
19
Inflammatory Stress and the Immune System
Leonard P. Kapcala1
Introduction
During the last several years, it has become apparent that important ''bidirectional'' communication occurs between the neuroendocrine and immune systems. More precisely this cross-talk occurs among the immune, endocrine, and central nervous systems and relates to perturbations in any of these systems (Besedovsky and del Ray, 1996). Communication is mediated by a variety of molecules, including neuropeptides, neurotransmitters, hormones, and cytokines which are contained within these systems along with their respective receptors. Learning about these interactions has particularly been made possible bassed on the discovery and characterization of cytokines (Besedovsky and del Ray, 1996). Cytokines are glycoproteins/proteins produced by many cell types, including macrophages and monocytes, B- and T-lymphocytes, endothelial cells, fibroblasts, neurons, glia (microglia and astrocytes), and some epithelial cells. Cytokines act on many cell types, exert redundant actions, induce the
production and secretion of other cytokines (e.g., stimulate a cascade of many cytokines), and often synergize with other cytokines to potentiate their actions.
One immune-neuroendocrine interaction that has been extensively investigated pertains to cytokine regulation of the hypothalamic-pituitaryadrenal axis (HPAA) (Bateman et al., 1989; Besedovsky and del Ray, 1996; Chrousos, 1995; Gaillard, 1994; Harbuz and Lightman, 1992; Koenig, 1991; Lilly and Gann, 1992; Reichlin, 1993; Rivier, 1995; Tilders et al., 1994). Although many cytokines appear to modulate HPAA activation, the most important cytokines involved in this regulation are interleukin (IL)-1, IL-6, and tumor necrosis factor-α (TNF-α). Major biochemical components in this neuroendocrine axis (Figure 19-1) include hypothalamic corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP), anterior pituitary adrenocorticotropin (ACTH), and adrenal glucocorticoids (cortisol in humans; corticosterone [CORT] in rodents) (Chrousos, 1995). In response to stress, HPAA activation occurs primarily by increased release of hypothalamic CRH and AVP (Harbuz and Lightman, 1992). Whereas AVP, a relatively weak secretagogue for ACTH secretion, markedly potentiates CRH stimulation and may be a critical factor in facilitating responses to recurrent stress (Harbuz and
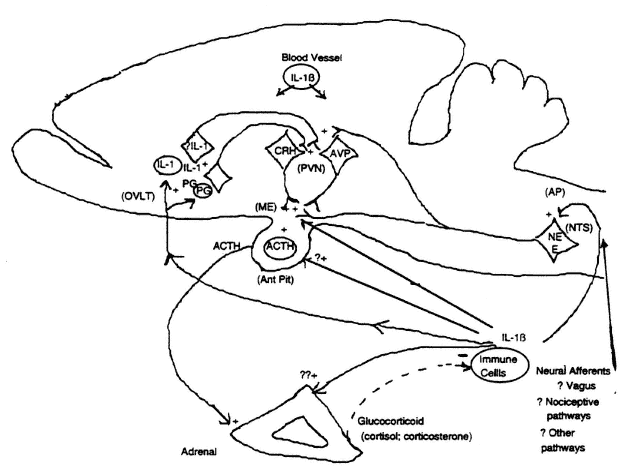
FIGURE 19-1
Schematic model of potential regulatory relationships between cytokines (using interleukin [IL]-1 as prototype) and the hypothalamic-pituitary-adrenal axis (HPAA) in the rat. AP, area postrema; AVP, arginine vasopressin; CRH, corticotropin-releasing hormone; E, epinephrine; ME, median eminence; NE, norepinephrine; NTS, nucleus tractus solitarius; OVLT, organum vasculosum of lamina terminalis; PG, prostaglandin; PVN, paraventricular nucleus. SOURCE: Adapted from Kapcala et al. (1995).
Lightman, 1992), CRH is the major stimulator of ACTH release. Stimulated ACTH secretion subsequently stimulates adrenocorticosteroid (CS, particularly glucocorticoid) secretion. Glucocorticoids in turn modulate and inhibit HPAA activation via negative feedback effects at suprahypothalamic, hypothalamic, and pituitary levels (Harbuz and Lightman, 1992).
Why does the HPAA become activated during various immune, inflammatory, and infectious insults? According to a hypothesis proposed by Munck and colleagues (1984) several years ago, such activation of the HPAA occurs so that glucocorticoids can suppress immune and inflammatory responses initiated by cytokines and thereby can modulate and dampen immune system activation. This dampening of immune system activation prevents more severe and excessive catabolic effects, including the ultimate deleterious effect, death. Consequently, immune system activation of the HPAA appears to occur to modulate excessive, deleterious effects of cytokines after they have produced their initial, beneficial effects (Urbaschek and Urbaschek, 1987; Vogel and Hogan, 1990) in facilitating an inflammatory response. A fine balance occurs relative to the level of cytokine activity. In general, relatively low levels of specific cytokines promote beneficial protective effects in helping the host respond to a perturbing immune, inflammatory, or infectious challenge. In contrast, uncontrolled production of specific cytokines resulting in relatively high circulating levels often results in severe pathological consequences, such as hypotension and lethal shock. Although responses to specific inflammatory, immune, or infectious insults are not necessarily identical, they provoke a similar central neuroendocrine response via the generation of similar cytokines and thus can all be viewed as "inflammatory stress." For purposes of discussion, these environmental perturbations can be viewed similarly not only because they induce a similar counterregulatory response (i.e., HPAA activation) but also because the consequences of this response expose the organism to similar immunosuppressive and anti-inflammatory actions following the initial induction of immune potentiating and inflammatory effects (Besedovsky and del Ray, 1996). These actions are aimed at controlling the disruption of homeostasis produced by the offending agent or stimulus.
Immune Regulation of the HPAA by Cytokines
IL-1, TNF-α, and IL-6 are the most important cytokines for stimulating the HPAA (Chrousos, 1995; Gaillard, 1994; Reichlin, 1993). Because IL-1 is the most potent (on a molar basis) cytokine that activates the HPAA, and the most frequently studied relative to the HPAA, stimulation of the HPAA by IL-1 is often viewed as a prototypical model for immune activation of the HPAA. Additional complexity is added by the fact that IL-1 exists in two forms (α, which is primarily membrane-associated, and β, which is primarily secreted); that there are at least two IL-1 receptors; and that IL-1 actions can be counterregulated by an endogenous receptor antagonist (IL-1ra) (Dinarello,
1992; Pruitt et al., 1995; Schöbitz et al., 1994). IL-1 effects may also be diminished by soluble receptors or a "decoy" receptor that is not coupled to a signal transduction message. Furthermore, IL-1 can exert a positive auto-feedback whereby it stimulates its own expression in the periphery (Dinarello et al., 1987) and brain (Gao et al., 1996; He et al., 1996) and stimulates hypothalamic transcription of its type 1 receptor (Gao et al., 1996, based on inhibition of an IL-1 stimulated increase in mRNA by a pharmacological inhibitor of transcription). Under acute circumstances, cytokines are thought to act centrally to stimulate hypothalamic CRH and perhaps AVP release (Bateman et al., 1989; Chrousos, 1995; Gaillard, 1994; Harbuz and Lightman, 1992; Lilly and Gann, 1992; Reichlin, 1993; Rivier, 1995), and subsequently ACTH and glucocorticoid secretion are stimulated.
Other studies suggest the possibility that direct stimulation of IL-1 at the pituitary level may also occur, but that stimulation at this level develops primarily during more prolonged cytokine activation (Chrousos, 1995; Gaillard, 1994; Koenig, 1991; Kehrer et al., 1988). However, it is not clear whether such stimulation is related to circulating cytokines or induction of IL-1, other cytokines, or other events within the pituitary via paracrine effects (Koenig et al., 1990). In addition, direct stimulation of IL-1 at the level of the adrenal has been suggested based on in vitro and in vivo studies (Andreis et al., 1991; Bateman et al., 1989; Chrousos, 1995; Gaillard, 1994; Gwosdow et al., 1992). This stimulation also appears to require prolonged exposure to IL-1. If such cytokine stimulation at these other levels of the axis are of physiological import, these mechanisms could be particularly significant during prolonged immune/cytokine system stimulation.
The major mechanism (Figure 19-1) mediating acute stimulation of ACTH secretion by IL-1 appears to involve CRH release (Bateman et al., 1989; Chrousos, 1995; Matta et al., 1990; Rivier, 1995; Sapolsky et al., 1987) and possibly AVP release (Nakatsuru et al., 1991; Watanobe and Takebe, 1994; Whitnall et al., 1992) from terminals at the median eminence. Based on many studies, it appears that the potency of the immune stimulus is directly related to the magnitude and duration of HPAA activation. This phenomenon of prolonged activation is manifested by higher levels of circulating ACTH or glucocorticoid for longer periods. IL-1 administered peripherally in relatively high doses or centrally also increases CRH mRNA (Brady et al., 1994; Ericsson et al., 1994; Harbuz et al., 1992b; Rivier, 1995; Suda et al., 1990) and immunocytochemical CRH (Ju et al., 1991; Rivest et al., 1992) in some parvocellular paraventricular nuclear (PVN) neurons, which are involved in stimulating the HPAA. Furthermore, IL-1 stimulates expression of mRNA and protein of an immediate-early gene (c-fos) in PVN and several other brain sites (Brady et al., 1994; Chang et al., 1993; Ericsson et al., 1994; Ju et al., 1991; Rivest et al., 1992; Veening et al., 1993). Together, these studies illustrate activation of the CRH perikaryon (i.e., cell body); however, the relationship between IL-1-induced c-fos production and activation of CRH gene expression
is not clear. Nevertheless, it appears that stimulation of perikarya producing CRH in PVN results in enhanced CRH synthesis when a sufficiently strong immune stimulus above a certain threshold induces significant cytokine production and secretion. Augmented CRH synthesis could maintain increased CRH release and therefore facilitate prolonged HPAA activation. In addition, centrally administered IL-1 increased AVP mRNA in PVN (Lee and Rivier, 1994), and it was found that endotoxin/lipopolysaccharide (LPS), which induces the production of cytokines, increased AVP mRNA in PVN when administered peripherally in a high dose (Kapcala et al., 1995). Increased release of AVP into the pituitary portal circulation has also been found in an animal model of inflammatory arthritis (Harbuz et al., 1992a), raising the possibility that different mechanisms may be involved in facilitating chronic activation of the HPAA in response to a chronic inflammatory stimulus.
Central Actions of IL-1
Although it is clear that peripherally generated cytokines activate the HPAA centrally, precise mechanisms by which this occurs have not been clearly established. Recognizing the presence of IL-1 receptors in many sites throughout the brain (Cunningham and DeSouza, 1994), including circumventricular organs, there are several putative mechanisms by which peripherally generated cytokines may communicate with the brain and more specifically regulate the HPAA (Figure 19-1). Although IL-1 transport into the brain has been described (Banks et al., 1991), stimulation via this mechanism is not widely held because IL-1 does not easily cross the blood-brain barrier (Coceani et al., 1988) under normal circumstances. However, with increased levels of circulating cytokines, the blood-brain barrier may be made more permeable to macromolecules such as cytokines (Burrought et al., 1992; Saija et al., 1995) and permit some cytokine entry into brain. Alternatively, a peripheral cytokine such as IL-1 might transduce brain signaling by stimulating endothelial IL-1 receptors (Cunningham and DeSouza, 1994; Dinarello, 1992; Tilders et al., 1994). Subsequently, a cytokine signal may spread throughout brain parenchyma (Breder et al., 1994; van Dam et al., 1995) by mechanisms that remain to be elucidated. Actions at circumventricular organs (e.g., the organum vasculosum of lamina terminalis [OVLT], median eminence, and area postrema—brain regions where a normal blood-brain barrier is not intact) could be important. This concept is consistent with stimulating CRH secretion from the median eminence (Matta et al., 1990) via IL-1 receptor stimulation of catecholamine release from axon terminals in this location. Additional evidence suggests peripheral IL-1 signaling of the brain and the HPAA particularly at the OVLT (Gaillard, 1994; Katsuura et al., 1990; Tilders et al., 1994). More specifically, it has been proposed (Katsuura et al., 1990) that IL-1 enters the OVLT and stimulates cells such as astrocytes to synthesize and release prostaglandins, which stimulate neuronal circuits that ultimately activate CRH
and AVP neurons in the PVN. Stimulation of presumably transcription of an immediate early gene such as c-fos in the nucleus tractus solitarius of the medulla, which sends noradrenergic projections to the median eminence and PVN, could reflect neuronal activation by IL-1 and would support peripheral IL-1 stimulation of the brain via the nearby area postrema. In consonance with this view, neuroanatomical cuts caudal to hypothalamus inhibit HPAA activation by intravenous (iv) IL-1β (Sawchenko et al., 1996).
Brain signaling by peripheral cytokines may also involve stimulation of afferent sensory circuits such as the vagus or peripheral nociceptive (pain-transmitting) neural pathways (Dantzer et al., 1994; Donnerer et al., 1992; Lilly and Gann, 1992; Wan et al., 1994). Subdiaphragmatic vagotomy inhibits the induction of many central effects produced by IL-1 or LPS particularly when administered intraperitoneally (Bluthe et al., 1994; Laye et al., 1995; Maier et al., 1993; Wan et al., 1994; Watkins et al., 1994a, b, 1995a). Of interest to the focus here, vagotomy inhibits stimulation of ACTH by intraperitoneal (ip) IL-1β (Kapcala et al., 1996) and LPS (Gaykema et al., 1995) and stimulation of CORT (Fleshner et al., 1995) secretion by ip IL-1β. Treatment with capsaicin (an alkaloid derivative of red pepper that inhibits the function of peripheral sensory afferents, including those mediating nociception) inhibits stimulation of plasma ACTH and CORT by iv IL-1 (Watanobe et al., 1994). Thus peripheral afferents may also play an important role in brain signaling by peripheral cytokines. Finally, it is also possible that invoking different mechanisms may not necessarily be mutually exclusive. Different mechanisms for brain signaling by peripheral cytokines may operate simultaneously or under specific circumstances depending on the body compartment in which the primary cytokine stimulus arises.
The Role of the Locus of Origin of IL-1
Increasingly, a consensus has been developing among researchers in the field that many mechanisms may be responsible for cytokine stimulation of the HPAA by an inflammatory, infectious, or immune stress and that the body compartment in which the stimulus originates may primarily dictate the main mechanism involved. Experimental corollaries of this view are that the administration of a cytokine such as IL-1 by a different route (e.g., iv, ip, intracerebroventricular [icv]) may result in the utilization of different mechanisms of activation of the HPAA (Rivier, 1995; Tilders et al., 1994).
Different modulatory effects of various regulatory factors on IL-1 activation of the HPAA have been reported depending on the route of IL-1 administration. Whereas inhibition of prostaglandin synthesis does not consistently and potently inhibit stimulation of ACTH or CORT by ip IL-1, this treatment virtually abolishes stimulation by iv IL-1 (Dunn and Chuluyan, 1992; Rivier, 1993). Lesioning of central noradrenergic pathways that modulate activity of the HPAA has different effects on stimulation of the HPAA
depending on whether IL-1 is administered centrally or peripherally (e.g., intra-arterially) (Barbanel et al., 1993). Removal of CS negative feedback by adrenalectomy abolished IL-1 stimulation of ACTH secretion when IL-1 was given centrally (Weidenfeld et al., 1989), but did not diminish stimulation when IL-1 was given peripherally (iv or ip) (Selmanoff et al., 1996). Finally, peripherally (ip or iv) administered IL-1 in doses that potently stimulated ACTH and CORT secretion did not stimulate gene expression of CRH and AVP in PVN as did centrally administered IL-1 (Lee and Rivier, 1994). Altogether, these observations support the likelihood of a developing overview that cytokine activation of the HPAA is quite complex and involves a multiplicity of mechanisms.
Use of LPS to Elucidate the Effects of IL-1
LPS, derived from the cell wall of gram-negative bacteria, induces the septic shock syndrome and is often used as a model for studying the sepsis syndrome in experimental animals or conditions associated with marked induction of cytokines. Not surprisingly, LPS is a potent activator of the HPAA, which has been recognized for many years. This action occurs via its stimulation of the production and release of cytokines (Bateman et al., 1989; Chrousos, 1995; 1993; Dunn, 1992; Ebisui et al., 1994; Perlstein et al., 1993; Vogel and Hogan, 1990), particularly IL-1 and TNF, but also perhaps IL-6. It has been proposed that LPS activates the HPAA mainly via generation of IL-1 (Rivier et al., 1989; Schotanus et al., 1993). It has also been proposed that induction of septic shock (Ohlsson et al., 1990; Pruitt et al., 1995; Russell and Tucker, 1995; Wakabayashi et al., 1991) by LPS may also be highly dependent on IL-1 because antagonism of IL-1 with IL-1ra was therapeutic. The level of circulating LPS, and correspondingly, the level of cytokines generated by LPS may also be important for determining the mechanism of HPAA stimulation. One study showed that low doses of LPS activated the HPAA mainly via TNF but that higher doses invoked an important role for IL-1 (Ebisui et al., 1994), suggesting that a different intensity of immune stimulation by the same factor may operate through different mechanisms. Other studies showed that macrophage depletion (DeRijk et al., 1991) and blockade of IL-6 actions (Perlstein et al., 1993) selectively attenuated relatively low-dose LPS stimulation. These results also support different mechanisms of HPAA stimulation depending on the magnitude of stimulation by LPS.
Despite beliefs that LPS stimulates the HPAA via induction of cytokines that acutely act primarily centrally, it seems likely that LPS may also stimulate ACTH and glucocorticoid secretion by generating cytokines that act directly on the anterior pituitary and possibly on the adrenal cortex. A variety of studies show that rats with hypothalamic resection, lesions, deafferentation, and pituitary stalk section (Elenkov et al., 1992; Makara et al., 1970, 1971) were still able to increase plasma CORT after LPS. Consequently, it appears that immune
stimulation of the stress axis may operate through several fail-safe mechanisms that facilitate at least a partial activation of this critically important response.
The Role of IL-1β
Expression of the IL-1β gene in brain can be induced by various stimuli including LPS, immobilization stress, ischemia, mechanical injury, and various pharmacological treatments (Ban et al., 1992; Dantzer et al., 1994; Higgins and Olschowka, 1991; Laye et al., 1995; Minami et al., 1991, 1992; Takao et al., 1993; van Dam et al., 1992, 1995; Yan et al., 1992). Although IL-1β mRNA has been reported to be present throughout several rat brain regions in the basal state (Bandtlow et al., 1990), and immunocytochemical IL-1β has been found in rat brain (Lechan et al., 1990), after central colchicine administration and in human brain (Breder et al., 1988) at autopsy, a consensus view among many investigators is that expression of brain IL-1β mRNA is minimal under basal, unstimulated conditions (Ban et al., 1992; Dantzer et al., 1994; Higgins and Olschowka, 1991; Laye et al., 1995; Minami et al., 1991, 1992; Takao et al., 1993; van Dam et al., 1992, 1995; Yan et al., 1992). Consistent with this perspective, this author (He et al., 1996) has found that peripherally (ip) administered LPS potently stimulates expression of IL-1β mRNA in specific brain regions including circumventricular organs (OVLT, median eminence, subfornical organ, area postrema) and parenchymal sites (PVN, arcuate-periarcuate region, vagal nucleus) that do not normally express this mRNA in the unstimulated state. Although peripherally administered IL-1β also induced IL-1β mRNA in these same regions, the intensity of stimulation was much weaker than LPS based on the number of cells expressing the mRNA and the signal intensity per cell. Considering that these stimuli activate the HPAA and that extremely small doses of centrally administered IL-1 potently activate the HPAA (Kapcala et al., 1995), induction of brain IL-1 expression particularly in hypothalamus by cytokines originating in the periphery has been considered as a potential mechanism by which amplification of immune activation of the HPAA could occur (Tilders et al., 1994) especially as a mechanism to prolong HPAA activation.
Immune System Activation of the HPAA Counterregulates Cytokines and Protects Against an Excessive Host Response to Inflammatory, Immune, and Infectious Insults
Lethality of IL-1 in Adrenalectomized or Hypophysectomized Animals
It has been proposed that CS secretion (Munck et al., 1984) protects against potentially deleterious, catabolic effects of cytokines produced during immune,
infectious, or inflammatory processes by downmodulating the production, release, and actions of cytokines. In support of this hypothesis is the markedly increased sensitivity to lethal effects in animals with a defective HPAA or surgically induced compromise of the HPAA (Table 19-1) after exposure to LPS, various inflammatory conditions that stimulate cytokine production, and cytokines themselves (Bertini et al., 1988; Butler et al., 1989; Harbuz et al., 1993; MacPhee et al., 1989; Nakano et al., 1987; Sternberg et al., 1989). Glucocorticoid treatment administered in some of these studies (Bertini et al., 1988; Butler et al., 1989; MacPhee et al., 1989; Nakano et al., 1987) protected against lethal effects (Table 19-1), which strongly suggests that the HPAA plays a protective role against immune or inflammatory stimuli.
Adrenalectomy (ADX) or hypophysectomy (HYPOX) (removal of the pituitary) also results in LPS stimulation of higher serum levels of IL-1 and TNF for longer periods, which indicates a modulatory role of the endogenous HPAA (Butler et al., 1989, Zuckerman et al., 1989). Increased sensitivity to lethal effects of IL-1 has been described in mice following ADX or HYPOX (Bertini et al., 1988; Butler et al., 1989), and enhanced lethal sensitivity to LPS has been reported in the rat (Nakano et al., 1987) (perhaps the most commonly studied
TABLE 19-1 Conditions Producing Lethal Effects in Animals with an Abnormal Hypothalamic-Pituitary-Adrenal Axis (HPAA)
|
Condition* |
Animal |
Abnormal HPAA |
Glucocorticoid Rx |
Reference |
|
IL-1β, TNF-α, LPS |
Mouse |
ADX |
DEX 30 mg/kg |
Bertini et al., 1988 |
|
IL-1β, TNF-α, LPS |
Mouse |
ADX, HYPOX |
DEX 10 mg/kg; CORT 100 mg/kg |
Butler et al., 1989 |
|
LPS |
Rat |
ADX |
DEX 0.05 mg/kg; CORT 5 mg/kg |
Nakano et al., 1987 |
|
Encephalitis (allergic) |
Rat |
ADX |
CORT 50 mg pellet |
MacPhee et al., 1989 |
|
Arthritis (streptococcal cell wall) |
Rat (Lewis) |
Central (CRH) hypoadrenalism |
|
Sternberg et al., 1989 |
|
Arthritis (adjuvant) |
Rat |
ADX |
|
Harbuz et al., 1993 |
|
NOTE: Rx, treatment; IL, interleukin; TNF, tumor necrosis factor; ADX, adrenalectomy; HYPOX, hypophysectomy; DEX, dexamethasone; CORT, corticosterone * Various doses of LPS, IL-1, or TNF were studied. SOURCE: Adapted from Kapcala et al. (1995). |
||||
laboratory animal) following ADX. However, this author was not aware that lethal effects of IL-1 had been reported in rats or in any other species (rabbits, monkeys) treated with relatively high doses of IL-1. Questions about the significance of lethal effects of IL-1 in isolated mouse studies were raised because mice show variable susceptibility to experimentally induced inflammatory conditions as a function of their genetic composition and activity of their HPAA (Mason et al., 1990) and can also exhibit idiosyncratic responses to specific stimuli that are not necessarily observed in other species. This laboratory was interested in determining whether lethality induced by IL-1 was a species-specific response exhibited solely in mice. Thus, rats were studied to place into perspective the physiological importance of immune activation of the HPAA and particularly to determine whether the increased sensitivity to lethal effects of IL-1 in mice is a general phenomenon associated with compromised CS secretion. The focus on IL-1 was especially influenced by the central role that IL-1 is thought to play in facilitating various immune, infectious, and inflammatory disorders (Dinarello and Wolff, 1993; Pruitt et al., 1995). This laboratory was additionally interested in determining the physiological significance of protection from lethality by glucocorticoid treatment because previous studies (Bertini et al., 1988; Butler et al., 1989) had shown protection against lethal effects of IL-1 only when pharmacological quantities of glucocorticoid were used.
The effects of ADX on lethal responses to IL-1β were studied in adult Sprague-Dawley rats and compared to effects of LPS. ADX rats exhibited dose-dependent lethality after IL-1β (generously provided by Janet Kerr and Maryanne Covington at DuPont-Merck) and ultimately 100 percent mortality at the highest dose studied (Table 19-2) (Kapcala et al., 1995). In contrast, rats with an intact HPAA did not show any lethal responses after similar doses of IL-1β. The treatment time after adrenal removal did not affect the lethal response to IL-1β based on studying rats at different times (2–23 days) after ADX. These results demonstrated that lethal effects produced by IL-1 in ADX mice were not species specific nor idiosyncratic to mice and clearly illustrated that IL-1β could be lethal to rats when CS secretion is compromised. Similar responses were seen after LPS. Rats with an intact HPAA tolerated a range of relatively high doses (4–40 mg/kg ip) of LPS during monitoring over 12 hours (Table 19-2). However, ADX rats were exquisitely sensitive to lethal effects of LPS (Table 19-2) and exhibited at least a 200-fold increased sensitivity.
Glucocorticoid Treatment in Adrenalectomized Animals
This laboratory also wanted to determine whether glucocorticoid treatment in physiological quantities reflecting stress-stimulated CS secretion could protect rats against cytokines, as does an intact HPAA. Treatment of ADX rats with CORT or dexamethasone in doses estimated to be equivalent to a physiological stress response was protective against lethal effects of IL-1β and
LPS (Table 19-2). Normal CORT replacement in the rat is approximately 2 to 3 mg/kg (Akana et al., 1985). Thus, it was estimated that CORT treatment would approximate a nearly maximal (10-fold increase of CORT secretion) stress response and that the dexamethasone dose would be the glucocorticoid equivalent of at least a moderate stress response and might possibly be equivalent to a maximal stress response. Conceivably, the lack of complete protection by CORT may have been related to the fact that the treatment was not optimized with CORT to mimic the normal kinetics of the CORT response shown by rats with an intact HPAA. Complete protection by treatment with dexamethasone compared with CORT may have been due to the longer plasma and biological half-life of dexamethasone. These unique observations clearly demonstrated that complete protection against lethality induced by IL-1β was afforded by glucocorticoid treatment in quantities approximating stress physiological CS secretion. Not only did these results show the lethal capability of a cytokine such as IL-1β in the face of a compromised HPAA, they also supported the hypothesis that physiological activation of the HPAA by immune-inflammatory response stimuli is a general means across species for counterregulating cytokine actions, modulating host defense immune-inflammatory reactions and ultimately protecting against potentially lethal actions of cytokines.
TABLE 19-2 Effects of IL-1β and Endotoxin/Lipopolysaccharide (LPS) on Percent Survival in Adrenalectomized or Hypophysectomized Rats (n = 4 to 5 rats per group)
|
Condition |
IL-1β (60 µg/kg ip) |
LPS (4 mg/kg ip) |
|
Sham ADX |
100 |
100 |
|
ADX |
0 |
0 |
|
ADX + DEX (0.2 mg/kg) |
100 |
100 |
|
ADX + CORT (24 mg/kg) |
75 |
67* |
|
Sham HYPOX |
100 |
100 |
|
HYPOX |
50† |
100 |
|
NOTE: ip, intraperitoneal; ADX, adrenalectomy; DEX, dexamethasone; CORT, corticosterone; HYPOX, hypophysectomy. * n = 3 in this group only. † Identical survival rate as ADX rats given same dose in this experiment. DEX or CORT were administered ip 1 hour prior to recombinant human IL-1β (DuPont-Merck) or LPS (E. coli serotype 055:B5). SOURCE: Adapted from Kapcala et al. (1995). |
||
Hypophysectomy and IL-1 Lethality
This laboratory next sought to determine the importance of the pituitary gland by studying lethal effects of IL-1β and LPS in HYPOX rats. HYPOX had been performed on the day prior to study to ensure that the adrenal cortex of HYPOX rats would still be responsive to direct stimulation as was observed in some rats stimulated with a biologically potent analogue of ACTH, ACTH1–24. Hypox rats also typically showed low basal CORT levels (< 8 µg/dl). HYPOX rats exhibited an increased sensitivity to lethal effects of IL-1β and LPS, similar to ADX rats (Table 19–2). These responses confirmed the importance of the pituitary component of the stress axis for modulating the immune-inflammatory response and protecting the organism from excessive host defensive responses that could ultimately be fatal. Regardless that in vitro studies (Andreis et al., 1991; Bateman et al., 1989; Gaillard, 1994; Gwosdow et al., 1992) had shown that IL-1 could directly stimulate CORT secretion from the adrenal after prolonged exposure, lethal responses to IL-1β in HYPOX rats suggested that direct stimulation of CORT secretion from the adrenal cortex by IL-1β was not sufficient in vivo to protect against lethal actions by IL-1β, and supported the idea that cytokine actions directly at the level of the adrenal were not physiologically important.
Earlier in this chapter, it was outlined that many studies (Bateman et al., 1989; Besedovsky and del Ray, 1996; Chrousos, 1995; Gaillard, 1994; Harbuz and Lightman, 1992; Rivier, 1995; Tilders et al., 1994) clearly illustrate that the primary locus of cytokine action for activating the HPAA occurs acutely at the level of the hypothalamus. However, given that cytokine effects on the pituitary (Chrousos, 1995; Gaillard, 1994; Kehrer et al., 1988; Koenig, 1991) have been described, the importance of the brain in facilitating a glucocorticoid stress response which protects organisms against deleterious host defense responses induced by immune-inflammatory stimuli was questioned. Therefore, this laboratory assessed the importance of the brain's influence, particularly the hypothalamic component of the HPAA for mediating a critical, protective stress response to immune-inflammatory stimuli. Rats which had undergone acute pituitary stalk section (PSS), and thereby disconnection of the hypothalamus from the pituitary, were tested to determine their ability to tolerate an LPS challenge. PSS rats treated with a relatively high dose of LPS (20 mg/kg) showed approximately a 25 percent lethality rate (Kapcala et al., 1995). PSS rats pretreated with a stress equivalent dose of glucocorticoid (0.2 mg/kg dexamethasone) were completely protected against the lethal effects of LPS. These results suggested that activation of at least the pituitary-adrenal axis by LPS may result in significant protection against potentially lethal effects of cytokines.
Protection provided by an intact pituitary-adrenal axis without the influence of the hypothalamus was not as complete as that produced by an intact HPAA. Nevertheless, it was remarkable and noteworthy that significant protection
occurred. These results indicate a functional significance to earlier reports in which LPS-stimulated CORT secretion in rats with hypothalamic resection, lesions, deafferentation, and PSS (Elenkov et al., 1992; Makara et al., 1970, 1971). Several potential mechanisms for stimulating the pituitary can be postulated based on previous investigations (DeSouza et al., 1993; Kehrer et al., 1988; Koenig et al., 1990). It is recognized that presumed stimulation of a pituitary-adrenal axis (without hypothalamic regulation) may not result in circulating glucocorticoid levels generated as rapidly or as high as those produced by an intact HPAA. Nevertheless, this stimulatory mechanism may still be important for counterregulation against immune, infectious, or inflammatory stress and can still provide a significant level of protection. Considering the vital importance of glucocorticoid secretion and the protection derived from such secretion, it seems teleologically reasonable that the stress axis might operate in a fail-safe, redundant manner with multiple back-up mechanisms for ultimately stimulating glucocorticoid secretion.
Inflammatory, Immune, and Infectious Stress Induces Peripheral Immunosuppression and Anti-Inflammatory Effects
Immune-neuroendocrine interactions and putative mechanisms mediating a protective neuroendocrine response against inflammatory, immune, or infectious stress have been described. Protection by the host against an excessive cytokine response is related to suppression of the immune system and modulation of cytokine responses by immune-inflammatory cells. Induction of related counterregulatory responses are believed to play an important role in modulating susceptibility to inflammatory disorders (Mason et al., 1990; Sternberg and Licinio, 1995; Sternberg et al., 1992; Wick et al., 1993). For general purposes of discussion, peripheral anti-inflammatory and immunosuppressive effects will be broadly considered without distinction. This stress-induced peripheral immunosuppression (Friedman and Irwin, 1995; Sternberg and Licinio, 1995; Sternberg et al., 1992) optimally develops via central mechanisms stimulating hypothalamic CRH neurons. Stimulation of CRH-producing neurons and related HPAA activation ultimately increases secretion of CS, which plays a pivotal role in this immunosuppression. Immune activation of central CRH neurons also stimulates the sympathetic nervous system, and this action produces additional peripheral immunosuppression independent of CS (Friedman and Irwin, 1995; Sundar et al., 1990). Although it is clear that the CS-dependent mechanism plays a critical role in suppressing and modulating the host's cytokine response, the physiological significance of the CS-independent immunosuppression remains to be more precisely determined.
Protective Role of Glucocorticoids
What mechanisms are involved in CS protection against an excessive inflammatory-immune response by the host and a potentially lethal result? CSs via their glucocorticoid potency exert anti-inflammatory actions on many cell types at many sites via multiple mechanisms. Glucocorticoids are potent inhibitors (Dinarello, 1992; Knudsen et al., 1987; Lee et al., 1988; Munck et al., 1984; Peretti et al., 1989; Pruitt et al., 1995; Staruch and Wood, 1985) of the production, secretion, and actions of cytokines and a variety of other mediators (e.g., prostaglandins, leukotrienes, platelet-activating factor, complement, histamine, and small peptides such as bradykinin, substance P), which participate in inflammatory and immune reactions (Munck et al., 1984; Williams and Yarwood, 1990). These molecules facilitate many inflammatory actions such as increasing vascular permeability, influx of leukocytes, and generation of other mediators that enhance the inflammatory cascade of events. Important targets on which glucocorticoids exert anti-inflammatory effects include (1) macrophages and monocytes, which produce many cytokines and mediate antigen presentation to immune cells; (2) B-lymphocytes, which produce antibodies and primarily mediate humoral immunity, and T-lymphocytes, which primarily facilitate cell-mediated immunity; and (3) endothelial cells, which produce cytokines, eicosanoids, and nitric oxide, a key molecule that mediates several features of LPS-induced septic shock.
Glucocorticoids inhibit the transcription of IL-1β mRNA, decrease mRNA stability, and diminish the efficiency of cytokine mRNA translation (Dinarello, 1992; Knudsen et al., 1987; Lee et al., 1988; Marx, 1995; Munck et al., 1984). Another mechanism that mediates counterregulatory effects of glucocorticoids involves lipocortins (Perretti, 1994; Pruitt et al., 1995; Williams and Yarwood, 1990). Lipocortins are a family of proteins that inhibit phospholipase A2 and thereby inhibit the release of arachidonic acid. This inhibition prevents the formation of important inflammatory mediators such as prostaglandins and thromboxanes via the cyclooxygenase pathway and leukotrienes via the lipoxygenase pathway (Perretti, 1994; Pruitt et al., 1995; Williams and Yarwood, 1990).
The significance of increased glucocorticoid secretion induced by LPS administration has also been investigated from a pathophysiological perspective by antagonizing glucocorticoid actions with RU 486 (a competitive glucocorticoid receptor antagonist). Treatment with RU 486 resulted in more severe hypotension and pathological changes in multiple organs and more striking elevations of phospholipase A2 activity and lipoperoxide (Fan et al., 1994), which implies that glucocorticoid secretion counterregulated these effects. In another study (Szabo et al., 1994), RU 486 treatment enhanced a pathological cardiovascular response (i.e., hypotension and vascular hyporeactivity to vasoconstriction by norepinephrine) to LPS and augmented the induction of nitric oxide synthase by LPS. Nitric oxide synthase produces
nitric oxide, which is intimately involved in mediating many cardiovascular effects of septic shock. Moreover, Szabo and colleagues (1994) suggested that glucocorticoids may play a key role in promoting endotoxin tolerance and protecting against the deleterious effects of excessive nitric oxide production that may occur in chronic inflammatory conditions.
Glucocorticoid treatment of dogs and nonhuman primates (baboons) dramatically improves survival (Hinshaw et al., 1982) when given prior to administration of LPS or gram-negative bacteria (E. coli) or shortly thereafter. Yet, in large studies of humans, a clear benefit of glucocorticoids for decreasing mortality in septic patients (Veterans Cooperative Group, 1987) has not been demonstrated. Although it had been questioned (Veterans Cooperative Group, 1987) whether glucocorticoids may benefit a subset of septic patients, if this is so, it is still not clear which septic patients should receive glucocorticoids, in what doses, and at what time. Considering that baboon studies (Hinshaw et al., 1982) clearly show that glucocorticoids enhance survival during the sepsis syndrome only when administered during a relatively narrow window of opportunity, it is likely that by the time the diagnosis of the sepsis syndrome has been made in humans, sepsis may have progressed too far for exogenous glucocorticoids to be of any significant benefit.
Protective Role of Sympathoadrenal Responses
Stress activation of hypothalamic CRH neurons can also produce peripheral immunosuppression independent of glucocorticoids via stimulation of the sympathoadrenomedullary system (Friedman and Irwin, 1995) (Figure 19-2). It has long been recognized that CRH regulates autonomic activity via axon terminals from the hypothalamus that project to the locus coeruleus, and that stimulation of these neurons increases peripheral sympathetic activity and adrenomedullary secretion resulting in increased plasma epinephrine levels (Besedovsky and del Ray, 1996; Friedman and Irwin, 1995). Peripheral immunosuppression occurs via neurohormonal modulation of immune cells in lymphoid organs and the circulation (Besedovsky and del Ray, 1996; Friedman and Irwin, 1995). This modulation is related to the presence of β-adrenergic receptors on immune cells and innervation of lymphoid organs such as spleen, thymus, lymph nodes, and bone marrow by sympathetic noradrenergic neurons.
Central administration of IL-1 decreases a humoral immune response as reflected by reduced antibody production in response to an antigen and also a cell-mediated immune response as reflected by reduced natural killer cell cytotoxicity, and mitogen-induced lymphocyte proliferation and IL-2 production (Saperstein et al., 1992; Sundar et al., 1990; Weiss et al., 1994). This central effect of IL-1 is mediated by CRH because it is blocked by antagonizing CRH (Saperstein et al., 1992) and can be replicated by central administration of CRH (Friedman and Irwin, 1995). That a significant component of this immunosuppressive effect is independent of glucocorticoid is demonstrated
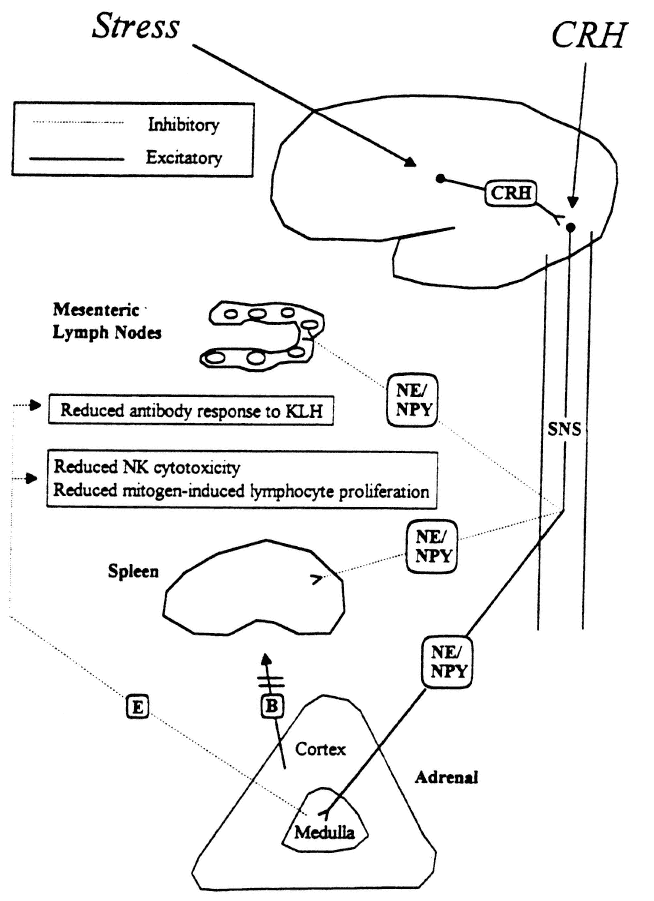
FIGURE 19-2
Proposed model for stress-induced modulation of immune function by corticotropin-releasing hormone (CRH) and the sympathetic nervous system. B, corticosterone; E, epinephrine NE, norepinephrine; NPY, neuropeptide Y; SNS, sympathetic nervous system.
SOURCE: Friedman and Irwin (1995), used with permission.
by the fact that it can still be induced after ADX (Sundar et al., 1990). Its dependence on the sympathetic nervous system is further illustrated by pharmacologically inhibiting the effect via blockade of sympathetic activity (Sundar et al., 1990). Central administration of LPS can also reproduce this peripheral immunosuppression, which is associated with an increase in central IL-1 activity (Weiss et al., 1994). Although the immunosuppressive actions of glucocorticoids are well known, the immunosuppressive effects mediated by the sympathoadrenomedullary system are not nearly as well recognized. Neither is the general concept appreciated that central effects mediating alterations in brain function can produce peripheral immunosuppression and anti-inflammatory actions.
Author's Conclusions and Recommendations
During recent years, an increasing number of immune-neuroendocrine interactions have been identified and characterized that illustrate important bidirectional communication between these systems (Besedovsky and del Ray, 1996). Perhaps the most important interaction relates to immune system activation of the HPAA via cytokines. Stimulation of this counterregulatory response is believed to play a critical role in preventing the host from mounting an excessive defense response involving cytokines against ''inflammatory stress,'' such as inflammatory, immune, and infectious insults. Because of these interactions, which have become appreciated relatively recently, regulatory relationships exist whereby behavioral stimuli and inflammatory stress can ultimately modulate the function of the immune system. Conversely, immune-inflammatory stimuli can ultimately modify behavioral output.
One interesting consequence of these interactions has been observed in animal studies but has not been investigated in humans. This phenomenon relates to the observation that pretreatment of animals with low doses of a cytokine or LPS, which induces cytokines, can diminish or abolish lethal or toxic effects of subsequent inflammatory-immune stimulation with a cytokine, LPS, radiation, or infectious agent (Evans et al., 1991; Morrissey et al., 1995; Neta et al., 1992, 1993; Shalaby et al., 1991; Sheppard and Norton, 1991). Mechanisms mediating these beneficial and protective effects are not clear. Conceivably, some beneficial effect may be related to stimulation of hypothalamic CRH neurons and subsequent activation of glucocorticoid-dependent and glucocorticoid-independent (sympathoadrenomedullary) anti-inflammatory-immunosuppressive mechanisms. Additional investigation of these observations, derived from animal studies, and understanding of mechanisms involved might ultimately lead toward developing treatment regimens that would protect humans against inflammatory, immune, or infectious insults under specific circumstances. Investigative studies utilizing pretreatment regimens with specific cytokines, LPS, or nontoxic immune stimulation could potentially be extremely valuable in designing novel
therapeutic approaches to minimize deleterious consequences of inflammatory, immune, or infectious insults.
Based on the information that has been accumulated and presented, major recommendations include the following:
- Continue to investigate and characterize interactions among the immune, endocrine, and nervous systems and learn about mechanisms mediating these interactions.
- Determine and characterize effects of nutritional abnormalities on interactions among the immune, endocrine, and nervous systems and whether nutritional supplements can beneficially enhance the organism's response to inflammatory stress.
- Investigate and determine protective mechanisms afforded by pretreatment of animals with LPS or a cytokine against lethal or toxic effects of inflammatory stress produced by an inflammatory (e.g., traumatic injury or radiation exposure), immune, or infectious insult.
- Investigate in humans whether pretreatment with a specific regimen (based on specific doses and times) with a cytokine, LPS, or immunotherapy can temporarily modulate the immune system in a beneficially, protective manner against detrimental effects of inflammatory stress induced by inflammatory, immune, or infectious insults.
- Determine whether pretreatment with a specific regimen of a cytokine, LPS, or immunotherapy can temporarily modulate the immune system in a beneficial manner to counterregulate the potentially detrimental effects of immunosuppression induced by stress (e.g., combat stress).
Acknowledgments
Grateful appreciation is extended to Drs. Lee Eiden, Thierry Chautard, and Robert Eskay for their contributions and participation in the lethality studies and to Connie Mack for secretarial assistance.
References
Akana, S.F., C.S. Cascio, J. Shinsako, and M.F. Dallman. 1985. Corticosterone: Narrow range required for normal body and thymus weight and ACTH. Am. J. Physiol. 249:R527-R532.
Andreis, P.G., G. Neri, A.S. Belloni, M. Giuseppina, A. Kasprzak, and L.G.G. Nussdorfer. 1991. Interleukin-1β enhances corticosterone secretion by acting directly on the rat adrenal gland. Endocrinology 129:53-57.
Ban, E., F. Haour, and R. Lenstra. 1992. Brain interleukin-1 gene expression induced by peripheral lipopolysaccharide administration. Cytokine 4:48-54.
Bandtlow, C.E., M. Meyer, D. Lindholm, M. Spranger, R. Heumann, and H. Thoenen. 1990. Regional and cellular codistribution of interleukin-1β and nerve growth factor
mRNA in the adult rat brain: Possible relationship to the regulation of nerve growth factor synthesis. J. Cell Biol. 111:1701-1711.
Banks, W.A., L. Ortiz, S.R. Plotkin, and A.J. Kastin. 1991. Human interleukin (IL)-1 (IL-1α) and murine IL-1β are transported from blood to brain in the mouse by a shared saturable mechanism. J. Pharm. Exp. Ther. 259:988-996.
Barbanel, G., S. Gaillet, M. Mekaouche, L. Givalois, G. Ixtart, P. Siaud, A. Szafarczyk, F. Malaral, and I. Assenmacher. 1993. Complex catecholaminergic modulation of the stimulatory effect of interleukin-1β on the corticotropic axis. Brain Res. 626:31-36.
Bateman, A., A. Singh, R. Kral, and S. Solomon. 1989. The immune-hypothalamic-pituitary-adrenal axis. Endocr. Rev. 10:92-112.
Bertini, R., M. Bianchi, and P. Ghezzi. 1988. Adrenalectomy sensitizes mice to the lethal effects of interleukin-1 and tumor necrosis factor. J. Exp. Med. 167:1708-1712.
Besedovsky, H.O., and A. del Rey. 1996. Immune-neuro-endocrine interactions: Facts and hypotheses. Endocr. Rev. 17(1):64-102.
Bluthe, R.M., V. Walter, P. Parnet, S. Laye, J. Lestage, D. Verrier, S. Poole, B.E. Stenning, K.W. Kelley, and R. Dantzer. 1994. Lipopolysaccharide induces sickness behavior in rats by a vagal mediated mechanism. C.R. Acad. Sci. Paris, Sciences de la via/Life sciences. 317:499-503.
Brady, L.S., A.B. Lynn, M. Herkenham, and Z. Gottesfeld. 1994. Systemic interleukin-1 induces early and late patterns of c-fos mRNA expression in brain. J. Neurosci. 14:4951-4964.
Breder, C.D., C.A. Dinarello, and C.B. Saper. 1988. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 240:321-324.
Breder, C.D., C. Hazuka, T. Ghayur, C. Klug, M. Huginin, K. Yasuda, M. Teng, and C.B. Saper. 1994. Regional induction of TNF-α expression in the mouse brain after systemic lipopolysaccharide administration. Proc. Nat. Acad. Sci. USA 91:11393-11397.
Burrought, M., C. Cabellos, S. Prasad, and E. Tuomanen. 1992. Bacterial components and the pathophysiology of injury to the blood-brain barrier: Does cell wall add to the effects of endotoxin in gram-negative meningitis? J. Infect. Dis. 165 (suppl. 1):S82-S85.
Butler, L.D., N.K. Layman, P.E. Riedl, R.L. Cain, and J. Shellhaas. 1989. Neuroendocrine regulation of in vivo cytokine production and effects: I. In vivo regulatory networks involving the neuroendocrine system, interleukin-1 and tumor necrosis factor-α. J. Neuroimmunol. 24:143-153.
Chang, S.L., T. Ren, and J.E. Zadina. 1993. Interleukin-1 activation of fos proto-oncogene protein in the rat hypothalamus. Brain Res. 617:123-130.
Chrousos, G.P. 1995. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332:1351-1362.
Coceani, F., J. Lees, and C.A. Dinarello. 1988. Occurrence of interleukin-1 in cerebrospinal fluid of the conscious cat. Brain Res. 446:245-250.
Cunningham, E.T., and E. DeSouza. 1994. Interleukin 1 receptors in the brain and endocrine tissues. Immunol. Today 14:161-176.
Dantzer, R., R.M. Bluthe, J.L. Bret-Dibat, S. Laye, R. Parnet, J. Lestage, D. Verrier, S. Poole, B. E. Stenning, and K.W. Kelley. 1994. The neuroimmune basis of sickness behavior [abstract]. P. 41 in Proceedings of the 25th Congress of the International Society for Psychoneuroendocrinology.
DeRijk, R., N. Van Rooijen, F.J.H. Tilders, H.O. Besedovsky, A. Del Rey, and F. Berkenbosch. 1991. Selective depletion of macrophages prevents pituitary-adrenal activation in response to subpyrogenic, but not to pyrogenic, doses of bacterial endotoxin in rats. Endocrinology 129:330-338.
DeSouza, E.B. 1993. Corticotropin-releasing factor and interleukin-1 receptors in the brainendocrine-immune axis. Ann. N.Y. Acad. Sci. 697:9-27.
Dinarello, C.A. 1992. The biology of interleukin-1. Mol. Biol. Immunol. 51:1-32.
Dinarello, C.A., and S.M. Wolff. 1993. The role of interleukin-1 in disease. N. Engl. J. Med. 328:106-113.
Dinarello, C.A., T. Ikejima, S.J.C. Warner, S.F. Orencole, and G. Lonnemann. 1987. Interleukin-1 induces interleukin-1. I. Induction of circulating interleukin-1 in rabbits in vivo and in human mononuclear cells in vitro. J. Immunol. 139:1902-1910.
Donnerer, J., R. Amann, G. Skofitsch, and F. Lembeck. 1992. Substance P afferents regulate ACTH-corticosterone release. Ann. N.Y. Acad. Sci. 632:296-303.
Dunn, A.J. 1992. The role of interleukin-1 and tumor necrosis factor-α in the neurochemical and neuroendocrine responses to endotoxin. Brain Res. Bull. 29:807-812.
Dunn, A.J., and H.E. Chuluyan. 1992. The role of cyclooxygenase and lipoxygenase in the interleukin-1-induced activation of HPA axis. Life Sci. 51:219-225.
Ebisui, O., J. Fukata, N. Murakami, H. Kobayashi, H. Segawa, S. Muro, I. Hanaoka, Y. Naito, Y. Masui, Y. Ohmmoto, H. Imura, and K. Nakao. 1994. Effects of IL-1 receptor antagonist and antiserum to TNF-α on LPS-induced plasma ACTH and corticosterone rise in rats. Am. J. Physiol. 29:E986-E992.
Elenkov, I.J., K. Kovacs, J. Kiss, L. Bertok, and E. S. Vizi. 1992. Lipopolysaccharide is able to bypass corticotropin-releasing factor in affecting plasma ACTH and corticosterone levels: Evidence from rats with lesions of the paraventricular nucleus. J. Endocrinol. 133:231-236.
Ericsson, A., K.J. Kovacs, and P.E. Sawchenko. 1994. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J. Neurosci. 14:897-913.
Evans. M.J., C.J. Kovacs, J.M. Gooya, and J.P. Harrell. 1991. Interleukin-1α protects against the toxicity associated with combined radiation and drug therapy. Int. J. Radiat. Oncol. Biol. Phys. 20:303-306.
Fan, J., X. Gong, J. Wu, Y. Zhang, and R. Xu. 1994. Effect of glucocorticoid receptor (GR) blockade on endotoxemia in rats. Circ. Shock 42:76-82.
Fleshner, M., L.E. Goehler, J. Hermann, J.K. Relton, S.F. Maier, and L.R. Watkins. 1995. Interleukin-1β induced corticosterone elevation and hypothalamic NE depletion is vagally mediated. Brain Res. Bull. 37:605-610.
Friedman, E.M., and M.R. Irwin. 1995. A role for CRH and the sympathetic nervous system in stress-induced immunosuppression. Ann. N.Y. Acad. Sci. 771:396-418.
Gaillard, R.C. 1994. Neuroendocrine-immune system interactions. Trends in Endocrinology and Metabolism 7:303-309.
Gao, Y., J.R. He, and L.P. Kapcala. 1996. Interleukin-1β and lipopolysaccharide regulation of interleukin-1β and interleukin-1 type 1 receptor in hypothalamic neurons and glia [abstract 336.3]. Soc. Neurosci. 21.
Gaykema, R.P.A., I. Dijkstra, and F.J.H. Tilders. 1995. Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology 136:4717-4770.
Gwosdow, A.R., N.A. O'Connell, J.A. Spencer, and M.S.A. Kuman. 1992. Interleukin-1-induced corticosterone release occurs by an adrenergic mechanism from rat adrenal gland. Am. J. Physiol. 263:E461-E466.
Harbuz, M.S., and S.L. Lightman. 1992. Stress and the hypothalamo-pituitary-adrenal axis: Acute, chronic, and immunological activation. J. Endocrinol. 134:327-339.
Harbuz, M.S., R.G. Rees, D. Eckland, D.S. Jessop, D. Brewerton, and S.L. Lightman. 1992a. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial
proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology. 130:1394-1400.
Harbuz, M.S., R.G. Rees, and S.L. Lightman. 1993. HPA axis responses to acute stress and adrenalectomy during adjuvant-induced arthritis in the rat. Am. J. Physiol. 264:R179-R185.
Harbuz, M.S., A. Stephanou, N. Sarlis, and S.L. Lightman. 1992b. The effects of recombinant human interleukin (IL)-1α, IL-1β, or IL-6 on hypothalamo-pituitary-adrenal axis activation. J. Endocrinol. 133:349-355.
He, J.R., Y. Gao, and L.P. Kapcala. 1996. Induction of interleukin-1β gene expression in specific brain sites by peripheral interleukin-1β and lipopolysaccharide [abstract 336.1]. Soc. Neurosci. 21.
Higgins, G.A., and J.A. Olschowka. 1991. Induction of interleukin-1β mRNA in adult rat brain . Mol. Brain Res. 9:143-148.
Hinshaw, L.B., B.K. Beller-Todd, and L.T. Archer. 1982. Current management of the septic shock patient: Experimental basis for treatment. Circ. Shock 9:543-553.
Ju, G., X. Zhang, B.Q. Jin, and C.S. Huang. 1991. Activation of corticotropin-releasing factor containing neurons in the paraventricular nucleus of the hypothalamus by interleukin-1 in the rat. Neurosci. Lett. 132:151-154.
Kapcala, L.P., T. Chautard, and R.L. Eskay. 1995. The protective role of the hypothalamic-pituitary-adrenal axis against lethality produced by immune, infectious, and inflammatory stress. Ann. N.Y. Acad. Sci. 771:419-437.
Kapcala, L.P., J.R. He, Y. Gao, J.O. Pieper, and L.J. DeTolla. 1996. Subdiaphragmatic vagotomy inhibits intra-abdominal interleukin-1β stimulation of adrenocorticotropin secretion. Brain Res. 728:247-254.
Katsuura, G., A. Arimura, K. Koves, and P.E. Gottschall. 1990. Involvement of organum vasculosum of lamina terminalis and preoptic area in interleukin 1β-induced ACTH release. Am. J. Physiol. 258:E163-E173.
Kehrer, P., D. Turnill, J.M. Dayer, A.F. Muller, and R.C. Gaillard. 1988. Human recombinant interleukin-1β and -α, but not recombinant tumor necrosis factor-α, stimulate ACTH release from rat anterior pituitary cells in vitro in a prostaglandin E2 and cAMP independent manner. Neuroendocrinology 48:160-166.
Knudsen, P.J., C.A. Dinarello, and T.B. Strom. 1987. Glucocorticoids inhibit transcriptional and post-transcriptional expression of IL-1 in U93F cells. J. Immunol. 139:4129-4134.
Koenig, J.I. 1991. Presence of cytokines in the hypothalamic-pituitary axis. Prog. Neuroendocrin. Immunol. 4:143-153.
Koenig, J.I., K. Snow, B.D. Clark, R. Toni, J.G. Cannon, A.R. Shaw, C.A. Dinarello, S. Reichlin, and S.L. Lee. 1990. Intrinsic pituitary interleukin-1β is induced by bacterial lipopolysaccharide. Endocrinology 126(6):3053-3058.
Laye, S., R.M. Bluthe, S. Kent, C. Combe, C. Medina, P. Parnet, K. Kelly, and R. Dantzer. 1995. Subdiaphragmatic vagotomy blocks induction of IL-1β mRNA in mice brain in response to peripheral LPS. Am. J. Physiol. 268:R1327-R1331.
Lechan, R.M., R. Toni, B.D. Clark, J.G. Cannon, A.R. Shaw, C.A. Dinarello, and S. Reichlin. 1990. Immunoreactive IL-1β localization in rat forebrain. Brain Res. 514:135-140.
Lee, S., and C. Rivier. 1994. Hypophysiotropic role and hypothalamic gene expression of corticotropin-releasing factor and vasopressin in rats injected with interleukin-1 systemically or into the brain ventricles. Neuroendocrinology 6:217-224.
Lee, S.W., A.P. Tsou, H. Chan, J. Thomas, K. Petrie, E.M. Eugui, and A.C. Allison. 1988. Glucocorticoids selectively inhibit the transcription of the interleukin-1β gene and decrease the stability of interleukin-1β mRNA. Proc. Natl. Acad. Sci. USA 85:1204-1208.
Lilly, M.P., and D.S. Gann. 1992. The hypothalamic-pituitary-adrenal-immune axis. Arch. Surg. 127:1463-1474.
MacPhee, I.A., F.A. Antoni, and D.W. Mason. 1989. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J. Exp. Med. 169:431-445.
Maier, S.F., E.P. Wiertelak, D. Martin, and L.R. Watkins. 1993. Interleukin-1 mediates the behavioral hyperalgesia produced by lithium chloride and endotoxin. Brain Res. 632:321-324.
Makara, G.B., E. Stark, and T. Meszaros. 1971. Corticotropin release induced by E. Coli endotoxin after removal of the medial hypothalamus. Endocrinology 88:412-414.
Makara, G.B., E. Stark, and M. Palkovits. 1970. Afferent pathways of stressful stimuli: Corticotropin release after hypothalamic deafferentation. J. Endocrinol. 47:411-416.
Marx, J. 1995. How the glucocorticoids suppress immunity. Science 270:232-233.
Mason, D.I., I.A. MacPhee, and F.A. Antoni. 1990. The role of the neuroendocrine system in determining genetic susceptibility to experimental allergic encephalomyelitis and implications for human inflammatory disease. Immunology 70:1-5.
Matta, S.G., J. Singh, R. Newton, and B.M. Sharp. 1990. The adrenocorticotropin response to interleukin-1β instilled into the rat median eminence depends on the local release of catecholamines. Endocrinology 127:2175-2182.
Minami, M., Y. Kuraishi, T. Yamaguchi, S. Nakai, Y. Hirai, and M. Satoh. 1991. Immobilization stress induces interleukin-1β mRNA in the rat hypothalamus. Neurosci. Lett. 123:254-256.
Minami, M., Y. Kuraishi, T. Yamaguchi, K. Yabuuchi, A. Yamazaki, and M. Satoh. 1992. Induction of Interleukin-1β mRNA in rat brain after transient forebrain ischemia. J. Neurochem. 58:390-392.
Morrissey, P.J., K. Charrier, and S.N. Vogel. 1995. Exogenous tumor necrosis factor-α and interleukin-1α increase resistance to Salmonella typhimurium: Efficacy is influenced by the Ity and LPs loci. Infect. Immun. 63:3196-3198.
Munck, A., P.M. Guyre, and N.J. Holbrook. 1984. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr. Rev. 5:25-44.
Nakano, K., S. Suzuki, and C. Oh. 1987. Significance of increased secretion of glucocorticoids in mice and rats injected with bacterial endotoxin. Brain Behav. Immun. 1:159-172.
Nakatsuru, K., S. Ohgo, Y. Oki, and S. Matsukura. 1991. Interleukin-1 (IL-1) stimulates arginine vasopressin (AVP) release from superfused rat hypothalamo-neurohypophyseal complexes independently of cholinergic mechanism. Brain Res. 554:38-45.
Neta, R., R. Perlstein, S.N. Vogel, G.D. Ledney, and J. Abrams. 1992. Role of interleukin 6 (IL-6) in protection from lethal irradiation and in endocrine responses to IL-1 and tumor necrosis factor. J. Exp. Med. 175:689-694.
Neta R., D. Williams, F. Selzer, and J. Abrams. 1993. Inhibition of c-kit ligand/steel factor by antibodies reduces survival of lethally irradiated mice. Blood 81(2):324-327.
Ohlsson, K., P. Bjork, M. Bergenfeldt, R. Hageman, and R.C. Thompson. 1990. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature 348:550-552.
Perlstein R.S., M.H. Whitnall, J.S. Abrams, E.H. Mougey, and R. Neta. 1993. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology 132:946-952.
Perretti, M. 1994. Lipocortin-derived peptides. Biochem. Pharmacol. 47(6):931-938.
Perretti, M., C. Becherucci, G. Scapigliati, and L. Parente. 1989. The effect of adrenalectomy on interleukin-1 release in vitro and in vivo. Br. J. Pharmacol. 98:1137-1142.
Pruitt J.H., E.M. Copeland, and L.L. Moldawer. 1995. Interleukin-1 and interleukin-1 antagonism in sepsis, systemic inflammatory response syndrome, and septic shock. Shock 3(4):235-251.
Reichlin, S. 1993. Neuroendocrine-immune interactions [review article]. New Engl. J. Med. 329(17):1246-1253.
Rivest, S., G. Torres, and C. Rivier. 1992. Differential effects of central and peripheral injection of interleukin-1β on brain c-fos expression and neuroendocrine functions. Brain Res. 587:13-23.
Rivier, C. 1993. Effect of peripheral and central cytokines on the hypothalamic-pituitary-adrenal axis of the rat. Ann. N.Y. Acad. Sci. 697:97-105.
Rivier, C. 1995. Influence of immune signals on the hypothalamic-pituitary axis of the rodent. Front. Neuroendocrinol. 16:151-182.
Rivier, C., R. Chizzonite, and W. Vale. 1989. In the mouse, the activation of the hypothalamic-pituitary-adrenal axis by a lipopolysaccharide (endotoxin) is mediated through interleukin-1. Endocrinology 125:2800-2805.
Russell, D.A., and K. K. Tucker. 1995. Combined inhibition of interleukin-1 and tumor necrosis factor in rodent endotoxemia: Improved survival and organ function. J. Infect. Dis. 171:1528-1538.
Saija, A., P. Princi, M. Lanza, M. Scalese, E. Aramnejad, and A. De Sarro. 1995. Systemic cytokine administration can affect blood-brain barrier permeability in the rat. Life Sci. 56:775-784.
Saperstein, A., H. Brand, T. Audhya, D. Nabriski, B. Hutchinson, S. Rosenzweig, and C. S. Hollander. 1992. Interleukin 1β mediates stress-induced immunosuppression via corticotropin-releasing factor. Endocrinology 130:152-158.
Sapolsky, R., C. Rivier, G. Yamamoto, P. Plotsky, and W. Vale. 1987. IL-1 stimulates the secretion of hypothalamic CRF. Science 238:522-524.
Sawchenko, P.E., E.R. Brown, R.K.W. Chan, A. Ericsson, H.-Y. Li, B.L. Roland, and K.J. Kovács. 1996. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Pp. 201-222 in The Emotional Motor System, G. Holstege, R. Bandler, and C.B. Saper, eds. Progress in Brain Research, vol. 107. Amsterdam: Elsevier.
Schöbitz, B., E.R. De Kloet, and F. Holsboer. 1994. Gene expression and function of interleukin-1, interleukin-6, and tumor necrosis factor in the brain. Prog. Neurobiol. 44:397-432.
Schotanus, K., F. Tilders, and F. Berkenbosch. 1993. Human interleukin-1 receptor antagonist prevents adrenocorticotropin, but not interleukin-6 response to bacterial endotoxin in rats. Endocrinology 132:1569-1576.
Selmanoff, M.K., L.P. Kapcala, J.R. He, Y. Gao, D.N. Darlington, and D.E. Carlson. 1996. Adrenocorticosteroid feedback is not necessary for adrenocorticotropin stimulation by peripheral interleukin-1β. Soc. Neurosci. 21:336.4.
Shalaby, M.R., J. Halgunset, O.A. Haugen, H. Aarset, L. Aarden, A. Waage, K. Matsushima, H. Kvithyll, D. Boraschi, J. Lamvik, and T. Espevid. 1991. Cytokine-associated tissue injury and lethality in mice: A comparative study. Clin. Immunol. Immunopathol. 61:69-82.
Sheppard, B.C., , and J.A. Norton. 1991. Tumor necrosis factor and interleukin-1 protection against lethal effects of tumor necrosis factor. Surgery 109:698-705.
Staruch, M.J., and D.D. Wood. 1985. Reduction of serum interleukin-1-like activity after treatment with dexamethasone. J. Leukocyte Biol. 37:193-207.
Sternberg, E.M., and J. Licinio. 1995. Overview of neuroimmune stress interactions: Implications for susceptibility to inflammatory disease. Ann. N.Y. Acad. Sci. 771:364-371.
Sternberg, E.M., G.P. Chrousos, R.L. Wilder, and P.W. Gold. 1992. The stress response and the regulation of inflammatory disease. Ann. Int. Med. 117:854-866.
Sternberg, E.M., J.M. Hill, G.P. Chrousos, T. Kamilaris, S.J. Listwak, P.W. Gold, and R. Wilder. 1989. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl. Acad. Sci. USA 86:2374-2378.
Suda, T., F. Tozawa, T. Ushiyama, T. Sumitomo, M. Yamada, and H. Demura. 1990. Interleukin-1 stimulates corticotropin-releasing factor gene expression in rat hypothalamus. Endocrinology 126:1223-1228.
Sundar, S.K., M.A. Cierpial, C. Kilts, J.C. Ritchie, and J.M. Weiss. 1990. Brain IL-1-induced immunosuppression occurs through activation of both pituitary-adrenal axis and sympathetic nervous system by corticotropin-releasing factor. J. Neurosci. 10:3701-3706.
Szabo, C., C. Thiemermann, C. Wu, M. Perretti, and J.R. Vane. 1994. Attenuation of the induction of nitric oxide synthase by endogenous glucocorticoids accounts for endotoxin tolerance in vivo. Proc. Natl. Acad. Sci. USA 91:271-275.
Takao, T., S.G. Culp, and E.B. DeSouza. 1993. Reciprocal modulation of interleukin-1β (IL-1β) and IL-1 receptors by lipopolysaccharide (endotoxin) treatment in the mouse brain-endocrine-immune axis. Endocrinology 132:1497-1504.
Tilders, F.J.H., R.H. DeRijk, A.M. vanDam, K. Schotanus, and T.H.A. Persoons. 1994. Activation of the hypothalamic-pituitary-adrenal axis by bacterial endotoxins: Routes and intermediate signals. Psychoneuroendocrinology 19:209-232.
Urbaschek, R., and B. Urbaschek. 1987. Tumor necrosis factor and interleukin-1 as mediators of endotoxin-induced beneficial effects. Rev. Infect. Dis. 9 (suppl. 5):S607-S615.
van Dam, A., M. Bauer, F.J.H. Tilders, and F. Berkenbosch. 1995. Endotoxin-induced appearance of immunoreactive interleukin-1β in ramified microglia in rat brain: A light and electron microscopic study . Neuroscience 65(3):815-826.
van Dam, A., M. Brouns, S. Louisse, and F. Berkenbosch. 1992. Appearance of interleukin-1 in macrophages and in ramified microglia in the brain of endotoxin-treated rats: A pathway for the induction of non-specific symptoms of sickness? Brain Res. 588:291-296.
Veening, J.G., M.J.M. van der Meer, H. Joosten, A.R.M.M. Hermus, C.E.M. Rijnnkels, L.M. Geeraedts, and C.G. Sweep. 1993. Intravenous administration of interleukin-1β induces fos-like immunoreactivity in corticotropin-releasing hormone neurons in the paraventricular hypothalamic nucleus of the rat. J. Chem. Neuroanat. 6:391-397.
Veterans Cooperative Group. 1987. Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N. Engl. J. Med. 317:659-665.
Vogel, S.N., and M.M. Hogan. 1990. Role of cytokines in endotoxin-mediated host responses. Pp. 238-258 in Immunophysiology, The Role of Cells and Cytokines in Immunity and Inflammation, J. Oppenheim and E. M. Shevach, eds. New York: Oxford University Press.
Wakabayashi, G., J.A. Gelfand, J. F. Burke, R.C. Thompson, and C.A. Dinarello. 1991. A specific receptor antagonist for interleukin-1 prevents Escherichia coli-induced shock in rabbits. FASEB J. 5:338-343.
Wan, W., L. Wetmore, C.M. Sorensen, A.H. Greenberg, and D.M. Nance. 1994. Neural and biochemical mediators of endotoxin and stress-induced c-fos expression in the rat brain. Brain Res. Bull. 34:7-14.
Watanobe, H., and K. Takebe. 1994. Effects of intravenous administration of interleukin-1β on the release of prostaglandin E2, corticotropin-releasing factor, and arginine
vasopressin in several hypothalamic areas of freely moving rat: Estimation by push-pull perfusion. Neuroendocrinology 60:8-15.
Watanobe, T., A. Morimoto, N. Tan, T. Makisumi, S.G. Shimuda, T. Nakamori, and N. Murakami. 1994. ACTH response induced in capsaicin-desensitized rats by intravenous injection of interleukin-1 or prostaglandin. Eur. J. Physiol. 475:139-145.
Watkins, L.R., L.E. Goehler, J.K. Relton, N. Tartaglia, L. Silbert, D. Martin, and S.F. Maier. 1995a. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: Evidence for vagal mediation of immune-brain communication . Neurosci. Lett. 183:27-31.
Watkins, L.R., E.P. Wiertelak, L.E. Goehler, K. Mooney-Heiberger, J. Martinez, L. Furness, K.P. Smith, and S.F. Maier. 1994a. Neurocircuitry of illness-induced hyperalgesia. Brain Res. 639:283-299.
Watkins, L.R., E.P. Wiertelak, L.E. Goehler, K.P. Smith, D. Martin, and S.F. Maier. 1994b. Characterization of cytokine-induced hyperalgesia. Brain Res. 654:15-26.
Weidenfeld, J., O. Abramsky, and H. Ovadia. 1989. Effect of interleukin-1 on ACTH and corticosterone secretion in dexamethasone and adrenalectomized pretreated male rats. Neuroendocrinology 50:650-654.
Weiss, J.M., N. Quan, and S.K. Sundar. 1994. Widespread activation and consequences of interleukin-1 in the brain. Ann. N.Y. Acad. Sci. 741:338-357.
Whitnall, M.H., R.S. Perlstein, E.H. Mougey, and R. Neta. 1992. Effects of interleukin-1 on the stress-responsive and -nonresponsive subtypes of corticotropin-releasing hormone neurosecretory axons. Endocrinology 131:37-44.
Wick, G., Y. Hu, and G. Krooemer. 1993. Immunoendocrine communication via the hypothalamic-pituitary adrenal axis in autoimmune diseases. Endocr. Rev. 14:539-563.
Williams, T.J., and H. Yarwood. 1990. Effect of glucocorticosteroids on microvascular permeability. Am. Rev. Respir. Dis. 141:S39-S43.
Yan, H.Q., M.A. Banos, P. Herregodts, and E.L. Hooghe. 1992. Expression of interleukin (IL)-1β, IL-6, and their respective receptors in the normal rat brain and after injury. Eur. J. Immunol. 22:2963-2971.
Zuckerman, S.H., J. Shellhaas, and L.D. Butler. 1989. Differential regulation of lipopolysaccharide-induced interleukin-1 and tumor necrosis factor synthesis: Effects of endogenous and exogenous glucocorticoids and the role of the pituitary-adrenal axis. Eur. J. Immunol. 19:301-305.
Discussion
ROBERT NESHEIM: Thank you, Dr. Kapcala. Questions? This has been a very interesting, in some ways, elucidating and, in some ways, confusing discussion.
DOUGLAS WILMORE: I enjoyed your presentation. It pretty much I think, in balance, mimics sort of Selye's point of view about the need for the adrenal gland in the stress response. I would just like to offer some other evidence that may indicate that this response may not be as important as we always thought.
First of all, there are knock-out animals. If you look at knock-out animals, all you need steroids for are to mature their lungs when they are born. It is
awfully hard to make those animals die any more than any other animals when they do not have adrenal gland function. You can do swimming and running and all sorts of other things relate to that.
LEONARD KAPCALA: I think the important thing to keep in mind is that those are stressors that would not necessarily be inducing a lot of cytokines. So what I think is important is, if you take animals that have a subnormal HPA-axis, I believe you can show increased susceptibility to severe illness and even the lethal effects, if you have enough of a cytokine challenge.
DOUGLAS WILMORE: Yes. If you have enough of a cytokine challenge, you can kill anything.
LEONARD KAPCALA: Right. But, as you can see, the intact animals survive tremendous cytokine challenges. So what we are looking at is a sensitivity. It is not an all-or-nothing phenomenon. If you have got enough of a challenge or an immune or inflammatory stimulus that comes along, a subnormal axis will put you at increased risk of dying. I think that that is the concept to keep in mind, not necessarily that animals will die in the basal state.
DOUGLAS WILMORE: Yes. Along with that same point of view is that, for example, in modern anesthesia [comment off mike] will turn off steroid stimulation to cortisol responses. That is really practiced all around the world now. It used to be that we always thought that people who had adrenal suppression needed high doses of stress steroids to go through an operative procedure. That in fact is not right.
What is right, however, is that a basal level of steroid administered through a general kind of response is needed—not a big peak response, but a basal steroid dose is needed.
We have done experiments where we put IL-1 into the head and looked at the catabolic response. There is a very nice catabolic-response model. You can get rodents, for example, to be catabolic for a week or so. Our thesis was very much like yours, that we could adrenalectomize those animals and do away with the response, because we were working down through the HPA-axis. The fact of the matter is that when we did adrenalectomize those animals, we gave them low-dose maintenance steroids at the same time. Those animals still had their stress response, still responded the same way, and it appeared to us that the redundancy of the system somehow had now turned on other mediators to do this. It particularly has to do with some of the brain work that you have done to show that the sympathetic nervous system is now being turned on and other hormonal regulation is coming to the fore.
So the only reason for the comment is that I think that we need to move ahead with sort of the view that we have to have an adrenal gland and a big adrenal response to function. I would be very interested, for example, to take some of your runners and suppress their adrenal elaboration and take them up to 80 percent ![]() O2max and see whether in fact you would get the same immunosuppression or not. Because I think that will help elucidate some of these mechanisms.
O2max and see whether in fact you would get the same immunosuppression or not. Because I think that will help elucidate some of these mechanisms.
LEONARD KAPCALA: At the end of some of these studies where you give these small cytokine challenges or endotoxin, and you boost the immune response to a lethal subsequent dose, it would be interesting to study them [small cytokine changes] in, say, animals that had undergone a stalk section because then you might be inhibiting a corticosterone response, but you would still allow the CRH sympathetic response and might be able to determine how much of that component might be important. I think that probably you would get the maximal response when you have both components working. But when you have at least the sympathoadrenal system functioning, that component gives you some protection.
DOUGLAS WILMORE: I think it is worth pointing out that Bill Beisel directed work here back in the 1960s or 1970s, if I remember the reference right—maybe the early 1970s—when IL-1 was really put into the head at a dose of about one-four hundredth of what had to be put into the systemic circulation to get a comparable kind of fever response.
The business of brain IL-1 cytokine responses, I think, is a tremendously important thing. There are a number of us in this room—Bruce [Bistrian], Joe [Cannon], myself, and others—who have worked until we are blue in the face to try to find these circulating factors in the blood stream, and they are awfully hard to recover. The business of stuff circulating through the blood stream and going to the brain and creating signals and looking for those specific cytokines has not been a real fruitful area unless you are terribly, terribly sick with infectious disease.
Other mediators, such as complement, and other sorts of inflammatory mediators, may well turn on central cytokines to then add these outflow kinds of responses. So I think we still have an awful lot to learn about the picture, and I just do not want us to get locked into the stereotypic Selye model of response.
WILLIAM BEISEL: When the steroid drugs were first introduced into medicine in about 1949 or thereabouts, every infectious disease on the books was used as a model to see if these corticosteroids would help in the treatment. The concept then was Selye's adrenal-response model. The thousands of papers published, unfortunately, showed that steroids in megadoses did not help that
much in the treatment of infectious diseases in people. Still, the clinical value of corticosteroids just did not appear to be present. When we began actually measuring the amount of the adrenal response in human volunteers in this building, who were being infected with various microorganisms, we were surprised that the adrenal response was minuscule compared to the immune response of surgical patients.
We did some of the same type of studies in animals that Dr. Wilmore just talked about. We found that a permissive effect of the steroids was necessary for a lot of responses by the animal and in adrenalectomized animals—the guy with the stress of just a small permissive dose would keep those animals going. I am still not convinced the large doses of steroids are of any protective value at all.
LEONARD KAPCALA: When our studies were performed, basically, what we were trying to do was to mimic what a maximal stress was, just showing that because, as you saw, the HPA axis of intact animals was completely protected so that these animals could tolerate very large doses.
I would just like to make one other comment. I know, in the medical literature right now, the pendulum has gone back and forth in terms of sepsis, and whether or not to give steroids. Right now it is in the camp that steroids have not been shown to be beneficial. I would just add one caveat though. I think that pharmacological steroids might be beneficial if they were given early enough because by the time you make the diagnosis, sepsis has advanced to a certain stage, and it is already too late, because animal models such as baboons, given sublethal or lethal doses of endotoxin can be protected. So I think the problem is that with humans, we just cannot really do that study. By the time we say this person is undergoing sepsis it is already too late, and the cytokine cascade has gone too far.
I would agree with your point about the importance of the permissive action of glucocorticoid in a certain situation such as the basal state. I think one reason we see relatively low cytokine levels in the basal, unstimulated state is because glucocorticoids are modulating some cytokine production and secretion. It has been shown that adrenalectomized animals will show an increased expression of cytokines just basally so that that is an important concept: that you do not necessarily need large amounts, but you do need larger amounts when the cytokine challenge becomes more significant.




























