
Page 193
7
Mechanisms of Toxicity
In this chapter, the subcommittee summarizes what is known about the mechanisms of toxicity for arsenic. The chapter is divided into two major sections-cancer and noncancer effects. In the cancer section, the subcommittee summarizes results from in vivo and in vitro bioassays designed to investigate the role of arsenic and metabolites of arsenic, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA), as tumor promoters and initiators. That summary is followed by a discussion of the data on modes of action for arsenic-induced carcinogenesis and how those data can help delineate the slope of the dose-response curve at low exposure concentrations. In the noncancer section, the subcommittee summarizes what is known about the mechanisms of action leading to noncancer effects. The potential relationships between mechanisms of arsenic-induced cell injury or cell death and carcinogenic processes are discussed with particular attention to the inter-relationships between arsenic-induced formation of reactive oxygen species or oxidative stress and chromosomal damage in target-cell populations. The roles of other documented arsenic-induced cellular responses, such as alterations in the heme biosynthetic pathway, alterations in cellular gene expression, and inhibition of DNA-repair enzyme activities, are discussed as components of the broad spectrum of cellular responses to arsenic exposure.
Cancer Effects
Results from Bioassays
Inorganic Arsenic
In general, long-term studies on the carcinogenicity of arsenic in labora-
Page 194
tory animals have yielded negative results. In one of the more extensive studies, which was carried out by the FDA, rats were administered sodium arsenite at concentrations up to 250 ppm or sodium arsenate at concentrations up to 400 ppm in the diet for 2 years. No increases in tumors were observed (Byron et al. 1967). A similar negative finding was noted in beagles maintained on diets containing sodium arsenate or sodium arsenite at 5-125 ppm for 2 years; however, the high dose proved to be lethal (Byron et al. 1967). In another study, rats were administered lead arsenite at concentrations of 463 or 1,850 ppm (arsenic at approximately 400 ppm) or sodium arsenite at a concentration of 416 ppm in the diet (based on body weight and food consumption data, equivalent to approximately 18-20 mg/kg of body weight per day) for 29 months with some indication of toxicity, but no increase in tumorigenicity when compared with controls (Kroes et al. 1974). In a study of 20 Cynomolgus monkeys, arsenic was administered by mouth as sodium arsenate at a dose of 0.1 mg/kg per day, 5 days per week, for 15 years. None of the animals developed malignant tumors (Thorgeirsson et al. 1994).
Animal studies on the carcinogenicity of arsenic administered via drinking water have also been conducted. In rats, sodium arsenite administered at a concentration of 5 mg/L for a lifetime did not cause an increase in tumors (Schroeder et al. 1968). The authors calculated that the concentration was equivalent to a dose of 0.38 mg/kg of body weight per day. Although some evidence of arsenic accumulation was found in the animals, it had no effect on survival (Schroeder et al. 1968). In a similar study, rats received sodium arsenate at a concentration of 5 mg/L (approximately 350 µg/kg of body weight per day) for a lifetime and had no indication of tumorigenicity (Kanisawa and Schroeder 1969). In a shorter study examining the effect of arsenic on mammary tumors, Schrauzer and Ishmael (1974) administered sodium arsenite to mice at 10 mg/L in drinking water for 16 weeks. The incidence of mammary tumors was lower in the tested animals (27%) than in the controls (82%). However, they found that arsenic increased the size of spontaneous mammary tumors. In a study, which has not yet been peer reviewed and is available only in abstract form, Ng et al. (1998) reported preliminary results indicating that the administration of sodium arsenate in drinking water (0.500 mg/L) to C57B1/6J and metallothionein knock-out mice for up to 26 months caused tumors of the gastrointestinal tract, lungs, liver, spleen, bone, skin, reproductive system, and eye. No tumors were observed in the control groups of the study.
Several animal studies have examined the interaction of arsenic with other chemicals to determine if it might be acting as a promoter rather than as an initiator of tumors. Baroni et al. (1963) administered arsenic trioxide as a 0.01 % solution in drinking water or sodium arsenate by skin application (two
Page 195
drops of a solution of 15.8 g/L twice weekly) to mice. Each of these treatments was tested in combination with skin application of croton oil (to test for promoting action), and after initiation with a single topical application of 7,12-dimethylbenz[a]anthracene or with administration of ethyl carbamate by intubation (to test for promoting action). There was no indication that either arsenic trioxide or sodium arsenate was acting as an initiator or promoter.
Kroes et al. (1974) found that the administration of arsenic in the diet of rats (lead arsenate at 463 ppm or sodium arsenate at 416 ppm) for 29 months did not enhance the carcinogenicity of diethylnitrosamine (DENA) (5 µg per day administered by intubation 5 days per week). When three strains of mice were administered arsenic trioxide (0.01%) in their drinking water for 8 weeks and then subjected to topical application of methylcholanthrene, arsenic decreased the number of papillomas produced in CxC3H mice (Milner 1969). When arsenic trioxide was administered at 2 ppm in the drinking water of mice for a lifetime with or without selenium at 2 ppm, arsenic alone caused no increase in the incidence of mammary tumors, but some increase occurred in the growth and multiplicity of the tumors (Schrauzer et al. 1978). Selenium was protective in that the incidence of mammary tumors in the group given selenium alone was significantly lower than the control group. However, no protection was observed when selenium was administered with arsenic because the incidence of mammary tumors increased in the co-exposed group compared with the control group.
Shirachi et al. (1983) administered sodium arsenite at a maximum tolerated dose of 160 ppm in drinking water to partially hepatectomized rats with or without a single dose of diethylnitrosamine (30 mg/kg of body weight intraperitoneally). Although 1 of 7 rats administered DENA without arsenic developed kidney tumors at the end of 25 weeks, 7 of 10 administered DENA with arsenic developed kidney tumors, suggesting that arsenic acted as a promoter. In a statistical reanalysis of the data, Smith et al. (1992) concluded that even in rats administered arsenic without DENA, liver and kidney tumors increased for all three forms of arsenic—arsenite, arsenate, and DMA.
Starting with newborn mice, Laib and Moritz (1989) administered sodium arsenite (2 mg/kg per day) or sodium arsenate (20 mg/kg per day) 5 days per week for 3 weeks with or without DENA (2 mg/kg) and found an increase in ATPase-deficient foci in the livers of the mice given arsenite with DENA when compared with the mice given DENA alone or arsenate with DENA, suggesting a cocarcinogenic effect of the arsenite but not the arsenate.
Monomethylarsonic Acid (MMA)
In a study submitted to the EPA Office of Pesticides Program, male and
Page 196
female Sprague-Dawley rats were fed MMA in the diet at concentrations of 0, 25, 50, 100, and 200 ppm for 2 years. There was an increase in thyroid tumors in the males but only at the highest dose (EPA 1981). In another report, when Fischer 344 (F344) rats were fed MMA at concentrations of 0, 50, 400, and 1,300 ppm (equivalent to 0, 3.2, 27, and 93 mg/g per day for the males and 0, 3.8, 33, and 101 mg/kg per day for the females) for 2 years, there was a suggestion of an increased incidence of parathyroid gland adenomas at 400 and 1,300 ppm in the male rats and in the females at 1,300 ppm, but the statistical significance depended upon the test used (EPA 1992a).
Another study showed no evidence of carcinogenicity in male and female B6C3F1 mice fed MMA in the diet at concentrations of 0, 10, 50, 200, and 400 ppm (equivalent to 0, 1.8, 9.3, 38, and 83 mg/kg per day for the males and 0, 2.2, 12, 46, and 104 mg/kg per day for the females) for 2 years (EPA 1992b).
Dimethylarsinic Acid (DMA)
The arsenic metabolite DMA, which is the herbicide cacodylic acid, has been tested for its effects in vivo and in vitro. In studies on the tumorigenicity of DMA, Innes et al. (1969) found no evidence of tumorigenicity after administering DMA orally to two strains of mice at a daily dose of 46.4 mg/kg of body weight (maximum tolerated dose) for 18 months.
In a study for EPA (1992c), male and female F344 rats were fed DMA at concentrations of 0, 2, 10, 40, and 100 ppm in the diet (equivalent to 0, 0.14, 0.73, 2.8, and 7.3 mg/kg per day for the males and 0, 0.16, 0.79, 3.2, and 8.0 mg/kg per day for the females) for 2 years. The results showed a carcinogenic response as evidenced by increases in transitional-cell papillomas and carcinomas at the highest dose in both sexes. In the bladder, urinary transitional-cell hyperplasia and vacuolar degeneration occurred in a doserelated manner at the concentrations of 40 and 100 ppm. The study was judged to be minimally acceptable by EPA (1992c, 1993).
When male and female B6C3F1 mice were fed DMA at concentrations of 0, 8, 40, 200, and 500 ppm for 2 years, vacuolar degeneration of the urinary bladder transitional epithelium increased at 200 and 500 ppm in both sexes. There were no increases in urinary bladder tumors. This submitted study was judged by EPA to be inadequate because it was decided that higher doses of DMA could have been used (EPA 1991, 1992d).
As reported in the Integrated Risk Information System (IRIS) of EPA, cacodylic acid (DMA) is classified as D—not classifiable as to human carcino-
Page 197
genicity. The basis for this classification by the agency is that there are ''no human data and inadequate data in animals" (EPA 1998).
In a complex study by Yamamoto et al. (1995), rats were administered DENA, N-methyl-N-nitrosourea, 1,2-dimethylhydrazine, N-butyl-N-(4hydroxybutyl)nitrosamine and N-bis(2-hydroxypropyl)nitrosamine and were given DMA in drinking water at concentrations of up to 400 ppm for 30 weeks. DMA alone was without effect, but in the five-carcinogen test group, it significantly enhanced the formation of tumors of the urinary bladder, kidney, liver, and thyroid gland and increased preneoplastic lesions in the liver. In a study by Yamanaka et al. (1996), DMA was administered at concentrations of 200 or 400 ppm in the drinking water of ddY mice for 25 weeks following initiation with 4-nitroquinoline 1-oxide (4NQO). DMA alone was without effect, but DMA at 400 ppm administered with 4NQO increased the number of lung tumors per animal when compared with 4NQO administered alone. In a further study, rats were administered N-(4-hydroxybutyl)nitrosamine for 4 weeks and then given DMA at 0, 2, 10, 25, 50, and 100 ppm in drinking water for 32 weeks (Wanibuchi et al. 1996). Again, no neoplastic lesions were observed in rats administered DMA alone. Urinary bladder tumors increased in rats given N-(4-hydroxybutyl)nitrosamine followed by DMA at 10 ppm or higher. The 5-bromo-2'-deoxyuridine labeling index also increased. Collectively, the studies suggest that DMA is not an initiator, but it might be a promoter.
Yamanaka et al. (1989) found that DMA administered orally at 1,500 mg/kg caused DNA strand breaks in mouse lung. That dose is extremely high. In fact, it exceeds the LD50 (the lethal dose for 50% of the test animals) of 1,200 mg/kg of body weight in mice reported by other investigators (Kaise et al. 1989). Similarly, incubation of human lung cells (L-132) with 10 mM of DMA for 10 hr caused single-strand breaks (Tezuka et al. 1993). Cross-linking between DMA and nuclear proteins was also observed when L-132 was incubated with 10 mM of DMA (Yamanaka et al. 1993). The investigators have suggested that those effects might be related to the formation of active oxygen species (Yamanaka et al. 1991; Rin et al. 1995). Other investigators have shown that DMA in vitro at concentrations ranging from 62.5 to 250 mg/L induces tetraploids in Chinese hamster cells in a concentration-dependent manner (Endo et al. 1992).
Relevance of Findings of Bioassays to Humans
The findings of the rodent bioassays for inorganic arsenic are generally uniformly negative. Given the unquestionable oncogenic activity of inorganic
Page 198
arsenic in humans, it would at first appear that these studies are of no value in helping to elucidate the carcinogenic mode of action of arsenic. However, in line with the evidence suggested in the sections that follow, such negative data may be supportive of a non-genotoxic mode of action. Certainly if arsenic were a direct and genotoxic carcinogen, one would expect to find positive results in at least one of these assays. The fact that there are species differences with rodents being resistant suggests that understanding the reason for this difference may shed some light on the mode of action.
The very high doses of DMA used in animal bioassays in vivo bring into question whether the low arsenic concentrations in human exposures exert their tumor-promoting effects via the metabolite DMA. At low exposure concentrations, humans would not metabolize arsenic to DMA at the concentrations needed to promote the tumor production observed in the animal studies. Compared with inorganic arsenic, the DMA formed would also be expected to be excreted rapidly (see Chapter 5). In addition, as discussed below (see Mode of Action for Carcinogenicity), in vitro studies suggest that arsenite is orders of magnitude more potent than DMA in the induction of chromatid breaks and gaps in cultured human fibroblasts (Oya-Ohta et al. 1996). Recent studies by M.M. Moore et al. (1997) also compared the relative potentials of sodium arsenite, sodium arsenate, MMA, and DMA for mutagenic and clastogenic activities by using the L5178Y/TK+/- mouse lymphoma assay and found the organic arsenicals to be orders of magnitude less potent in producing genotoxicity.
Mode of Action
For the purposes of cancer risk assessment, it is important to distinguish between genotoxicity and mutagenicity when discussing mechanisms underlying the formation of tumors. Genotoxicity is the broader term that also encompasses cellular effects that are not themselves heritable. Those effects include DNA and protein adducts, sister chromatid exchanges (SCEs) and unscheduled DNA synthesis. In addition, abnormalities in DNA methylation, although not strictly a genotoxic effect, also could be included as a form of potentially reversible DNA modification. Mutagenicity specifically describes the production of changes in DNA that can be transmitted from generation to generation (for organisms and cells). Cancer is a genetic disease that requires the accumulation of mutations in several genes (most notably, oncogenes and tumor-suppressor genes) in a single cell to progress to a tumor. Thus, mutations, either directly or indirectly produced by a chemical, are the most pertinent indicator of the potential for carcinogenicity. The spectrum of
Page 199
mutational classes, their frequencies, and their mechanisms of formation are all important in establishing the nature of the dose response for tumors induced by a particular chemical of interest at exposure concentrations below those for which tumor data can be obtained. These various aspects of mutagenicity for arsenicals are discussed in this section in the context of arsenic-induced carcinogenicity.
To characterize dose-response relationships, it is also appropriate to distinguish between mode of action and mechanism of action. It is much more feasible to establish a mode of action, because the only requirement is identifying the necessary (but not sufficient) steps whereby a particular agent causes tumor development. In contrast, the requirements for understanding the mechanism of action whereby an agent induces a tumor are identifying the necessary steps and characterizing their specific nature (e.g., the specific genes involved). Thus, considerable research efforts are required to move from a mode of action to a mechanism of action for tumor induction. The present state of knowledge for arsenic clearly necessitates that the mode of action be considered.
Genotoxic Effects Induced by Arsenic Compounds
Genetic alterations induced in cells by chemicals can be of several different types. Broadly these are point mutations that involve alterations of a single base pair in the DNA, chromosomal alterations that include deletions within a chromosome arm, interchanges between two chromosomes and losses or gains of whole chromosomes (aneuploidy).
Arsenic does not induce point mutations in bacterial or mammalian cells (Jacobson-Kram and Montalbano 1985). A recent report by M.M. Moore et al. (1997) showed that sodium arsenite and sodium arsenate induced mutations in L5178Y/TK+/- cells at concentrations of 1-2 µg/mL and 10-14 µg/mL, respectively. In contrast, the methylated metabolites MMA and DMA were mutagenic only at much higher concentrations, 2,500-5,000 µg/mL and almost 10,000 µg/mL, respectively. The organic arsenicals are much less mutagenic than the inorganic arsenicals. Of particular note for the present discussion, all four arsenicals induced deletion mutations and not point mutations at the TK locus.
Hei et al. (1998) showed that arsenic exposure induced deletion mutations of human chromosome 11 in a human-hamster hybrid cell. Analysis at the molecular level showed that all mutants had lost one or more markers and that
Page 200
the frequency of very large deletions increased with exposure concentration. Arsenicals produce a range of chromosomal alterations in mammalian cells in vitro, rodents in vivo, and humans exposed to relatively high concentrations of arsenic in drinking water. A discussion of many of these studies and their relevance to dose-response assessment can be found in Rudel et al. (1996) and in the comprehensive review by Rossman (1998).
In vivo and in vitro studies of rodents and humans have reported chromosomal aberrations, including the induction of micronuclei (Larramendy et al. 1981; IARC 1987; Jha et al. 1992; Warner et al. 1994; Dulout et al. 1996; Gonsebatt et al. 1997; L.E. Moore et al. 1996, 1997).
Arsenic-induced aneuploidy has also been demonstrated in vivo and in vitro in human lymphocytes and in exfoliated bladder cells but not consistently in buccal cells from exposed individuals (Gonsebatt et al. 1997; Warner et al. 1994; Vega et al. 1995; Dulout et al. 1996). SCEs have been induced in vitro, but evidence for their occurrence in exposed humans is equivocal (Nordenson et al. 1978; Larramendy et al. 1981; Lerda 1994; Rasmussen and Menzel 1997). The relative potency of arsenic compounds for clastogenicity in normal human fibroblasts in vitro is the following: (1) arsenite, (2) arsenate, and (3) DMA (Oya-Ohta et al. 1996). For example, more than 7 mM of DMA is required for clastogenicity, whereas only 0.8 µM of arsenite is needed to induce chromosomal alterations.
There are reports that arsenic potentiates the mutagenic effects of alkylating agents (Li and Rossman 1989a, 1991; Yang et al. 1992), UV radiation (Rossman 1981; Lee et al. 1985), X-rays and DNA cross-linking agents, 8-methoxypsoralen plus ultraviolet (UV) light and cis-platinum (Lee et al. 1986), and diepoxybutane (Wiencke and Yager 1992).
Plasmid shuttle vector experiments indicated marked potentiation of point mutagenesis by short-wave UV at a concentration of 1 µM of sodium arsenite, an exposure that itself did not affect the cell viability of normal human fibroblasts (Wiencke et al. 1997); arsenic's effects were largely attributed to enhancement of small and large deletions and rearrangements. The enhancement of genotoxicity that has been observed for clastogenic effects and point mutations has not been found for induced SCEs. Arsenite (5 µM) was found to enhance by sixfold the neoplastic transformation of C3H/10T/1/2 cells by bovine papilloma virus (Kowalski et al. 1996).
The role of these reported comutagenic effects on arsenic-induced carcinogenicity are as yet unclear given the somewhat artificial nature of the interac-
Page 201
tions studied. Further discussion is provided below on the influence of these comutagenic effects on DNA repair.
Methylation changes in genes or their control regions could result in altered gene expression and perhaps carcinogenesis (Baylin et al. 1998). Further, nonmutagenic carcinogens, such as arsenic, could be carcinogenic via this mechanism (Costa 1995). In an initial study to test that hypothesis, Mass and Wang (1997) showed that exposure of human lung adenocarcinoma A549 cells to sodium arsenite (0.08-2 µM) or sodium arsenate (30-300 µM) produced dose-responsive hypermethylation within a 341-base-pair fragment of the promoter of p53. In contrast, DMA (2-2,000 µM) did not produce hypermethylation. Some data suggested that such hypermethylation might be a genomic change and not just a region-specific methylation change (Mass and Wang 1997). However, other explanations can be found for the phenomenon described including the possibility of selection of cells that were hypermethylated before arsenic exposure.
Recently, Zhao et al. (1997) showed that chronic exposure to low concentrations of arsenic caused transformation of a rat liver epithelial cell line into one that was capable of causing tumors in nude mice. Hyperexpression of the metallothionein gene was also detected. The authors hypothesized that the methylation of arsenic by methyltransferases could cause a decrease in the Sadenosyl-methionine (SAM) available for methylation of DNA, leading to hypomethylation and a resultant aberrant gene express. They concluded that hypomethylation of DNA is a tenable epigenetic mechanisms for arsenic-induced carcinogenicity.
Given that effects on cellular methylation status could have important consequences for gene expression patterns, levels of methylation donors in the diet could modulate the effects of arsenic on DNA metabolism.
Some evidence supports the concept that arsenite induces oxidative stress in mammalian cells and that the induced oxidative damage can result in genotoxicity. For example, the concept is supported indirectly by the finding that adding superoxide dismutase to the culture medium reduces the frequency of arsenite-induced SCE in human lymphocytes (Nordenson and Beckman 1991). Similarly, vitamin E can protect human fibroblasts from arsenic
Page 202
toxicity (Lee and Ho 1994). Arsenite can also increase the concentrations of a number of proteins that can protect against oxidative stress—e.g., metallothionein (Albores et al. 1992) and heme oxygenase (Keyse and Tyrrell 1989). Although those studies and other similar ones suggest that arsenic induces oxidative stress and results in genotoxicity, more direct evidence clearly needs to be provided. Recent studies by Hei et al. (1998) suggest that reactive oxygen species are involved in the formation of deletion mutations of human chromosome 11 in a human-hamster hybrid cell following arsenic treatment. The log-linear shape of the survival curve for these cells is noted to be supportive evidence for an effect of arsenic on DNA repair. This conclusion is supported, in part, by the data of Gurr et al. (1998), who showed that arsenic induced micronuclei in cells in vitro at concentrations above 10 µM. The induction of micronuclei was reduced by treatment with NO synthase inhibitors and superoxide dismutase, as well as calcium chelators and uric acid.
An alternative explanation for arsenic's possible role in the induction of genotoxicity is that arsenic could affect the repair of endogenously produced oxidative DNA damage. However, without specific studies to address that hypothesis, it remains speculative.
Sodium arsenite can increase the incorporation of 3H-thymidine into the DNA of human keratinocytes in vitro as well as increase cell number at low concentrations (0.001-0.002 µM) indicating increased cell proliferation. At higher concentrations, sodium arsenite was cytotoxic (Germolec et al. 1997). In support of arsenic-inducing cell proliferation, Germolec et al. (1997) showed that sodium arsenite induced increased mRNA transcripts of keratinocyte growth factors, including granulocyte macrophage-colony-stimulating factor (GM-CSF), transforming growth factor a (TGF-a), and the inflammatory cytokine tumor necrosis factor a (TNF-a) in primary human epidermal keratinocytes in vitro. In addition, c-myc expression, as an indicator of proliferation was increased. These effects on transcription were seen at sodium arsenite concentrations of 0.5-4 µM, with an approximately linear response over this range. While these data have the potential for providing input into the mode of action via the cell proliferation pathway, at this time they are observational rather than informative. Arsenicals can also cause cell proliferation in target organs in vivo, most likely as a regenerative response subsequent to induced toxicity.
Oral administration of high concentrations of DMA to rats and mice after exposure to various genotoxic carcinogens caused increases in tumors in lung,
Page 203
bladder, liver, kidney, and thyroid (Yamamoto et al. 1995; Wanibuchi et al. 1996; Yamanaka et al. 1996) (see the previous section Results from Bioassays). With that particular protocol, DMA is presumably acting as a promoting agent that would be supportive of a cell-proliferation effect. However, the specific experimental design can create some difficulties in interpretation.
Arsenic-Induced Carcinogenicity and the Shape of Dose-Response Curve—A Mode-of-Action Approach
The preceding section provides an overview of the various genotoxic effects of arsenicals in vitro and in vivo. The aim of this section is to discuss how some or all of those effects might be involved in arsenic-induced carcinogenicity, and further, how that information can provide information on the shape of the dose-response curve at low exposure concentrations.
As discussed above, cancer is a genetic disease that requires the accumulation of a series of mutations in a single cell during the progression from a normal cell to a cancer cell. Those mutations can be point (gene) mutations, deletion mutations, other structural chromosomal alterations (e.g., inversions or rearrangement), or chromosomal numerical alterations (chromosomal losses or gains). On the basis of the mutagenicity profile for arsenicals discussed above, chromosomal alterations rather than point mutations are more likely to be involved in arsenic carcinogenicity. These chromosomal aberrations could feasibly be induced by direct or indirect interaction of arsenic with the DNA. Although the latter is much more likely to be based on available experimental evidence, direct interaction cannot be ruled out.
Chromosomal structural alterations can arise via one of two basic pathways, direct interaction with DNA and indirect DNA effects. However, on the basis of the published data on mutational spectra for arsenic and the present discussion on other cellular responses to arsenic, arsenic-induced effects on cellular housekeeping processes are more likely to result ultimately in the formation of chromosomal alterations. Such indirect effects are predicted to lead to sublinear dose responses for chromosomal alterations, as supported by the experimental data reviewed by Rudel et al. (1996). Such a sublinear dose response is predicted for the less likely scenario of production of chromosomal aberrations following direct interaction of arsenic with DNA. This conclusion is based on the fact that the majority of structural chromosomal aberrations (with the possible exception of terminal deletions) require at least two independent events at the DNA level for their formation. Chromosomal alterations result from errors of DNA repair at the molecular level, or during DNA replication. The evidence in support of this hypothesis is
Page 204
described in the following paragraph. Chromosomal gains and losses result from failure of chromosomes to separate at anaphase or from failure of chromosomes to move to the poles at anaphase. Either of those processes could result in a threshold dose response.
Chromosomal aberrations are formed by errors in DNA repair of induced or endogenous DNA damage, errors in DNA replication on an altered template, or inhibition of enzymes, such as topoisomerases. Such alterations result in chromosomal damage. For the great majority of chemicals, aberrations result from errors in DNA replication on an adducted template. There is no evidence to support those errors as a mode of action for arsenic other than the possibility that arsenic induces oxidative DNA damage. It is perhaps more likely that the aberrations are produced by errors of DNA repair induced by direct or indirect interference of the process by arsenic. Direct effects could be initiated by oxidative DNA damage, indirect effects by alterations to the repair proteins. The outcome would lead to a sublinear or possibly threshold response whether the effects on repair fidelity were direct or indirect.
Early studies supported the idea that DNA-repair enzymes are the molecular target mediating arsenic's genotoxic and comutagenic effects; studies indicated that relatively high concentrations of sodium arsenite inhibit DNA ligases I and II (Li and Rossman 1989b; Lee-Chen et al. 1994). Evidence supporting an earlier step in DNA repair has also been presented (Okui and Fujimara 1986; Hartwig et al. 1997). Studies indicating that arsenic alters the mutational spectra of UV radiation (Yang et al. 1992; Wiencke et al. 1997) are consistent with interference in either early or late steps in DNA repair. Extension of the concepts of mutational spectra analysis to human tumors in arsenic-exposed populations could provide information on the question of arsenic's mode of action and might be useful in risk assessment (Clewell et al. in press). A study of the pattern of p53 mutations in bladder cancers arising in a blackfoot-disease endemic area in Taiwan suggested that the etiology of the tumors might have involved a mechanism that increased the extent of DNA damage per mutational event (Shibata et al. 1994). The relationship of human tumor data to arsenic's effects on DNA repair requires further study. Recent studies, however, using purified human DNA-repair enzymes indicated that micromolar concentrations of arsenite increase the activities of DNA polymerase beta, O6-methylguanine-DNA methyltransferase, and DNA ligases I, II, and III (Hu et al. 1998). Inhibition occurred at higher doses (i.e., 1-5 mM of sodium arsenite). Those results cast doubt on the direct inhibition of those components of repair by arsenic. Other enzymatic activities involved in DNA repair have been reported to be inhibited by arsenite; nontoxic concentrations of arsenite inhibit human poly(ADP-ribose)polymerase (PARP) activity (Yager
Page 205
and Wiencke 1997). Although inhibition of PARP might be related to SCE induction, that effect alone is unlikely to account for the magnitude of the synergistic effects of arsenic or the extensive range of agents whose genotoxicity is augmented by arsenic (i.e., mono- and bi-functional alkylating agents, ionizing radiation, and cross-linking agents).
Biochemical data, however, indicate that preferential binding of arsenic at low micromolar concentrations will target proteins with specific structural features. Although arsenite has long been recognized as highly reactive with protein sulfhydryl groups (Joshi and Hughes 1981; Knowles and Benson 1984; Squibb and Fowler 1983), it can be highly selective in reacting with only a small number of closely spaced (vicinal) dithiol groups in proteins at low concentrations (Wiencke and Yager 1992). For example, the selective inhibition of dexamethasone binding to glucocorticoid receptors by arsenite is thought to be due to the formation of a stable dithioarsenite complex with one vicinal dithiol group within the steroid-binding domain of the glucocorticoid receptor (Lopez et al. 1990). Arsenite also blocks DNA binding by the receptor, presumably through similar interactions with vicinal dithiols present in the DNA-binding domain of the protein (Simons et al. 1990). The number of proteins containing vicinal dithiols is relatively small, but it is important that this structural feature is common among DNA-binding proteins, transcription factors, and DNA-repair proteins (Berger 1985). Closely spaced thiol groups within the DNA-binding proteins complex with zinc and can form so-called "zinc fingers, " which are thought to be positioned into the major groove of the DNA double helix and mediate DNA-protein interactions. Proteins involved in DNA repair that contain putative zinc fingers include the UVRA protein (Husain et al. 1986), PARP (Cherney et al. 1987; Uchida et al. 1987), the RAD-18 protein (Jones et al. 1988), and the XPAC protein (Tanaka et al. 1990).
In support of the concept of preferential binding of arsenic with vicinal dithiols is the inhibition of PARP, which contains two zinc-finger motifs essential to its enzymatic function. Also supportive is the observation that arsenite inhibits the excision of thymine dimers and potentiates the cytotoxicity of 254 nanometers (nm) of UV light in wild-type human fibroblasts but does not affect the cytotoxic effects of UV light in excision-defective xeroderma pigmentosum group A cells (Okui and Fujiwara 1986). The XPAC protein has been shown to contain motifs of both C4 and C2H2 classes of zinc-finger proteins (Tanaka et al. 1990). These studies provide some reasonable support for a role of arsenic in altering the fidelity or kinetics of DNA repair, but clearly further research is needed to address the issue more directly.
With respect to arsenic's mode of action in the induction of aneuploidy, the studies of Ramirez et al. (1997) are pertinent. The researchers examined
Page 206
the aneuploidy-inducing effects of arsenite in cultured human lymphocytes by using a variety of techniques, including fluorescence in situ hybridization (FISH) with DNA probes for chromosomes 1 and 7, immunostaining of the lymphocyte spindle apparatus, and an in vitro assay measuring the polymerization and depolymerization of tubulin. Dose-related increases in hyperdiploidy were seen in lymphocytes treated with arsenite at concentrations ranging from 0.001 to 0.1 µM. Examination of the spindle apparatus using an anti-b-tubulin antibody indicated that arsenite might disrupt spindle formation by interaction with microtubules. In addition, in vitro assays using purified tubulin indicated that arsenite inhibited microtubule assembly and induced tubulin depolymerization. Although these studies are of potential importance, they must be considered in light of the in vivo human data indicating that the predominant effect of arsenic in humans in vivo is clastogenesis and not aberrant chromosomal segregation. As an example, one of the proposed mechanisms for the involvement of alterations in methylation patterns in carcinogenesis involves the induction of chromosomal instability (Gonzalgo and Jones 1997). Hypermethylation of CpG sites within the 5' region of the p16INK4a tumor-suppressor gene (Ahuja et al. 1997) and the DNA mismatch repair gene hMLH1 (Kane et al. 1997) was recently found to be closely associated with microsatellite instability in colorectal cancer cells. It is conceivable that such instability could also be manifested by gross alterations in chromosomal structure, such as those observed in arsenic-exposed cells. However, if arsenic's effects on chromosomal aberrations is mediated by aberrant methylation, a sublinear dose response is likely to result for these effects at low exposure concentrations.
Increased or dysregulated cell proliferation plays a necessary, although not sufficient, role in the development of tumors (Butterworth et al. 1995). Cytotoxicity and regenerative cell proliferation demonstrate sublinear or, most frequently, threshold responses, irrespective of the mechanism by which the cytotoxicity is induced.
In summary, investigations of mode of action for carcinogenicity are conducted to predict the shape of cancer dose-response curves below the level of direct observation of tumors. For arsenic carcinogenicity, the mode of action has not been established, but the several modes of action that are considered plausible (namely, indirect mechanisms of mutagenicity) would lead to a sublinear dose-response curve at some point below the point at which a significant increase in tumors is observed (see EPA (1997) for further discussion). However, because a specific mode (or modes) of action has not yet been identified, it is prudent not to rule out the possibility of a linear response.
Page 207
Noncancer Effects
Inhibitory Effects on Cellular Respiration
The mechanisms of arsenical toxicity to individual cell types have historically centered around the inhibitory effects on cellular respiration at the level of the mitochondrion (Fluharty and Sanadi 1960, 1962; Packer 1961). Hepatotoxicity, as evidenced by porphyrinuria, has been seen in arsenic-poisoned humans following acute high-dose exposures and, to a milder extent, following chronic low-dose exposures. That effect is a major health effect related to decreased cellular respiration and mitochondrial toxicity (Fowler 1977). The preferential effects of arsenicals on nicotinamide adenine dinucleotide (NAD)-linked mitochondrial respiration (Fowler et al. 1979) have historically been linked to formation of stable arsenical complexes with the vicinal dithiols of the lipoic acid cofactor for the pyruvate dehydrogenase complex, but alterations in mitochondrial protein synthesis and inner-membrane structural integrity might also play an important role following in vivo exposures (Fowler et al. 1979, 1987). Disruption of oxidative phosphorylation and concomitant decreases in cellular levels of ATP (Chen et al. 1986, Figure 7-1) are thought to be important central events in the onset of cellular injury and cell death, because ultrastructural morphometric alterations in mitochondrial structure, disruption of mitochondrial respiratory function, and hepatic porphyrinuria are closely correlated (Figure 7-2). The hepatic porphyrinuria is dominated by increased amounts of uropophyrins (octa- and hepta-carboxyl isomers have also been observed for arsine gas (Fowler et al. 1989), inorganic arsenic, and indium arsenide (Conner et al. 1995)) with lesser amounts of coproporphyrin III. Those porphyrinurias have also been demonstrated in human populations in Mexico (Garcia-Vargas et al. 1991, 1994; Garcia-Vargas and Hernandez-Zavala 1996) and inner Mongolia (Yamauchi et al. 1998) when proper handling procedures (Woods et al. 1991) for protecting urine samples against photo-oxidation of porphyrins are followed.
Mitochondrial swelling has been reported in human renal proximal tubule cells following acute high-dose poisoning (Frejaville et al. 1972) and in rats following prolonged oral exposure to arsenate in drinking water (Brown et al. 1976). In the latter studies, these ultrastructural effects were associated with inhibition of mitochondrial respiratory function similar to that observed in liver cells. Overt clinical manifestations of nephropathy in humans have not been reported in relation to chronic oral intake of arsenic in drinking water.
Page 208
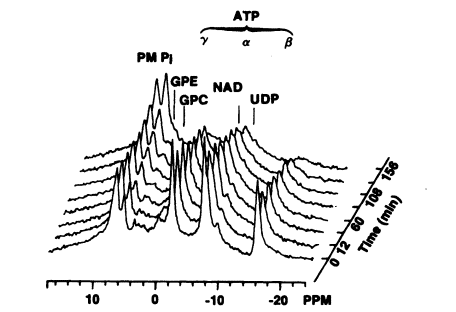
FIGURE 7-1
In vivo 31P nuclear magnetic resonance scans for hepatic ATP showing progressive decreases over time following
a single intravenous injection of sodium arsenite to a rat at a concentration of 10 mg/kg. Source: Chen et al. 1986.
Reprinted with permission from Biochemical and Biophysical Research Communications; copyright 1986, Academic Press.
Alterations in the Heme and Porphyrin Biosynthetic Pathways
Alterations in the heme biosynthetic and degradative pathway also occur (Woods and Fowler 1978; Cebrian et al. 1983; Garcia-Vargas et al. 1991, 1994; Conner et al. 1995; Garcia-Vargas and Hernandez-Zavala 1996; Yamauchi et al. 1998), indicating that this essential pathway is susceptible to arsenical disturbance. The increased urinary excretion of carboxyl uroporphyrins VI, VII, and VIII and carboxyl coproporphyrin IV are the main measurable effects in experimental animal models following subchronic exposure to arsine gas (Fowler et al. 1989) in arsenic or sodium arsenite (Conner et al. 1995). As noted above, proper handling of urine samples is essential to obtain correct porphyrin analyses and avoid co-elution of uroporphyrin peaks under the carboxyl coproporphyrin IV peak.
More recent studies by Garcia-Vargas and Hernandez-Zavala (1996) have confirmed the presence of arsenic uroporphyrinuria-coproporphyrinuria in both human populations following prolonged exposure to arsenic in drinking
Page 209
water. The conditions for appropriate handling of urine samples for highpressure liquid chromotography are clear and detailed.
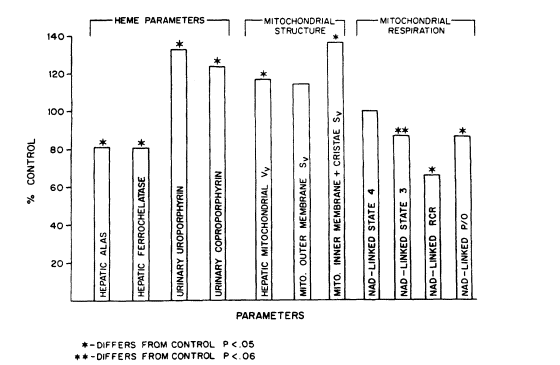
FIGURE 7-2
Summary of statistical correlations between ultrastructural morphometric alterations in
mitochondrial substructure, respiratory function, mitochondrial heme pathway enzymes,
and increased porphyrin excretion patterns in rats exposed to arsenate at 40 ppm in drinking water for 6 weeks. ALAS, delta-aminoleulinic acid synthetase; Vv, volume density; Sv,
surface density; RCR, respiratory control ratio; NAD, linked substrates such as pyruvate
and malate; P/O, phosphate-to-oxygen ratio. Source: Fowler et al. 1987. Reprinted with
permission from the Annals of the New York Academy of Sciences; copyright 1987.
Alterations in Cellular Gene Expression-Stress Protein Induction
Exposure of cells to arsenicals either in vitro (Caltibiano et al. 1986; Taketani et al. 1989; Aoki et al. 1990; Van Delft et al. 1993; Van Wijk et al. 1993) at concentrations ranging from 10-6 to 10-3 M or in vivo at concentrations of 0.3 mg/kg and 3.0 mg/kg (Conner et al. 1993) has been shown to
Page 210
cause the induction of a number of the major stress proteins including members of the 32, 60, 70, and 90 stress-protein families. Clearly, in vitro exposure of cells to arsenite in a variety of model systems (Chin et al. 1990; Wu and Welsh 1996) results in a broad spectrum stress-protein response. In addition, there is down regulation of a number of constitutively expressed proteins. The increased expression of the major stress proteins suggests the presence of oxidative damage to essential proteins perhaps secondary to uncoupling of mitochondrial oxidative phosphorylation and increased intracellular production of H202 and subsequent production of reactive oxygen species (ROS). In particular, the increased presence of the 32-kilodalton stress protein known to be heme oxygenase following arsenical exposure (Taketani et al. 1989; Keyse and Tyrrell 1989) provides further support for this concept. Given the capacity of arsenicals to produce oxidative stress, which includes induction of heme oxygenase, it is clear that reactive oxygen species, such as nitrate oxide and hydroxyl radicals, might play important roles in mediating the observed alterations in gene-expression patterns noted in many studies.
Altered cytokine gene expression was recently reported in relation to the development of keratoses and might play a role in the development following chronic arsenical exposure (Germolec et al. 1998).
Arsenic Toxicity and Metallothionein
Tolerance to arsenic toxicity exists, and because metallothionein (MT) is known to be responsible for the tolerance to cadmium toxicity, the interactions of cadmium and MT have been examined.
MT is a low-molecular-weight, cysteine-rich, metal-binding protein. MT has been proposed to play an important role in the homeostasis of essential metals, in the detoxication of heavy metals, and in the scavenging of free radicals (Kägi 1993; Sato and Bremner 1993). Moreover, MT is a small protein easily induced by heavy metals, hormones, acute stress, and a variety of chemicals (Hamer 1986; Kägi 1993). The cysteine contents and the capacity of these MT isoforms to bind metals are similar.
Arsenicals are effective inducers of MT in mice (Maitani et al. 1987; Kreppel et al. 1993) and rats (Albores et al. 1992; Hochadel and Waalkes 1997). The ability of different arsenical forms to induce MT varies markedly: As(III) is a potent MT inducer, as it took approximately 3 times more As(V), 50 times more MMA, and 120 times more DMA to induce MT (Kreppel et al. 1993) (see Figure 7-3). In contrast, MMA is the most effective MT inducer (80-fold), followed by As(III) (30-fold), As(V) (25-fold), and DMA (10-fold)
Page 211
(Kreppel et al. 1993). The induction of MT is also observed following oral administration. Again, the doses of organic arsenicals (MMA and DMA) required for MT induction are one order higher than those of inorganic arsenicals (As(III) and As(V)) (Maitani et al. 1987). Induction of MT by arsenicals appears to be mediated at the transcription level, because MT-I and MT-II mRNA levels are increased by As(III) and As(V), As(III) being more effective (Albores et al. 1992; Kreppel et al. 1993). However, arsenicals are unable to induce MT in cultured hepatocytes (Bauman et al. 1993; Kreppel et al. 1993), suggesting that induction of MT by arsenicals might occur indirectly. The potency of arsenicals to induce MT (As(III) > As(V) > MMA > DMA) parallels the toxicity of arsenicals (Peoples 1975; Maitani et al. 1987; Klaassen 1995). That implies that MT induction might be, at least in part, due to a toxic event associated with arsenical administration and implies that MT might play a role in the detoxication of arsenicals.
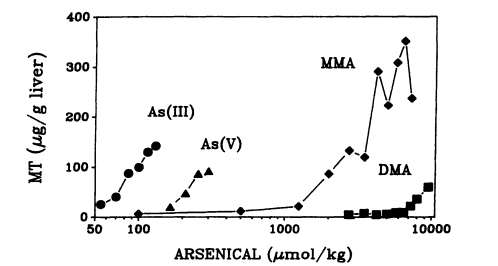
FIGURE 7-3
MT concentration in liver of mice exposed to various doses of arsenic(III), arsenic(V), MMA, and DMA.
Mice were exposed subcutaneously with approximately equally toxic doses of the different arsenicals, and liver MT concentration was determined 24 hr after exposure.
Data represent the mean of two mice per dose. Source: Kreppel et al. 1993. Reprinted with permission from Fundamental and Applied Toxicology; copyright 1993, Society of Toxicology.
MT induction has been proposed as one of the adaptive mechanisms for tolerance to arsenic toxicity (Kreppel et al. 1988; Cherian 1995; Silver and Phung 1996). However, how MT protects against arsenic toxicity is unclear.
Page 212
The affinity of arsenic for MT in vitro is markedly lower than the affinity of zinc or cadmium for MT (Waalkes et al. 1984). In intact animals, only a small portion of dosed arsenic is found to be associated with the MT fraction (Maitani et al. 1987; Albores et al. 1992; Kreppel et al. 1994; Chen and Whanger 1994). That suggests that, unlike the proposed detoxication mechanism of MT for cadmium (Goering et al. 1995), MT does not protect against arsenic toxicity by ''sequestering the metal"; rather, MT might function as an antioxidant against arsenic-induced oxidative injury.
Evidence has been accumulated that oxidative damage is an important mechanism for arsenic toxicity (see above). Because of its high sulfhydryl content, MT has also been suggested to react with free radicals and electrophiles (Klaassen and Cagen 1981; Basu and Lazo 1990). Indeed, MT can serve as a sacrificial scavenger for hydroxyl radicals in vitro (Thornalley and Vasäk 1985) and thus protect against free radical-induced DNA damage (Abel and De Ruiter 1989; Chubatsu and Meneghini 1993; Schwarz et al. 1995). MT can also assume the function of superoxide dismutase (zinc and copper SOD) in yeast (Tamai et al. 1993) and protect against lipid peroxidation in erythrocyte ghosts produced by xanthine oxidase-derived superoxide anion, and hydrogen peroxide (Thomas et al. 1986). MT is induced by oxidative stress-producing chemicals (Bauman et al. 1991) and alkylating agents (Kotsonis and Klaassen 1979) and has been shown to protect against oxidative damage (Sato and Bremner 1993) and the toxicity of alkylating anticancer drugs (Lazo and Pitt 1995). Determining whether MT protects against arsenic toxicity via its antioxidant role warrants further investigation.
Thus, in conclusion MT is thought to have a protective effect against arsenic toxicity and to be responsible for at least part of its self-induced tolerance. However, the data supporting that conclusion are indirect, and the availability of genetically engineered mice that lack MT should be examined to determine with greater confidence the role of MT in modifying the toxicity of arsenic.
Arsine Gas Toxicity
The toxicity of arsine gas (AsH3) is directed primarily toward the red blood cells, producing hemolysis following acute, short-term, or prolonged exposure (Fowler et al. 1989; Blair et al. 1990a,b). Of interest is the apparent involvement of ROS and glutathione depletion in the production of red-cell destruction, providing support for the idea that arsenicals might form radical species in the presence of oxygen by some as yet undefined mechanism within the red cells.
Page 213
Interactions Between Arsenic and Other Common Toxic Metals
Studies that examined lead, cadmium, and arsenic (Mahaffey and Fowler 1977; Fowler and Mahaffey 1978; Mahaffey et al. 1981) showed a number of interactions between these common toxic trace elements. The interactive effects in rodents are observed to occur in a well-tolerated intermediate-dose range in rodents for each of the elements studied. A number of the interactions involved marked increases in renal concentrations of copper (Mahaffey et al. 1981) in the arsenic exposure groups. Those interactions could be related to arsenic-induced metallothionein. The most dramatic effect was the mixture-specific increases in porphyrins with additive exacerbation of the arsenic-specific porphyrinuria pattern by concomitant lead and cadmium exposures (Mahaffey et al. 1981). (See discussion of selenium and other interactions in Chapters 8 and 9.)
Summary And Conclusions
· In vivo studies in rats and mice to determine the ability of inorganic arsenic to act as a cocarcinogen or as a promoter have produced mixed results. Studies on DMA suggest that this metabolite is not an initiator but might act as a promoter. The studies carried out to date, however, used very high doses, making interpretation of the results difficult, especially if DMA is formed in situ following the administration of inorganic arsenic.
· The mode of action for arsenic carcinogenicity has not been established. Inorganic arsenic and its metabolites have been shown to induce deletion mutations and chromosomal alterations (aberrations, aneuploidy, and SCE) but not point mutations. Other genotoxic responses that can be pertinent to the mode of action for arsenic carcinogenicity are comutagenicity, DNA methylation, oxidative stress, and cell proliferation; however, data on those genotoxic responses are insufficient to draw firm conclusions. The most plausible and generalized mode of action for arsenic carcinogenicity is that it induces structural and numerical chromosomal abnormalities without acting directly with DNA.
· Data on the modes of action for carcinogenicity can help to predict the shape of cancer dose-response curves below the level of direct observation of tumors. For arsenic carcinogenicity, the mode of action has not been established, but the several modes of action that are considered most plausible (namely, indirect mechanisms of mutagenicity) lead to a sublinear dose-response at some point below the level at which a significant increase in tumors is observed. However, because a specific mode (or modes) of action
Page 214
has not been identified at this time, it is prudent not to rule out the possibility of a linear response.
· In vitro studies of human and animal cells show that the genotoxic effects occur at submicromolar concentrations of arsenite that are similar to those found in urine of humans consuming drinking water at about the current MCL.
· The inhibitory effects of arsenicals on mitochondrial respiratory function are well known and highly specific to respiration supported by the NAD-linked substrates, such as pyruvate. The mechanism appears to involve inhibition of the pyruvate dehydrogenase complex via arsenic complexation with the vicinal dithiol-dihydrolipoic acid cofactor for that enzyme complex.
· Inhibition of mitochondrial respiration results in decreased cellular production of ATP and increased production of hydrogen peroxide. Those effects could cause formation of ROS, resulting in oxidative stress.
· Oxidative stress produced by ROS formation results in the observed induction of the major stress protein families.
· Intracellular production of ROS results in the inhibition of the heme biosynthetic pathway enzyme uroporphyrinogen decarboxylase. That in turn results in the observed uroporphyrinuria. The observed increases in coproporphyrin in the urine are secondary to the arsenical inhibition of this mitochondrial enzyme.
· There is clear quantitative evidence of deleterious ultrastructural morphometric and biochemical alterations in liver mitochondria associated with the observed porphyrinuria in experimental animals, indicating that the porphyrinuria is a good candidate as a putative biomarker of arsenical-induced hepatotoxicity.
· The role of arsenical-induced oxidative stress in mediating DNA damage is not completely clear, but the intracellular production of ROS might play an initiating role in the carcinogenic process by producing DNA damage.
· MT is thought to have a protective effect against arsenic toxicity and to be responsible for at least part of its self-induced tolerance. However, the data supporting that conclusion are indirect.
Recommendations
Identification of proximate markers of arsenic-induced cancers and their application in carefully designed epidemiological studies might better define the cancer dose-response curves at low concentrations. Molecular and cellular characterization of neoplasms from arsenic exposed populations and appropriate controls might aid in identifying the mechanism by which arsenic induces
Page 215
tumors. Chronic low-dose studies in a suitable animal model (mouse, hamster, or rabbit) might increase our understanding of the mode of action of arsenic carcinogenicity, particularly the potential role of chromosomal alterations.
There is a pressing need to understand the sequence of cellular arsenic exposure, the methylation processes, inhibition of mitochondrial function, and formation of ROS.
Other studies of less critical importance for characterizing risks but nonetheless needed to fill important data gaps include the following:
— Studies to examine the relationships between ROS formation, oxidative stress, induction of the stress protein response and necrosis or apoptosis.
— Studies to examine the relationships between arsenical-induced oxidative stress, DNA damage, induction of proto-oncogenes, inhibition of DNA mechanisms, and cancer.
— Investigations of the potential role of arsenic as a cocarcinogen, preferably in defined human populations.
— Studies to identify and characterize mechanism-based biomarkers of arsenical effects in human target-organ systems.
— Studies using genetically engineered mice that lack MT to determine with greater confidence the role of MT in modifying the toxicity of arsenic.
References
Abel, J., and N. De Ruiter. 1989. Inhibition of hydroxyl-radical-generated DNA degradation by metallothionein. Toxicol. Lett. 47:191-196.
Ahuja, N., A.L. Mohan, Q. Li, J.M. Stolker, J.G. Herman, S.R. Hamilton, S.B. Baylin, and J.P. Issa. 1997. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 57:3370-3374.
Albores, A., J. Koropatnick, M.G. Cherian, and A.J. Zelazowski. 1992. Arsenic induces and enhances rat hepatic metallothionein production in vivo. Chem.-Biol. Interact. 85:127-140.
Aoki, Y., M.M. Lipsky, and B.A. Fowler. 1990. Alteration of protein synthesis in primary cultures of rat kidney proximal tubule epithelial cells by exposure to gallium, indium and arsenite. Toxicol. Appl. Pharmacol. 106:462-468.
Baroni, C., G.J. van Esch, and U. Saffiotti. 1963. Carcinogenesis tests of two inorganic arsenicals. Arch. Environ. Health 7:668-674
Basu, A., and J.S. Lazo. 1990. A hypothesis regarding the protective role of metallothioneins against the toxicity of DNA interactive anticancer
Page 216
drugs. Toxicol. Lett. 50:123-135.
Bauman, J.W. , J. Liu, Y.P. Liu, and C.D. Klaassen. 1991. Increase in metallothionein produced by chemicals that induce oxidative stress. Toxicol. Appl. Pharmacol. 110:347-354.
Bauman, J., J. Liu, and C.D. Klaassen. 1993. Production of metallothionein and heat-shock proteins in response to metals. Fundam. Appl. Toxicol. 21:15-22.
Baylin, S.B., J.G. Herman, J.R. Graff, P.M. Vertino, and J.P. Issa. 1998. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res. 72:141-196.
Berger, N.A. 1985. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 101:4-15.
Blair, P.C., M.B. Thompson, M. Bechtold, R.E. Wilson, M.P. Moorman, and B.A. Fowler. 1990a. Evidence for oxidative damage to red blood cells in mice induced by arsine gas. Toxicology 63:25-34.
Blair, P.C., M.B. Thompson, R.E. Morrissey, M.P. Moorman, R.A. Sloane, and B.A. Fowler. 1990b. Comparative toxicity of arsine gas in B6C3F1 mice, Fischer 344 rats and Syrian golden hamsters: System organ studies and comparison of clinical indices of exposure. Fundam. Appl. Toxicol. 14:776-787.
Brown, M.M., B.C. Rhyne, R.A. Goyer, and B.A. Fowler. 1976. The intracellular effects of chronic arsenic exposure on renal proximal tubule cells. J. Toxicol. Environ. Health 1:505-514.
Butterworth, B.E., R.B. Conolly, and K.T. Morgan. 1995. A strategy for establishing mode of action of chemical carcinogens as a guide for approaches to risk assessments. Cancer Lett. 93:129-146.
Byron, W.R., G.W. Bierbower, J.B. Brouwer, and W.H. Hansen. 1967. Pathologic changes in rats and dogs from two-year feeding of sodium arsenite or sodium arsenate. Toxicol. Appl. Pharmacol. 10:132-147.
Caltibiano, M.M., T.P. Koestler, G. Poste, and R.G. Greig. 1986. Induction of the 32- and 34-kDA stress proteins by sodium arsenite, heavy metals and thiol reactive reagents. J. Biol. Chem. 261:13381-13386.
Cebrian, M., A. Albores, M. Aguilar, and E. Blakely. 1983. Chronic arsenic poisoning in the north of Mexico. Hum. Toxicol. 2:121-133.
Chen, C.L., and P.D. Whanger. 1994. Interaction of selenium and arsenic with metallothionein: Effect of vitamin B12. J. Inorg. Biochem. 54:267276.
Chen, B., C.T. Burt, P.L. Goering, B.A. Fowler, and R.E. London. 1986. In vivo 31-P nuclear magnetic resonance studies of arsenite induced changes in hepatic phosphate levels. Biochem. Biophys. Res. Commun. 139:228-234.
Page 217
Cherian, M.G. 1995. Metallothionein and its interaction with metals. Handb. Exp. Pharmacol. 115:121-138.
Cherney, B.W., O.W. McBride, D. Chen, H. Alkhatib, K. Bhakia, P. Hensen, and M.E. Smulson. 1987. cDNA sequence, protein structure, and chromosomal location of the human gene for poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA 84:8370-8374.
Chin, K.V., S. Tanaka, G. Darlington, I. Pastan, and M.M. Gottesman. 1990. Heath shock and arsenite increase expression of the multi-drug resistance (MDR1) gene in human renal carcinoma cells. J. Biol. Chem. 265:221-226.
Chubatsu, L.S., and R. Meneghini. 1993. Metallothionein protects DNA from oxidative damage. Biochem. J. 291:193-198.
Clewell, H.J., P.R. Gentry, H.A. Barton, A.M. Shipp, J.W. Yager, and M.E. Andersen. In press. Requirements for a biologically realistic cancer risk assessment for inorganic arsenic. Int. J. Toxicol.
Conner, E.A., H. Yamauchi, B.A. Fowler, and M. Akkerman. 1993. Biological indicators for monitoring exposure/toxicity to III-V semiconductors. J. Exp. Anal. Epidemiol. 3:431440.
Conner, E.A., H. Yamauchi, and B.A. Fowler. 1995. Alterations in the heme biosynthetic pathway from the III-V semiconductor metal, indium arsenide (InAs). Chem.-Biol. Interact. 96:273-285.
Costa, M. 1995. Model for the epigenetic mechanism of action of nongenotoxic carcinogens. Am. J. Clin. Nutr. 61(Suppl.):666S-669S.
Dulout, F.N., C.A. Grillo, A.I. Seoane, C.R. Maderna, R. Nilsson, M. Vahter, F. Darroudi, and A.T. Natarajan. 1996. Chromosomal aberrations in peripheral blood lymphocytes from native Andean women and children from northwestern Argentina exposed to arsenic in drinking water. Mutat. Res. 370:151-158.
Endo, G., K. Kuroda, A. Okamoto, and S. Horiguchi. 1992. Dimethylarsenic acid induces tetraploids in Chinese hamster cells. Bull. Environ. Contam. Toxicol. 48:131-137.
EPA (U.S. Environmental Protection Agency). 1981. Methane Arsonic Acid (MAA): 2-Year Chronic/Oncogenic Rat Feeding Study. Memorandum from W. Dykstra, Toxicology Branch, Health Effects Division, to R. Mountfort, Registration Division. May 13. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1991. Cacodylic AcidCarcinogenicity Study in Mice. Memorandum from L.L. Taylor, Toxicology Branch II, Section II, Health Effects Division, to B. Crompton, Generic Chemical Support Branch, Special Review and
Page 218
Reregistration Division. Memorandum 008891. Dec. 2. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1992a. Review of Toxicology Studies with Methanearsonic Acid/Methanearsonic Acid, Monosodium to Support Reregistration of the Test Substance. Memorandum from S.L. Malish, Toxicology Branch II, Review Section IV, Health Effects Division, to B. Briscoe, Special Review and Reregistration Division, Health Effects Division. Memorandum 009478. April. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1992b. Review of Toxicology Studies with Methanearsonic Acid to Support Reregistration of the Test Substance. Memorandum from S.L. Malish, Toxicology Branch II, Review Section IV, Health Effects Division, to B. Briscoe, Special Review and Reregistration Division, Health Effects Division. Memorandum 009382. March. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1992c. Review of Toxicology Studies with Cacodylic Acid to Support Registration of Test Substance. Memorandum from S.L. Malish, Toxicology Branch II, Review Section IV, Health Effects Division, to B. Briscoe, Special Review and Reregistration Division, Health Effects Division. Memorandum 009391. March. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1992d. Review of Toxicology Study with Cacodylic Acid to Support Registration of Test Substance. Memorandum from S.L. Malish, Toxicology Branch II, Review Section IV, Health Effects Division, to B. Briscoe, Special Review and Reregistration Division, Health Effects Division. Memorandum 009775. October. U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1993. Cacodylic Acid: Registrant's Response to Agency's Review of Toxicology Data. Memorandum from S.L. Malish, Toxicology Branch II, Review Section IV, Health Effects Division, to V. Dietrich, Product Manager, Registration Division. Memorandum 010550. September. U.S. Environmental Protection Agency, Office of Prevention, Pesticides and Toxic Substances, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1997. Report on the Expert Panel on Arsenic Carcinogenicity: Review and Workshop. Prepared by
Page 219
the Eastern Research Group, Lexington, Mass., for the U.S. Environmental Protection Agency, National Center for Environmental Assessment, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1998. Cacodylic acid. In Integrated Risk Information System (IRIS). Online. Entry last revised Dec. 1, 1996. Available: http://www.epa.gov/iris/subst/0587.htm#II. U.S. Environmental Protection Agency, National Center for Environmental Assessment, Cincinnati, Ohio.
Fluharty, A., and D.R. Sanadi. 1960. Evidence for a vicinal dithiol in oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 46:608-616.
Fluharty A.L., and D.R. Sanadi. 1962. On the mechanism of oxidative phosphorylation IV. Mitochondrial swelling caused by arsenite in combination with 2,3-dimercaptopropanol and by cadmium ion. Biochemistry 1:276-281.
Fowler, B.A. 1977. Toxicology of environmental arsenic. Pp. 79-122 in Toxicology of Trace Elements. Advances in Modern Toxicology, Vol. 2, R.A. Goyer and M.A. Mehlman, eds. Washington, D.C.: Hemisphere.
Fowler, B.A. and K.R. Mahaffey. 1978. Interactions among lead, cadmium, and arsenic in relation to porphyrin excretion patterns. Environ. Health Perspect. 25:87-90.
Fowler, B.A., J.S. Woods. and C.M. Schiller. 1979. Studies of hepatic mitochondrial structure and function: Morphometric and biochemical evaluation of in vivo perturbation by arsenate. Lab. Invest. 41:313-320.
Fowler, B.A., A. Oskarsson, and J.S. Woods. 1987. Metal- and metalloidinduced porphyrinurias: Relationships to cell injury. Ann. N.Y. Acad. Sci. 514:172-182.
Fowler, B.A., M.P. Moorman, B. Adkins, W.E. Bakewell, P.C. Blair, and M.B. Thompson. 1989. Arsine: Toxicity Data from Acute and Shortterm Inhalation Exposures. Pp 85-89 in Hazard Assessment and Control Technology in Semiconductor Manufacturing. American Conference of Governmental Industrial Hygienists. Boca Raton, Fla.: Lewis.
Frejaville, J.P., J. Bescol, J.P. Leclerc, L. Gulliam, P. Crabie, F. Consco, P. Gervais, and M. Gaultier. 1972. Acute poisoning with arsenic derivatives ( apropos of 4 personal cases); hemostasis disorders; ultramicroscopic study of the liver and kidney [ in French]. Ann. Med. Interne 123:713-722.
Garcia-Vargas, G.G., and A. Hernandez-Zavala. 1996. Urinary porphyrins and heme biosynthetic enzyme activities measured by HPLC in arsenic toxicity. Biomed. Chromatogr. 10(6):278-284.
Garcia-Vargas, G.G., A. Garcia-Rangel, M. Aguilar-Romo, J. Garcia-
Page 220
Salcedo, L.M. Del Razo, P. Ostrosky-Wegman, C.C. DeNava, and M.E. Cebrian. 1991. A pilot study of the urinary excretion of porphyrins in human populations chronically exposed to arsenic in Mexico. Hum. Exp. Toxicol. 10:189-194.
Garcia-Vargas, G.G., L.M. Del Razo, M.E. Cebrian, A. Albores, P. Ostrosky-Wegman, R. Montero, M.E. Gonsebatt, C.K. Lim, and F. DeMatteis. 1994. Altered urinary porphyrin excretion in a human population chronically exposed to arsenic in Mexico. Hum. Exp. Toxicol. 13:839-847.
Germolec, D.R, J. Spalding, G.A. Boorman, J.L. Wilmer, T. Yoshida, P.P. Simeonova, A. Bruccoleri, F. Kayama, K. Gaido, R. Tennant, F. Burleson, W. Dong, R.W. Lang, and M.I. Luster. 1997. Arsenic can mediate skin neoplasia by chronic stimulation of keratinocyte-derived growth factors. Mutat. Res. 386:209-218.
Germolec, D.R., J. Spalding, H.S. Yu, G.S. Chen, P.P. Simeonova, M.C. Humble, A. Bruccoleri, G.A. Boorman, J.F. Foley, T. Yoshida, and M.I. Luster. 1998. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am. J. Pathol. 153:1775-1785.
Goering, P.L., M.P. Waalkes, and C.D. Klaassen. 1995. Toxicology of cadmium. Handb. Exp. Pharmacol. 115:189-213.
Gonsebatt, M.E., L. Vega, A.M. Salazar, R. Montero, P. Guzman, J. Blas, L.M. Del Razo, G. Garcia-Vargas, A. Albores, M.E. Cebrian, M. Kelsh, and P. Ostrosky-Wegman. 1997. Cytogenetic effects in human exposure to arsenic. Mutat. Res. 386:219-228.
Gonzalgo, M.L., and P.A. Jones. 1997. Mutagenic and epigenetic effects of DNA methylation. Mutat. Res. 386:107-118.
Gurr, J.R., F. Liu, S. Lynn, and K.Y. Jan. 1998. Calcium-dependent nitric oxide production is involved in arsenite-induced micronuclei. Mutat. Res. 416:137-148.
Hamer, D.H. 1986. Metallothionein. Annu. Rev. Biochem. 55:913-951.
Hartwig, A., U.D. Groblinghoff, D. Beyersmann, A.T. Natarajan, R. Filon, and L.H. Mullenders. 1997. Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis 18:399-405.
Hei, T.K., S.X. Liu, and C. Waldren. 1998. Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. USA 95:8103-8107.
Hochadel, J.F., and M.P. Waalkes. 1997. Sequence of exposure to cadmium and arsenic determines the extent of toxic effects in male Fischer rats. Toxicology 116:89-98.
Hu, Y., L. Su, and E.T. Snow. 1998. Arsenic toxicity is enzyme specific and
Page 221
its affects on ligation are not caused by the direct inhibition on DNA repair enzymes. Mutat. Res. 408:203-218.
Husain, I., B. van Houten, D.C. Thomas, and A. Sancar. 1986. Sequences of Escherichia coli uvrA gene and the protein reveal two potential ATP binding sites. J. Biol. Chem. 261:4895-4901.
IARC (International Agency for Research on Cancer). 1987. Arsenic and arsenic compounds. Pp. 100-206 in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs 1-42, Suppl. 7. Lyon, France: International Agency for Research on Cancer.
Innes, J.R.M., B.M. Ulland, M.G. Valerio, L. Petrucelli, L. Fishbein, E.R. Hart, A.J. Pallotta, R.R. Bates, H.L. Falk, J.J. Gart, M. Klein, I. Mitchell, and J. Peters. 1969. Bioassay of pesticides and industrial chemicals for tumorigenicity in mice: A preliminary note. J. Natl. Cancer Inst. 42:1101-1114.
Jacobson-Kram, D., and D. Montalbano. 1985. The Reproductive Effects Assessment Group's report on the mutagenicity of inorganic arsenic. Environ. Mutagen. 7:787-804.
Jha, A.N., M. Noditi, R. Nilsson, and A.T. Natarajan. 1992. Genotoxic effects of sodium arsenite on human cells. Mutat. Res. 284:215-221.
Jones, J.S., S. Weber, and L. Prakash. 1988. The Saccharomyces cerevisiae RAD 18 gene encodes a protein that contains potential zinc finger domains for nucleic acid binding and a putative nucleotide binding sequence. Nucleic Acids Res. 16:7119-7131.
Joshi, S., and J.B. Hughes. 1981. Inhibition of coupling factor B activity by cadmium ion, arsenite-2,3-dimercaptopropanol, and phenylarsine oxide, and preferential reactivation by dithiols. J. Biol. Chem. 256:11112-11116.
Kägi, J. H. R. 1993. Evolution, structure and chemical activity of class I metallothioneins: An overview. Pp 29-56 in Metallothionein III: Biological Roles and Medical Implications, K.T. Suzuki, N. Imura, and M. Kimura, eds. Berlin: Birkhauser Verlag.
Kaise, T., H. Yamauchi, Y. Horiguchi, T. Tani, S. Watanabe, T. Hirayama, and S. Fukui. 1989. A comparative study on acute toxicity of methylarsonic acid, dimethylarsinic acid, and trimethylarsine oxide in mice. Appl. Organomet. Chem. 3:273-277.
Kane, M.F., M. Loda, G.M. Gaida, J. Lipman, R. Mishra, H. Goldman, J.M. Jessup, and R. Kolodner. 1997. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH 1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 57:808-811.
Page 222
Kanisawa, M., and H.A. Schroeder. 1969. Life term studies on the effect of trace elements on spontaneous tumors in mice and rats. Cancer Res. 29:892-895.
Keyse, S.M., and R.M. Tyrrell. 1989. Heme oxygenase is the major 32-Kda stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 86:99-103.
Klaassen, C.D. 1995. Heavy Metals and heavy-metal antagonists. Pp. 16491672 in Goodman and Gilman's Pharmacological Basis of Therapeutics, 9th Ed., J.G. Hardman, L.E. Limbird, P.M. Molinoff, R.W. Ruddon and A.G. Gilman. eds. New York: McGraw-Hill.
Klaassen, C.D., and S.Z. Cagen. 1981. Metallothionein as a trap for reactive organic intermediates. Adv. Exp. Med. Biol. 136(Pt A):633-646.
Knowles, F.C., and A.A. Benson. 1984. The enzyme inhibitory form of inorganic arsenic. Z. Gesamte. Hyg. 30:625-626.
Kotsonis, F., and C.D. Klaassen. 1979. Increase in hepatic metallothionein in rats treated with alkylating agents. Toxicol. Appl. Pharmacol. 51:1927.
Kowalski, L.A., S.S. Tsang, and A.J. Davison. 1996. Arsenic and chromium enhance transformation of bovine papillomavirus DNAtransfected C3H/01T1/2 cells. Cancer Lett. 103:65-69.
Kreppel, H., K. Kolb, F.X. Reichl, B. Fichtl, and W. Forth. 1988. Pretreatment with low doses of cadmium or zinc decreases lethality in mice acutely poisoned with arsenic. Pp. 594-600 in Trace Element Analytical Chemistry in Medicine and Biology, Vol. 5, P. Brätter and P. Schramel, eds. Berlin: de Gruyter.
Kreppel, H., J. Bauman, J. Liu, J.M. McKim, Jr., and C.D. Klaassen. 1993. Induction of metallothionein by arsenicals in mice. Fundam. Appl. Toxicol. 20:184-189.
Kreppel, H., J. Liu, Y.P. Liu, F.X. Reichl, and C.D. Klaassen. 1994. Zincinduced arsenite tolerance in mice. Fundam. Appl. Toxicol. 23:32-37.
Kroes, R., M.J. van Logten, J.M. Berkvens, T. De Vries, and G.J. van Esch. 1974. Study on the carcinogenicity of lead arsenate and sodium arsenate and on the possible synergistic effect of diethylnitrosamine. Food Cosmet. Toxicol. 12:671-679.
Laib, R.J., and H. Moritz. 1989. Investigation of tumor initiating and/or cocarcinogenic properties of arsenite and arsenate with the rat liver foci bioassay. Exp. Pathol. 37:231-233.
Larramendy, M.L., N.C. Popescu, and J.A. DiPaolo. 1981. Induction by inorganic metal salts of sister chromatid exchanges and chromosome aberrations in human and Syrian hamster cell strains. Environ. Mutagen.
Page 223
3:597-606.
Lazo, J.S., and B.R. Pitt. 1995. Metallothioneins and cell death by anticancer drugs. Annu. Rev. Pharmacol. Toxicol. 35:635-653.
Lee, T.C., and I.C. Ho. 1994. Differential cytotoxic effects of arsenic on human and animal cells. Environ. Health Perspect. 102(Suppl 3):101-105
Lee, T.C., R.Y. Huang, and K.Y. Jan. 1985. Sodium arsenic enhances the cytotoxicity, clastogenicity, and 6-thioguanine-resistant mutagenicity of ultraviolet light in Chinese hamster ovary cells. Mutat. Res. 148:83-89.
Lee, T.C., K.C. Lee, Y.J. Tzeng, R.Y. Huang, and K.Y. Jan. 1986. Sodium arsenite potentiates the clastogenicity and mutagenicity of DNA crosslinking agents. Environ. Mutagen. 8:119-128.
Lee-Chen, S.F., C.T. Yu, D.R. Wu, and K.Y. Jan. 1994. Differential effects of luminol, nickel and arsenite on the rejoining of ultraviolet light and alkylation-induced DNA breaks. Environ. Mol. Mutagen. 23:116120.
Lerda, D. 1994. Sister-chromatid exchange (SCE) among individuals chronically exposed to arsenic in drinking water. Mutat. Res. 312:111120.
Li, J.H., and T.G. Rossman. 1989a. Mechanism of co-mutagenesis of sodium arsenite with N-methyl-N-nitrosourea. Biol. Trace Elem. Res. 21:373-381.
Li, J.H., and T.G. Rossman. 1989b. Inhibition of DNA ligase activity by arsenite: a possible mechanism of its comutagenesis. Mol. Toxicol. 2:19.
Li, J.H., and T.G. Rossman. 1991. Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells. Biol. Metals 4:197200.
Lopez, S., Y. Miyashita, and S.S. Simons. 1990. Structurally based selective interaction of arsenite with steroid receptors. J. Biol. Chem. 265:16039-16042.
Mahaffey, K.R., and B.A. Fowler. 1977. Effects of concurrent administration of lead; cadmium, and arsenic in the rat. Environ. Health Perspect. 19:165-171.
Mahaffey, K.R., S.G. Capar, B.C. Gladen, and B.A. Fowler. 1981. Concurrent exposure to lead, cadmium, and arsenic: effects on toxicity and tissue metal concentrations in the rat. J. Lab. Clin. Med. 98:463-481.
Maitani, T., N. Saito, M. Abe, S. Uchiyama, and Y. Saito. 1987. Chemical form-dependent induction of hepatic zinc-thionein by arsenic administration and effect of co-administered selenium in mice. Toxicol. Lett. 39:63-70.
Mass, M.J. and L. Wang. 1997. Arsenic alters cytosine methylation patterns
Page 224
of the promoter of the tumor suppressor gene p53 in human lung cells: A model for a mechanism of carcinogenesis. Mutat. Res. 386:263-277.
Milner, J.E. 1969. The effect of ingested arsenic on methylcholanthreneinduced skin tumors in mice. Arch. Environ. Health 18:7-11.
Moore, L.E., M.L. Warner, A.H. Smith, D. Kalman, and M.T. Smith. 1996. Use of the flourescent micronucleus assay to detect the genotoxic effects of radiation and arsenic exposure in exfoliated human epithelial cells. Environ. Mol. Mutagen. 27:176-184.
Moore, L.E., A.H. Smith, C. Hopenhayn-Rich, M.L. Biggs, D.A. Kalman, and M.T. Smith. 1997. Micronuclei in exfoliated bladder cells among individuals chronically exposed to arsenic in drinking water. Cancer Epidemiol. Biomarkers Prev. 6:31-36.
Moore, M.M., K. Harrington-Brock, and C.L. Doerr. 1997. Relative genotoxic potency of arsenic and its methylated metabolites. Mutat. Res. 386:279-290.
Ng, J.C. A.A. Seawright, L. Qi, C.M. Garnett, M.R. Moore, and B. Chriswell. 1998. Tumors in mice induced by chronic exposure of high arsenic concentration in drinking water [abstract]. P. 28 in Book of Abstracts of the Third International Conference on Arsenic Exposure and Health Effects, July 12-15, San Diego, Calif.
Nordenson, I., and L. Beckman. 1991. Is the genotoxic effects of arsenic mediated by oxygen free radicals? Hum. Hered. 41:71-73.
Nordenson, I., G. Beckman, L. Beckman, and S. Nordstrom. 1978. Occupational and environmental risks in and around a smelter in northern Sweden. II. Chromosomal aberrations in workers exposed to arsenic. Hereditas 88:47-50.
Okui, T., and Y. Fujiwara. 1986. Inhibition of human excision DNA repair by inorganic arsenic and the co-mutagenic effect in V79 Chinese hamster cells. Mutat. Res. 172:69-76.
Oya-Ohta, Y., T. Kaise, and T. Ochi. 1996. Induction of chromosomal aberrations in cultured human fibroblasts by inorganic and organic arsenic compounds and the different roles of glutathione in such induction. Mutat. Res. 357:123-129.
Packer, L. 1961. Metabolic and structural states of mitochondria. II. Regulation by phosphate. J. Biol. Chem. 236:214-220.
Peoples, S.A. 1975. Review of arsenical pesticides. Pp. 1-12 in Arsenical Pesticides, ACS Symp. Ser 7, E.A. Woolson, ed. Washington, D.C.: American Chemical Society.
Ramirez, P., D.A. Eastmond, J.P. Laclette, and P. Ostrosky-Wegman. 1997. Disruption of microtubule assembly and spindle formation as a mechanism for the induction of aneuploid cells by sodium arsenite and vanadium
Page 225
pentoxide. Mutat. Res. 386:291-298.
Rasmussen, R.E., and D.B. Menzel. 1997. Variation in arsenic-induced sister chromatid exchange in human lymphocytes and lymphoblastoid cells lines. Mutat. Res. 386:299-306.
Rin, K., K. Kawaguchi, K. Yamanaka, M. Tezuka, N. Oku, and S. Okada. 1995. DNA-strand breaks induced by dimethylarsinic acid, a metabolite of inorganic arsenics, are strongly enhanced by superoxide anion radicals. Biol. Pharm. Bull. 18:45-48.
Rossman, T.G. 1981. Enhancement of UV-mutagenesis by low concentrations of arsenite in E. coli. Mutat. Res. 91:207-211.
Rossman, T.G. 1998. Molecular and genetic toxicology of arsenic. Environ. Toxicol. 17:171-187.
Rudel, R., T.M. Slayton, and B.D. Beck. 1996. Implications of arsenic genotoxicity for dose response of carcinogenic effects. Regul. Toxicol. Pharmacol. 23:87-105.
Sato, M., and I. Bremner. 1993. Oxygen free radicals and metallothionein. Free Radicals Biol. Med. 14:325-337.
Schrauzer, G.N., and D. Ishmael. 1974. Effects of selenium and arsenic on the genesis of spontaneous mammary tumors in inbred C3H mice. Ann. Clin. Lab. Sci. 4:441-447.
Schrauzer, G.N., D.A. White, J.E. McGinness, C.J. Schneider, and L.J. Bell. 1978. Arsenic and cancer: Effects of joint administration of arsenite and selenite on the genesis of mammary adenocarcinoma in inbred female C3H/St mice. Bioinorganic Chem. 9:245-253.
Schroeder, H.A., M. Kanisawa, D.V. Frost, and M. Mitchener. 1968. Germanium, tin and arsenic in rats: effects on growth, survival, pathological lesions and life span. J. Nutrition 96:37-45.
Schwarz, M.A., J.S. Lazo, J.C. Yalowich, W.P. Allen, M. Whitmore, H.A. Bergonia, E. Tzeng, T. Billiar, P.D. Robbins, J.R. Lancaster, and B.R. Pitt. 1995. Metallothionein protects against the cytotoxic and DNAdamaging effects of nitric oxide. Proc. Natl. Acad. Sci. USA 92:4452-4456.
Shibata, A., P.F. Ohneseit, Y.C. Tsai, C.H. Spruck, P.W. Nichols, H.S. Chiang, M.K. Lai, and P.A. Jones. 1994. Mutational spectrum in the p53 gene in bladder tumors from the endemic area of blackfoot disease in Taiwan. Carcinogenesis 15:1085-1087.
Shirachi, D.Y., M.G. Johansen, J.P. McGowen, and S.H. Tu. 1983. Tumorigenic effect of sodium arsenite in rat kidney. Proc. West. Pharmacol. Soc. 26:413-415.
Silver, S., and L.T. Phung. 1996. Bacterial heavy metal resistance: New surprises. Annu. Rev. Microbiol. 50:753-789.
Page 226
Simons, S.S., P.K. Chakraborti, and A.H. Cavanaugh. 1990. Arsenite and cadmium (II) as probes of glucocorticoid receptor structure and function. J. Biol. Chem. 265:1938-1945.
Smith, A.H., C. Hopenhayn-Rich, M.N. Bates, H.M. Goeden, I. HertzPicciotto, H.M. Duggan, R. Wood, M.J. Kosnett, and M.T. Smith. 1992. Cancer risks fron arsenic in drinking water. Environ. Health Perspect. 97:259-267.
Squibb, K.S., and B.A. Fowler. 1983. The toxicity of arsenic and its compounds. Pp 233-270 in Biological and Environmental Effects of Arsenic, B.A. Fowler, ed. Amsterdam: Elsevier.
Taketani, S., H. Kohno, T. Yoshinaga, and R. Tokunaga. 1989. The human 32-kDa stress protein induced by exposure to arsenite and cadmium ions is heme oxygenase. FEBS Lett. 245:173-176.
Tamai, K.T., E.B. Gralla, L.M. Ellerby, J.S. Valentine, and D.J. Thiele. 1993. Yeast and mammalian metallothioneins functionally substitute for yeast copper-zinc superoxide dismutase. Proc. Natl. Acad. Sci. USA 90:8013-8017.
Tanaka, K., N. Miura, I. Satokata, I. Miyamoto, M.C. Yoshida, Y. Satoh, S. Kondo, A. Yasui, H. Okayama, and Y. Okada. 1990. Analysis of a human DNA excision repair gene in group A xeroderma pigmentosum and containing a zinc-finger domain. Nature 348:73-76.
Tezuka, M., K. Hanioka, K. Yamanaka, and S. Okada. 1993. Gene damage induced in human alveolar type II (L-132) cells by exposure to dimethylarsinic acid. Biochem. Biophys. Res. Commun. 191:1173-1183.
Thomas, J., G. Bachowski, and A. Girotti. 1986. Inhibition of cell membrane lipid peroxidation by cadmium- and zinc-metallothioneins. Biochim. Biophys. Acta 884:448-461.
Thorgeirsson, U.P., D.W. Dalgard, J. Reeves, and R.H. Adamson. 1994. Tumor incidence in a chemical carcinogenesis study of nonhuman primates. Regul. Toxicol. Pharmacol. 19:130-151.
Thornalley, P.J., and M. Vasäk. 1985. Possible role for metallothionein in protection against radiation-induced oxidative stress: kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim. Biophys. Acta 27:36-44.
Uchida, K., T. Morita, T. Sato, T. Ogura, S. Yamashita, S. Noguchi, H. Suzuki, H. Nyunoya, M. Miwa, and T. Sugimura. 1987. Nucleotide sequence of a full-length cDNA for human fibroblast poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 148:617-622.
Van Delft, S., P. Coffer, W. Kruijer, and R. Van Wijk. 1993. c-fos induction by stress can be mediated by the SRE. Biochem. Biophys. Res. Commun. 197:542-548.
Page 227
Van Wijk, R., M. Welters, J.E. Souren, H. Overlgonne, and F.A. Wiegant. 1993. Serum-stimulated cell cycle progression and stress protein synthesis in C3H1OT1/2 fibroblasts treated with sodium arsenite. J. Cell Physiol. 155:265-272.
Vega, L., M.E. Gonsebatt, and P. Ostrosky-Wegman. 1995. Aneugenic effect of sodium arsenite on human lymphocytes in vitro: An individual susceptibility effect detected. Mutat. Res. 334:365-373.
Waalkes, M.P., M.J. Harvey, and C.D. Klaassen. 1984. Relative in vitro affinity of hepatic metallothionein for metals. Toxicol. Lett. 20:33-39.
Wanibuchi, H., S. Yamamoto, H. Chen, K. Yoshida, G. Endo, T. Hori, and S. Fukushima. 1996. Promoting effects of dimethylarsinic acid on Nbutyl-N-(4-hydroxybutyl)nitrosamine-induced urinary bladder carcinogenesis in rats. Carcinogenesis 17:2435-2439.
Warner, M.L., L.E. Moore, M.T. Smith, D.A. Kalman, E. Fanning, and A.H. Smith. 1994. Increased micronuclei in exfoliated bladder cells of individuals who chronically ingest arsenic-contaminated water in Nevada. Cancer Epidemiol. Biomarkers Prevent. 3:583-590.
Wiencke, J.K., and J.W. Yager. 1992. Specificity of arsenite in potentiating cytogenetic damage induced by the DNA crosslinking agent diepoxybutane. Environ. Mol. Mutagen. 19:195-200.
Wiencke, J.K., J.W. Yager, A. Varkonyi, M. Hultner, and L.H. Lutze. 1997. Study of arsenic mutagenesis using the plasmid shuttle vector pZ189 propagated in DNA-repair proficient human cells. Mutat. Res. 386:335-344.
Woods, J.S. and B.A. Fowler. 1978. Altered regulation of mammalian hepatic heme biosynthesis and urinary porphyrin excretion during prolonged exposure to sodiium arsenite. Toxicol. Appl. Pharmacol. 43:361-371.
Woods, J.S., M.A. Bowers, and H.A. Davis. 1991. Urinary porphyrin profiles as biomarkers of trace metal exposure and toxicity: Studies on urinary porphyrin excretion patterns in rats during prolonged exposure to methyl mercury. Toxicol. Appl. Pharmacol. 110:464-476.
Wu, W., and M.J. Welsh. 1996. Expression of the 25-kDa heat shock protein (HSP27) correlates with resistance to the toxicity of cadmium chloride, mercuric chloride, cis-platinum(II)-diamine dichloride, or sodium arsenite in mouse embryonic stem cells transfected with sense or antisense HSP27 cDNA. Toxicol. Appl. Pharmacol. 141:330-339.
Yager, J.W., and J.K. Wiencke. 1997. Inhibition of poly(ADP-ribose) polymerase by arsenite. Mutat. Res. 386:345-351.
Yamamoto, S., Y. Konishi, T. Matsuda, T. Murai, M.A. Shibata, I. MatsuiYuasa, S. Otani, K. Kuroda, G. Endo, and S. Fukushima. 1995. Cancer
Page 228
induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid) in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res. 55:1271-1276.
Yamanaka, K., A. Hasegawa, R. Sawamura, and S. Okada. 1989. Dimethylated arsenics induce DNA strand breaks in lung via the production of activated oxygen species in mice. Biochem. Biophys. Res. Commun. 165:43-50.
Yamanaka, K., A. Hasegawa, R. Sawamura, and S. Okada. 1991. Cellular response to oxidative damage in lung induced by the administration of dimethylarsinic acid, a major metabolite of inorganic arsenics, in mice. Toxicol. Appl. Pharmacol. 108:205-213.
Yamanaka, K., M. Tezuka, K. Kato, A. Hasegawa, and S. Okada. 1993. Crosslink formation between DNA and nuclear proteins by in vivo and in vitro exposure of cells to dimethylarsinic acid. Biochem. Biophys. Res. Commun. 191:1184-1191.
Yamanaka, K., K. Ohtsubo, A. Hasegawa, H. Hayashi, H. Ohji, M. Kanisawa, and S. Okada. 1996. Exposure to dimethylarsinic acid, a main metabolite of inorganic arsenics, strongly promotes tumorigenesis initiated by 4-nitroquinoline 1-oxide in the lungs of mice. Carcinogenesis 17:767-770.
Yamauchi, H., T. Yoshida, H. Aikawa, F. Kayama, H. Aminaka, K. Yoshida, M. Akkerman, and B.A. Fowler. 1998. Metabolism and biological monitoring of arsenic poisoning following chronic arsenic exposure in Inner Mongolia, China [abstract]. Toxicological Sciences Supplement. Toxicologist 42(1-S):321.
Yang, J.L., M.F. Chen, C.W. Wu, and T.C. Lee. 1992. Post-treatment with sodium arsenite alters the mutational spectrum induced by ultraviolet light irradiation in Chinese hamster ovary cells. Environ. Mol. Mutagen. 20:156-164.
Zhao, C.Q., M.R. Young, B.A. Diwan, T.P. Coogan, and M.P. Waalkes. 1997. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. 94:10907-10912.




































