysis for cost-effectiveness of FDG-PET in the staging and management of non-small-cell lung carcinoma. J Nucl Med 1996; 37:1428-1436.
Discussant
Harvey R. Herschman
Department of Biological Chemistry and Medicinal Pharmacology
University of California at Los Angeles
U.S. Department of Energy
Laboratory of Structural Biology and Molecular Medicine
Molecular Biology Institute
UCLA Center for the Health Sciences
Los Angeles, California
Michael Phelps has described the principles of positron emission tomography (PET) and demonstrated how this technique can be used to monitor biochemical processes in living subjects. Our goal is to marry this powerful imaging procedure with modem molecular and cellular techniques to develop new paradigms that can be used both in basic research and in clinical applications. I will summarize the essentials of PET analysis necessary for the understanding of the proposed research, describe some of the striking accomplishments in studying the consequences of altering the genomes of experimental animals, describe some of the methods currently used by molecular and cell biologists to image the expression of reporter genes, and describe the attempt to merge these molecular and cellular procedures by using reporter genes for noninvasive imaging of gene expression in living animals.
PET IN ANALYZING BIOCHEMICAL FUNCTIONS IN LIVING SUBJECTS
PET imaging uses positron-emitting molecules that are injected intravenously in trace quantifies to allow us to observe and measure the biochemical functionality of tissues in living subjects. Typically, either positron-labeled ligands for cellular receptors or positron-labeled substrates for intracellular enzymes are used as PET probes. Retention of the positron-labeled PET probe in target tissues is detected with the tomograph, as a result either of the radiolabeled ligand binding to a receptor or of the conversion of the radiolabeled enzyme substrate to a ''trapped'' metabolic product. In the case of positron-labeled ligands for receptors—such as 3-(2'-[18F] fluoroethyl) spiperone ([18F]FESP), a ligand for the dopamine (DA) D2 receptor—the ligand is injected intravenously, and images are collected dynamically in the tomograph. Tissues rich in D2 receptor bind and retain the [18F]FESP ligand and are identified in living subjects with tomographic analysis. In the case of positron-labeled substrates for enzymes, the injected substrate is converted to a product that cannot escape from cells. For example, [18F]FDG is transported into, phosphorylated in, and retained in tissues in proportion to the glycolytic rate.
ADVANCES IN UNDERSTANDING OF GENE EXPRESSION IN VIVO AND OF CONSEQUENCES OF ALTERING PATTERNS OF GENE EXPRESSION IN LIVING ANIMALS
Role of Transgenic Mice in Study of Normal And Abnormal Phenotypes
It is now possible to add genes to mouse embryos to create mice that can express human genes. One of the first and most-dramatic examples is a mouse that overexpresses the growth-hormone gene.1 Overexpression of a human growth-hormone gene in the germ line of the mouse caused mice to grow to sizes substantially greater than those of littermates that did not have the transgene.
If we place genes that are altered in human diseases into mouse embryos, we can often make murine models of human diseases. For example, many patients suffer from leukemia as the result of a rearrangement of the genes that encode for antibodies. When a rearranged "oncogene" is placed in the germ line of mice, the animals develop leukemia. 2 Such experiments satisfy a modem version of Koch's postulates, in that the rearranged gene is a causal agent in the induction of leukemia.
Use of Mice Carrying Human Genes Responsible for Diseases to Test Potential Therapies Without Ethical and Legal Problems Encountered in Studies of Human Patients
The gene for amyotrophic lateral sclerosis (Lou Gehrig disease) causes a similar degenerative disease of the peripheral nervous system in mice.3 Studies on these mice have accelerated the regulatory approval of several drugs to modulate the effects of the human disease.4
The altered portion of the human gene responsible for Huntington disease has recently been transferred into mouse embryos. The animals carrying the Huntington mutation develop tremors, weight loss, and so on, and appear to develop a disease similar to that observed in humans.5 Plans are under way to begin tests of a variety of therapies for the mouse disease.
Homologous Recombination to Substitute Altered Sequences for Endogenous Genes of Mouse Germ Line: An Important New Method to Analyze Gene Function in Living Animals
We now have the ability to change individual nucleotides or base pairs of a given gene in a mouse embryo (as opposed to inserting a human gene) to create mice that have the chosen substitutions—mice that will pass on a synthetic mutation to their progeny. We can directly alter the 1 or 2 nucleotides of our choice in the 3 billion base pairs that make up the mouse genome. To illustrate the nature of this accomplishment, if each nucleotide were 0.1 in. long, the line that represents the mouse genome would wrap twice around the earth—50,000 mi. By using our ability to make such specific substitutions to create mice with these changes in their genes, we can model many human diseases. For example, we can create mice that have the gene for cystic fibrosis. Those mice can be used to study responses to therapy.6
Sometimes we get surprised by the results of experiments. Michael Greenberg and his associates were studying the role of a gene called FosB, and they wanted to know whether this gene is essential for viability. When they made a mouse strain with a mutation in the FosB gene,7 they found the animals to be normal in all respects that they assayed, except one: The mother mice would not nurture their young, and the babies perished unless transferred to foster mothers. That has led to the beginnings of molecular analysis of an entire behavioral process—nurturing behavior in mothers.
Those are just a few examples of how current cell biology and molecular biology have been extended into of the study of whole-animal physiology. How can these techniques be coupled with PET to allow the noninvasive study of the consequences of altering gene expression in living animals?
IMAGING TECHNIQUES CURRENTLY USED IN MOLECULAR AND CELL BIOLOGY TO STUDY REPORTER-GENE EXPRESSION
The light microscope revolutionized biology. Investigators could now see what had previously been only intellectual constructs. The electron microscope revealed entirely new dimensions of cell structure. Guided by the electron microscope, the field of cell biology emerged from biochemistry. The development of dynamic, noninvasive imaging techniques, such as PET, led to the marriage of imaging and biochemistry and allowed researchers and clinicians to study biochemical reactions in living animals and in patients.
Current Methods For Imaging Genome Function In Vertebrates
Developmental and behavioral biologists interested in imaging the expression of individual endogenous genes are restricted to immunochemical detection of protein products or in situ hybridization of mRNA. One must biopsy or, more commonly, sacrifice an animal in either case. Consequently, one usually obtains only a single "snapshot" from each subject.
Researchers studying the molecular basis of transcriptional gene activation use chimeric fusion-gene constructs in which the promoter-regulatory region of the gene under investigation is fused to a "reporter gene" whose protein product (such as luciferase or b-galactosidase) can be assayed biochemically or imaged with immu
nochemical or histochemical techniques. The fusion genes are placed in the germ line of "transgenic" mice, rats, pigs, or sheep to create new strains of animals. Using site-directed mutagenesis, researchers alter the promoter-regulatory regions of fusion genes, create transgenic animals, and characterize the regulatory elements that control expression of individual genes in vivo. With these procedures, investigators can study the mechanisms of regulation of the individual genes that make up the genome. However, using current enzymatic, immunochemical, or histochemical analysis of reporter-gene products, one must sacrifice the animal; again, one obtains only a single snapshot from each animal.
Green Fluorescent Protein And Monitoring Of Gene Expression In Living Preparations
All current methods to image expression either of endogenous genes or of transgenic reporter genes in rodents, primates, or humans qualitatively or quantitatively require the use of fixed tissue, either from biopsies or from sacrificed animals. Use of green fluorescent protein (GFP) as a reporter gene has recently provided a means to monitor expression of transplanted DNA in living cells repeatedly.8 However, the utility of GFP as a reporter is limited to biologic preparations that are transparent to visible light, such as tissue-cultures cells, yeast, and Caenorhabditis elegans. Our goal is to develop imaging technology to monitor expression of reporter genes repeatedly in living experimental vertebrates and in patients.
DEVELOPING PET-BASED IMAGING TECHNIQUES TO EXAMINE REPORTER-GENE EXPRESSION IN LIVING SUBJECTS
Pet Reporter-Gene-Pet Reporter-Probe Systems For Imaging Reporter-Gene Expression In Living Animals
We are developing a means to "mark" transplanted cells or DNA delivery systems with PET reporter genes (PRGs)—genes whose protein products are either receptors for positron-labeled PET reporter probes (PRPs) or enzymes that metabolize PRPs. When animals or patients that express a PRG are given an injection of the corresponding PRP, cells that express the gene will sequester the probe. In contrast, cells that do not express the reporter gene will not retain the reporter probe (figures 1 and 2). Tomographic imaging will demonstrate the PRG
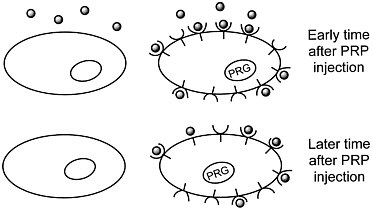
Figure 1 Sequestration of a positron-labeled PET reporter-ligand probe by a cell containing a PET reporter gene that encodes the ligand-binding receptor.
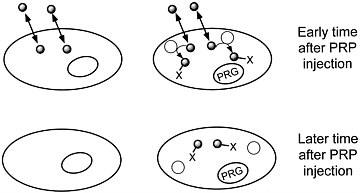
FIGURE 2 Sequestration of a positron-labeled PET reporter-substrate probe by a cell containing a PET reporter gene that encodes an enzyme that metabolizes the substrate.
dependent sequestration of the PRP. Experimenters and physicians will be able repeatedly to monitor, in living subjects, the tissue localization and level of expression of the PRG. Ideal PRGs should encode enzymes or receptors not normally present in mammalian tissues.
Herpes Simplex Virus Thymidine Kinase Enzyme and its Substrates for PRG-PRP Systems
The herpes simplex virus thymidine kinase enzyme (HSV tk) phosphorylates both thymidine and a number of guanine analogues (such as acyclovir and gancyclovir). In contrast, murine and human cellular thymidine kinases phosphorylate the guanine analogues only minimally. J.G. Tjuvajev and co-workers9 have demonstrated the utility of HSV tk as a reporter gene in the mouse autoradiographically, using several radiolabeled guanosine analogues as substrates. We are working on procedures to use HSV tk and 18F-labeled substrates as PRG-PRP combinations to image gene expression in vivo repeatedly and noninvasively.
DA D2 Receptor And FESP as a PRG-PRP System
We have used the DA D2 receptor as an alternative PRG first, because its normal expression at high concentrations is limited primarily to the brain; second, because a DA D2 receptor reporter gene will not be immunogenic; and third, because the nuclear medicine group the University of California, Los Angeles (UCLA), developed [18F]FESP to image the DA D2 receptor in vivo in rodents, nonhuman primates, and humans.10 The use of [18F]FESP as a PRP requires no further research on probe development. Moreover, the mathematical tracer-kinetic model for this PRG-PRP system was developed at UCLA. We have developed the DA D2 receptor and [18F]FESP as a PRG-PRP combination to image gene expression in vivo repeatedly.
Potential Role of Repeated Noninvasive Imaging of Gene Expression in Patient Care and Biomedical Research
The most-immediate potential practical application of the PRG-PRP procedure is in gene therapy. In 1 gene-therapy approach, cultured cells are transfected with a therapeutic gene and then transferred to the patient. In this adoptive-cell gene-therapy paradigm, it is difficult to determine whether the transplanted cells reach their target site, how long the transplanted cells remain viable, and whether expression of the therapeutic gene is later
compromised. In a second gene-therapy approach, DNA packaging and delivery systems (such as viruses and liposomes) that contain therapeutic genes are introduced directly into the patient; the packaging system is intended to target the therapeutic gene to the appropriate cells. Once again, after administration, the physician has no way to monitor localization or expression of the therapeutic gene. PRGs, administered in conjunction with therapeutic genes, will permit physicians to use PRPs and tomographic analysis for repeated monitoring of the localization, proliferation, and function both of cells used in adoptive-cell gene therapy and of DNA-delivery systems administered directly to patients. HSV tk, in conjunction with pharmacologic concentrations of gancyclovir, is being used in many active human-gene-therapy protocols. The availability of patients undergoing clinical trials for HSV-tk-based therapeutic protocols will facilitate investigation of the clinical utility of HSV tk as a PRG.
Reporter genes have been used in transgenic animals to analyze the relationship between genomic regulatory DNA sequences and gene expression and the effects of developmental and environmental manipulation on gene expression. In both contexts, analysis of reporter genes, such as b-galactosidase and luciferase, is limited to measurements at single times in biopsies or in tissue preparations from sacrificed animals. With PRGs and positron-labeled PRPs in transgenic animals, and new, high-resolution PET scanners dedicated to small-animal imaging, it will be possible to monitor repeatedly, in the same animal, time-dependent developmental, environmental, and experimental influences on gene expression.
REFERENCES
1. Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, Rosenfeld MG, Birnberg NC, Evans RM. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature 1982;300:611-615.
2. Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eµ-myc transgenie mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 1988;167:353-371.
3. Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994; 145:1271-1279.
4. Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Annals of Neurology 1996;39:147-157.
5. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996;87:493-506.
6. Clarke LL, Gawenis LR, Franklin CL, Harline MC. Increased survival of CFTR knockout mice with an oral osmotic laxative. Lab. Anim. Sci. 1996;46:612-618.
7. Brown JR, Ye H, Bronson RT, Dikkes P, Greenberg ME. A defect in nurturing in mice lacking the immediate early gene fosB. Cell 1996;86:297-309.
8. Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science 1994;263:802-805.
9. Tjuvajev JG, Stockhammer G, Desai R, Uehara H, Watanabe K, Gansbacher B, Blasberg RG. Imaging the expression of transfected genes in vivo . Cancer Research 1995;55:6126-6132.
10. Bahn MM, Huang SC, Hawkins RA, Satyamurthy N, Hoffman JM, Barrio JR, Mazziotta JC, Phelps ME. Models for in vivo kinetic interactions of dopamine D2-neuroreceptors and 3-(2'-[18F]fluoroethyl) spiperone examined with positron emission tomography. J. Cereb. Blood Flow Metab. 1989;9:840-849.

























