
4
Mechanisms of Developmental Toxicity
This chapter presents a historical perspective of the field of developmental toxicology and then an analysis of various mechanisms by which agents cause developmental toxicity, as currently understood by developmental toxicologists. The chapter was prepared in response to the first charge to the committee to evaluate the evidence supporting hypothesized mechanisms of developmental toxicity.
HISTORY OF DEVELOPMENTAL TOXICOLOGY: GROWTH OF A NEW FIELD
Teratology, the study of abnormal development, has a long history, much of which is shared with developmental biology. Progress in experimentally determining the causes of abnormal development began in earnest in the nineteenth century and continued through the first half of the twentieth century. By then it was recognized that genetic, nutritional (e.g., cretinism), infectious (e.g., congenital rubella syndrome), and chemical factors caused congenital anomalies in humans and that such manifestations of perturbed development could also be experimentally elicited in various animal systems.
Much of the early research into chemical causes of abnormal development used the same animal models and approaches that were in common use in experimental developmental biology. For example, Dareste elucidated the concept of critical periods of susceptibility by treating chick embryos at various developmental stages with hypoxia, which was induced by coating various fractions of the egg’s surface with wax (Dareste 1877, as cited in Wilson and Fraser 1977). The author concluded that the developmental stage during which treatment oc-
curred is the critical factor in determining which organ was affected. The concept of developmental phase specificity and other core principles of experimental developmental toxicology grew out of that early work. Knowledge about normal developmental processes was applied to understand abnormal development, the pathogenesis resulting in malformations. In the course of such investigations, various chemical and physical insults were used to perturb development to elucidate the underlying normal processes.
In the 1950s, approximately at the time when basic scientific research in normal and abnormal embryo development was expanding, phocomelia and amelia of the arms and legs, two very rare manifestations of human limb defects, occurred in infants born in several European countries and Australia. Independently and almost simultaneously, two alert physicians, one in Australia and one in Germany, concluded that the sudden increase in those defects was attributable to treatment with the pharmaceutical thalidomide early in the pregnancies of the mothers of the affected babies (McBride 1961; Lenz 1961; Lenz and Knapp 1962). Until that time, thalidomide was thought to be a “nontoxic” sedative/hypnotic. The removal of thalidomide from the international market brought the phocomelia epidemic to an end. By various estimates, 7,000 to 10,000 babies were affected. Animal studies later confirmed that thalidomide has toxicological properties that affect development in some species (for reviews, see Neubert and Neubert 1997 and Stephens 1988).
Mass media coverage of the thalidomide tragedy brought the field of developmental toxicology to the attention of the public. For the first time, it was tragically demonstrated that a chemical agent had the potential to profoundly affect human development. Responsible scientists and members of the regulatory community recognized that developmental toxicity might not be unique to thalidomide. It was soon realized that the toxicity testing methods for pharmaceutical products in use at that time were focused primarily on the adult and were inadequate to predict the response or susceptibility of the embryo or fetus. An ad hoc committee of scientists, including many of the charter members of the Teratology Society, was assembled by the U.S. Food and Drug Administration (FDA) to develop guidelines for assessing the hazard potential of new therapeutic agents for developmental toxicity. Other regulatory agencies in the United States and around the world have since adopted similar guidelines, which are applied not only to pharmaceuticals but to all classes of chemicals with significant human exposure potential.
The recognition that environmental factors, whether chemical, physical, or biological, could elicit malformations resulted in a new emphasis on identifying and characterizing other agents that might cause adverse impacts. The disciplines of developmental biology, pharmacology, toxicology, and obstetrics and gynecology were brought together to address these research questions. As our ability to understand the cellular and macromolecular actions of chemicals grew, so did the field of developmental toxicology, which increasingly focuses on un-
derstanding the mechanisms underlying chemically induced developmental defects. Perhaps the most influential review and assessment of the state of knowledge published in that expansionary phase of teratology was J.G. Wilson’s landmark book Environment and Birth Defects (1973). To this day, Wilson’s book is useful as a summary of the principles of teratogenesis (described below) that were developed from the work of teratologists such as Josef Warkany, Lauri Saxen, Robert Brent, Jan Langman, and David Smith. This time period was also one in which numerous clinical discoveries were made of chemical agents that produce abnormal development in humans. Fetal alcohol syndrome, undoubtedly already a problem since antiquity, was first recognized and described in the scientific literature (Lemoine et al. 1968; Jones and Smith 1973; Jones et al. 1973). Unfortunately, in spite of the intervening years and educational efforts, this syndrome remains all too common and is currently estimated as affecting 1.95 of every 1,000 live births in the United States (Abel 1995). A retinoic-acid-induced human embryopathy was described soon after Accutane (13-cis retinoic acid) was introduced as an efficacious drug to treat severe cystic acne (Lammer et al. 1985). Anticonvulsant drugs were also recognized as associated with abnormal development (Finnell et al. 1997a). A large body of work on the heavy metal lead, a ubiquitous contaminant, has shown that it can produce subtle effects on neurobehavioral development (for a review, see Bellinger 1994), emphasizing the fact that abnormal development can manifest itself as subtle functional deficits and not just structural changes. Another heavy metal, mercury, also was identified as a human developmental toxicant after an epidemic of cerebral palsy with microcephaly in Minamata, Japan, was associated with the ingestion of fish contaminated with methyl mercury (Harada and Noda 1988).
PRINCIPLES OF TERATOLOGY
Prior to and shortly after the thalidomide crisis, a body of data had accumulated showing that many chemical, biological, and physical agents can induce malformations in mammalian species, such as mice, rats, rabbits, and guinea pigs. On the basis of those accumulated data, Wilson in the 1970s formulated six principles of teratology that have guided research in developmental toxicology to this day (Wilson 1973). These principles are relevant to cite because they provide the context for the specific mechanisms of toxicity considered later in the chapter.
1. “The access of adverse environmental influences to developing tissues depends on the nature of the influences (agent).” This is to say, developmental toxicants can be accessible to the conceptus (the embryo or fetus, plus the embryo-derived extra-embryonic tissues) in two ways, directly or indirectly. Examples of the former include ionizing radiation, microwaves, and ultrasound, which travel directly through maternal tissues without modification and then interact with the conceptus. Most known developmental toxicants gain access to
the conceptus indirectly. In the maternal body, they are subject to potential metabolic alterations (e.g., biotransformations in the liver), distribution, storage, and excretion that either enhance or diminish their potential to affect the conceptus adversely. The net result of all these interventions is that some level of active developmental toxicant is available to cross the placenta and eventually reaches target sites in the conceptus. Although it is frequently assumed that the developmental toxicant must reach targets in the conceptus to disrupt development, it should be noted that adverse effects on growth and development can be mediated indirectly through effects on accessory tissues, such as the yolk sac and placenta, or on maternal tissues.
2. “The final manifestations of abnormal development are death, malformation, growth retardation, and functional disorder.” This principle highlights the now well-known fact that structural malformations are not the only possible outcome after the conceptus is exposed to a developmental toxicant. In fact, it is now known that in many cases the outcomes are interrelated. For example, at a relatively high dose of a developmental toxicant, the conceptus might suffer a high level of cell death that cannot be replenished by available repair and compensatory mechanisms. This, in turn could result in growth retardation if the induced cell death is widespread, and in death of the conceptus if the cell death compromises organ systems essential for viability of the conceptus. At lower doses particular malformations and functional disorders might occur. Which outcome, or combination of outcomes, will occur depends on the dose and chemical characteristics of the developmental toxicant (discussed in the third and fifth principle, respectively) and the developmental stage of the conceptus at the time of exposure (discussed in the fourth principle).
3. “Manifestations of deviant development increase in degree as dosage increases from the no-effect level to the totally lethal level.” Sufficient evidence was available in the 1970s to support the relationship of dose with the incidence of structural malformations, death, and, to a lesser degree, growth retardation. Evidence accumulated since then extends it to functional deficits as well. It is also important to point out that the relationship between dose and response, although monotonic, does not have to be linear. It can be a steep S-shaped curve for developmental toxicants, sometimes going from a no-effect level to maximal effects within a doubling of the dose.
4. “Susceptibility to teratogenic agents varies with the developmental stage at the time of exposure.” The change of susceptibility was originally published by Wilson as a hypothetical curve in which the degree of sensitivity to developmental toxicant-induced structural malformations was low during the pre-implantation phase, maximal during organogenesis, and low during fetal development. This shape of the developmental sensitivity curve reflects results from many stud-
ies of a variety of developmental toxicants, when developmental defects are scored at birth. The curve highlights the general conclusion that organ systems are most susceptible to pertubation by developmental toxicants, just prior to and during the overt phase of organ formation and differentiation, which occurs in mammals after the period of implantation, streak formation, and streak regression.
Several caveats need to be addressed, however. First, the pre-implantation period should not be viewed as a refractory period in terms of induced structural malformations (Rutledge 1997; Dwivedi and Iannaccone 1998). For example, ethylene oxide (EtO) can induce structural abnormalities in mice when administered during pre-implantation stages of embryogenesis (Generoso et al. 1987). Results from this study are particularly instructive because they show that agents can induce skeletal effects when administered to the pregnant dam at the zygote stage of development, long before skeletogenesis begins. Moreover, the spectrum of skeletal defects observed after exposure at the zygote stage differs from those observed after exposure during organogenesis. The mechanisms underlying that stage-specific effect of EtO on skeletal development are unknown.
The second caveat is that, although the susceptibility curve generally reflects the reality for structural defects, it is not a good generalization for developmental-toxicant-induced death, growth retardation, or functional deficits. For example, toxicant-induced death tends to occur most frequently at pre- and peri-implantation stages. As many as 30% of fertilized human oocytes are estimated to die during those early stages (see Chapter 2), and the role of developmental toxicants in that human loss is largely unknown.
The final point to be made is that development from fertilization to birth is a progressive process so that any adverse outcome (i.e., death, growth retardation, malformation, or functional deficits) after exposure to developmental toxicants will be dictated, in part, by the set of developmental processes active at the time of exposure.
5. “Teratogenic agents act through specific mechanisms on developing cells and tissues to initiate abnormal embryogenesis (pathogenesis).” Recent research focusing on how exogenous chemicals interact with endogenous molecular targets has increased our understanding of the mechanisms of action of toxicants. A detailed discussion on the mechanisms of action of toxicants follows this section.
6. “Susceptibility to teratogenesis depends on the genotype of the conceptus and the mother in which this interacts with environmental factors.” This principle was originally based on the knowledge that different species and strains of animals respond differently to a developmental toxicant. For example, it was already known in the 1970s that mouse embryos are unusually susceptible to the induction of cleft palate by glucocorticoids, and other mammalian species are resistant. Also, some mouse strains are sensitive to hyperthermia-induced neural
tube defects and others are not. The particular genotype of individual offspring in the same mouse uterus was shown to be associated with benzo[a]pyrene-induced birth defects (Shum et al. 1979; Nebert 1989). A more recent example is the finding that oral clefting is more common in the offspring of mothers who smoke and who have a variant allele of the transforming growth factor (TGF) gene. The correlation implicates direct or indirect interactions between constituents in tobacco smoke and TGF, a secreted protein that binds to the epidermal-growth-factor receptor and is known to be expressed in palatal epithelium before and during palatal closure (Hwang et al. 1995; see Chapter 5). Examples such as these, together with a wealth of data indicating that many developmental defects of unknown etiology exhibit a multifactorial pattern of inheritance, have led to the conclusion that gene-gene and gene-environment interactions play a significant role in the etiology of many developmental defects.
MECHANISMS OF TOXICITY
An understanding of how exposure to a toxicant can result in an adverse developmental outcome is needed to develop intervention and preventive public health practices. Risk assessors seek to obtain mechanism-based toxicity results from animal tests in order to make justifiable extrapolations to humans. The process by which a toxicant can produce dysmorphogenesis, growth retardation, lethality, and functional alterations commonly is referred to as the “mechanism” by which developmental toxicity is produced. In general, it has been difficult to analyze mechanisms in sufficient detail and depth for risk assessment purposes. There are four reasons.
1. Normal development is extremely complex, and it is possible that there is a myriad of points at which a toxicant might interact with an important molecular component and cause developmental toxicity. Information about molecular components and processes of development has only been available in the past few years, largely through the study of developmental mutants of invertebrate model organisms, such as Drosophila and Caenorhabditis elegans. As highlighted by Wilson’s principles, an understanding of mechanisms would be greatly enhanced by identifying critical key events altered by toxicants. Recent advances in research on signaling pathways and genetic regulatory circuits in development might have identified especially critical processes, ones that, if studied for their alteration by developmental toxicants, might provide exciting new clues for mechanistic investigations (see discussion in Chapters 6 and 7). For now, such insights are available in only a few cases, such as toxicant interactions with components of the nuclear hormone-receptor family of signal receptors and gene regulators.
2. Environmental toxicants include a wide range of chemical, physical, and biological agents that initiate a wide variety of mechanisms. Some agents are
specific for one or a few targets in the development or physiology of the conceptus, and others have a broad effect on many targets at different times and places in the conceptus and mother. Thus, the developmental toxicologist who focuses on these agents is probably faced with a wide variety of mechanisms.
3. Some toxicants might affect only a fraction of individuals in the population, probably because of genetic differences or differences in health history (diseases, nutrition, or other exposures). The differences add considerable complexity.
4. A mechanistic understanding of developmental toxicity involves understanding at several levels of biological organization. Once a toxicant interacts with a molecular component of the cell, it presumably affects its immediate function, so the function and alteration must be known. Then, the consequence for the altered function for the completion of a developmental process must be known. For example, in order to link specific branchiofacial defects with the action of a suspected toxicant, it is necessary to characterize the migratory events, proliferation control processes, and patterns of differentiation-promoting signal systems that affect neural crest cells from the time of their emigration from the neural tube. Other kinds of toxicants might alter specialized functions of organs of the fetus (e.g., the heart) and thus manifest impacts at the organ level. Yet other toxicants might cause cell death in the conceptus at a variety of times and locations and have multiple impacts.
A developmental toxicologist must understand the potential action of toxicants at many levels of biological organization to understand the overall processes of developmental toxicity (Faustman et al. 1997). Recognizing this dilemma, the U.S. Environmental Protection Agency (EPA) and the International Programme on Chemical Safety (IPCS) have defined chemical “modes of action” in addition to “mechanisms.” Modes of action are described in the proposed EPA Guidelines for Carcinogen Risk Assessment (1996b), and in the IPCS guidelines for international applications (IPCS Workshop on Developing a Conceptual Framework for Cancer Risk Assessment, 16-18 February 1999, Lyon, France). In these definitions, “… mechanism is taken to infer detailed molecular knowledge of the initial events that result in an adverse response in the organism, whereas “mode of action” refers to the cascade of major changes that occurs during the development of the adverse event.” Mode of action is contrasted to mechanism of action in that the latter usually implies a more detailed understanding of molecular and cellular events than the former. Furthermore, the altered process by which the initial molecular interactions lead to a structural or functional deficit is called pathogenesis. Pathogenesis can involve altered pathology at the cellular, tissue, and organ functional level.
To preserve the full range of causes and effects relevant to risk assessment of human developmental toxicity, the committee has sought to use “mechanism of
action” in the most inclusive sense, to include all events from initial molecular interactions to the developmental defect itself. Such an explanation would include the following types of mechanistic information:
-
The toxicant’s kinetics and means of absorption, distribution, metabolism, and excretion within the mother and conceptus.
-
Its interaction (or those of a metabolite derived from it) with specific molecular components of cellular or developmental processes in the conceptus or with maternal or extraembryonic components of processes supporting development.
-
The consequences of the interactions on the function of the components in a cellular or developmental process.
-
The consequences of the altered process on a developmental outcome, namely, the generation of a defect.
In Chapter 8, the committee discusses “levels of information” needed to understand inclusive mechanisms. The information is obtainable from various model systems (including in vitro and cell culture, nonmammalian animals, and mammals). Hypotheses about toxicant action in humans, based on the information from animal models, can then be strengthened or dismissed using information obtained from various levels of human data.
General Kinds of Initial Interactions of Toxicants with Cellular Molecules
Receptor-Ligand Interactions
Some chemicals interact directly with endogenous receptors for hormones, growth factors, cell-signaling molecules, and other endogenous compounds. They can activate the receptor inappropriately (agonists), inhibit the ability of the endogenous ligand to bind the receptor (antagonists), act in a way that activates the receptor but produces a less than maximal response (partial agonist), or act in a way that causes a decrease from the normal baseline in an activity under the control of the receptor (negative agonist). Receptors can be broadly classified as cytosolic/nuclear or membrane bound. Cytosolic/nuclear receptors reside within the cell and have ligands that are small and generally hydrophobic so that they can pass easily through the cell membrane. After the ligand binds to these receptors, the complex translocates to the nucleus where it interacts directly with specific sequences of DNA to activate or inactivate the expression of specific genes. Examples of cytosolic receptors are the estrogen receptors (ERa,ERb,ERR), retinoic acid receptors (RAR and RXR), and aryl hydrocarbon receptor (AHR). Agents that interact with one or more of these receptors and are known to produce abnormal development include retinoic acid and synthetic retinoids, glucocorti-
coids, androgens, and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). A detailed description of what is currently known about the mechanisms by which retinoic acid and TCDD perturb development can be found below. Table 4-1 describes several cytosolic receptors (in this case, nuclear hormone receptors) that are involved in receptor-mediated developmental toxicity.
Membrane receptors (i.e., trans-membrane proteins) are diverse and interact with a wide variety of molecules, from small molecules, such as glutamate and acetylcholine, and small proteins, such as insulin, to large proteins, such as Wingless-Int (WNT), Sonic Hedgehog (SHH), TGFβ, and Delta signals (discussed in Chapter 6). Signaling molecules interact with a portion of the receptor on the cell’s exterior. The binding of a ligand to a membrane receptor leads to a cascade of events within the cell membrane and cell known as signal transduction, which often involves five or more steps, including second messengers (intracellular signaling compounds) (described further in Chapter 6). It is conceivable that toxicants could affect any of these steps. For example, toxicants could interfere with receptor interactions or alter the activity of intermediates of the signal-transduction cascade. The number of agents known to exert developmental toxicity via interaction with membrane receptors is smaller than that for cytosolic receptors. Several membrane receptors—the Hedgehog receptor Patched, endothelin receptors A and B, and the cation channel delayed-rectifying Ikr—are known to play a role in mediating developmental toxicity and are highlighted in Table 4-1.
Despite the few examples of toxicant interactions with membrane receptors, the mechanism might be important in understanding how certain chemicals disrupt development. Most normal developmental processes involve cell-cell signaling and are mediated by trans-membrane receptors, including inductions, cell-matrix interactions, cell proliferation, cell movement, and autocrine and paracrine effects. The potential is great, therefore, that these mechanisms are significant in developmental toxicity.
Covalent Binding
Covalent binding occurs when the exogenous molecule chemically reacts with an endogenous molecule (e.g., forming a DNA or protein adduct). Among the kinds of reactive chemicals are aldehydes, epoxides, quinonimines, free radicals, acylating agents, and alkylating agents. Exposure to these chemicals might then result in abnormal transcription or replication of DNA, or abnormal function of the adducted protein. Phosphoramide mustard, a reactive metabolite of cyclophosphamide, is an example of a developmental toxicant that forms DNA adducts (alkylation) in embryos (Cushnir et al. 1990). Many chemicals that are not initially reactive are converted by DMEs (e.g., cytochromes P450) to “potentiated” reactive derivatives. An example of a developmental toxicant that forms both DNA and protein adducts in embryos is diphenylhydantoin, whose mechanism is described below.
TABLE 4-1 Receptor-Mediated Developmental Toxicity
|
Receptor (official namea) |
Endogenous Ligands |
Developmentally Toxic Ligand and Modifier |
Typical Effects |
Recent References |
|
Basic Helix-Loop-Helix Transcription Superfamily |
||||
|
Aryl hydrocarbon AHR |
Unknown |
Agonists: TCDD and related polycyclics |
Cleft palate, hydronephrosis |
Couture et al. 1990; Mimura et al. 1997; Abbott et al. 1998 |
|
Nuclear Hormone Receptors |
||||
|
Androgen AR (NR3C4) |
Testosterone Dihydro-testosterone |
Agonists: 17 α-ethinyl-testosterone and related progestins Antagonistsb: Flutamide |
Agonists: Masculinization of female external genitals Antagonists: Inhibition of Wolffian duct and prostate development and feminization of external genitals in males |
Kassim et al. 1997 |
|
Estrogen |
Estradiol |
Agonist: DES |
Agonist: various genital-tract defects in males and females |
Cunha et al. 1999 |
|
ERa, ERb (NR3A1 and 2) |
Antagonist: tamoxifen, clomiphene—weak |
|||
|
Glucocorticoid GR (NR3C1) |
Cortisol |
Agonists: cortisone, dexamethasone, triamcinolone |
Cleft palate |
Fawcett et al. 1996 |
|
Retinoic acid |
||||
|
RARα, β, and γ (NR1B1, 2, and 3) |
All-trans & 9-cis retinoic acids |
Agonists: numerous natural and synthetic retinoids Antagonists: BMS493, AGN 193109 and others |
Almost all organ systems can be affected |
Collins and Mao 1999; Chazaud et al. 1999; Kochhar et al. 1998; Elmazar et al. 1997. |
|
RARα, β, and γ (NR1B1, 2, and 3) |
9-cis retinoic acid |
|
|
|
|
Thyroid hormone TRa and b (NR1A1 and NR1A2) |
Thyroxine (T4 and T3) |
Antagonist: nitrophen |
Lung, diaphragm, harderian-gland defects |
Brandsma et al. 1994 |
|
Receptor (official namea) |
Endogenous Ligands |
Developmentally Toxic Ligand and Modifier |
Typical Effects |
Recent References |
|
Hedgehog Receptor |
||||
|
Patched |
Sonic, Desert, and Indian Hedgehogs |
Veratrum alkaloids: cyclopamine (mechanism unclear) |
Cyclopia, holoprosencephaly |
Incardona et al. 1998; Cooper et al. 1998 |
|
Membrane |
||||
|
Endothelin receptors A and B |
Endothelins 1, 2, and 3 |
Antagonists: L-753, 037, SB-209670, SB-217242 |
Craniofacial, thyroid, cardiovascular, intestinal aganglionosis (Hirschsprung’s disease) |
Spence et al. 1999; Treinen et al. 1999; Gershon 1999a,b |
|
Cation Channels |
||||
|
Delayed-rectifying IKr |
Potassium ion |
Inhibitors: almokalant, dofetilide, d-sotalol |
Digit, cardiovascular, orofacial clefts |
Webster et al. 1996; Wellfelt et al. 1999 |
|
aNuclear Receptors Committee 1999. bAlso, five α reductase inhibitors (e.g., finasteride) affect prostate and external genitals (Clarke et al. 1993). |
||||
Peroxidation of Lipids and Proteins
Some chemicals exist as free radicals or generate free radicals during their metabolism. Free radicals are highly reactive and will oxidize proteins or lipids, changing their structure. The developmental toxicity of hydroxyurea is at least partially mediated by free radicals (DeSesso 1979; DeSesso and Goeringer 1990; DeSesso et al. 1994) and that of niridazole appears to be entirely mediated by radical production (Barber and Fantel 1993). Physical agents such as ionizing radiation also produce this type of oxidative damage, as does the body itself during reperfusion after an ischemic episode. See recent reviews by Fantel (1996) and Wells and Winn (1996) for a more detailed discussion on this topic.
Interference with Sulfhydryl Groups
Sulfhydryl groups often play an important role in maintaining the tertiary structure and, therefore, the biological activity of proteins, especially in the disulfide linkages of secreted proteins. In some proteins, sulfhydryl groups are functional groups of the active (catalytic) site. Metals like mercury and cadmium are examples of developmental toxicants that cause oxidative stress and bind strongly to sulfhydryl groups and interfere with function (see reviews by Clarkson 1993; Stohs and Bagchi 1995; Quig 1998). The mechanism of one form of mercury, methylmercury, toxicity is described in detail below.
Inhibition of Protein Function
This is a broad category. Protein function occurs at catalytic sites (catalysis), regulatory sites (regulation of protein activity), macromolecule binding sites (such as specific DNA binding), or protein-protein association sites (as in aggregation of ribosomal proteins).
Some agents interfere with enzymes whose catalytic function is important in development, somewhat similar to an antagonist binding to a receptor. For example, methotrexate, a cancer chemotherapeutic agent mimics a substrate of dihydrofolate reductase, and its inhibitory binding results in a functional folate deficiency, which is developmentally adverse (DeSesso and Goeringer 1991, 1992). The mechanism is described in more detail below. Angiotensin-converting-enzyme (ACE) inhibitors are another example of agents that interfere with development by blocking enzyme action. These drugs block the conversion of angiotensin I to angiotensin II. Angiotensin II is a potent vasoconstrictive agent controlling blood pressure in adults. In the human fetus and neonate, it is needed to maintain renal perfusion and glomerular filtration. When angiotensin II levels are reduced in the fetus, glomerular filtration pressure and urine production are
reduced, causing fetal hypotension and oligohydramnios (reduced volume of amniotic fluid). Those primary mechanisms lead to fetal death and stillbirth, middle- to late-trimester onset of oligohydramnios, and intrauterine growth restriction followed by delivery of infants with hypotension and renal failure (Barr 1997).
Other chemicals block protein polymerization, such as colchicine and colcimid blocking tubulin polymerization to microtubules or cytochalasins blocking actin aggregation to microfilaments. Those drugs bind to protein-protein association sites. There are examples of chemicals also binding at other kinds of sites. All fit the geometry and weak bonding properties of the site and competitively interfere with the binding of the normal cell component (substrate or ligand). Chelators of essential elements may interfere with protein function by limiting the availability of metal co-factors. Examples of proteins that require metals to function are metalloproteinases and several other enzymes, and zinc-finger transcription factors.
Maternally Mediated Effects
All the mechanisms discussed above occur within the embryo. However, there are examples in which developmental toxicity is the consequence of toxicity in the mother. Effects on the embryo occur secondarily, as a result of some effect on the pregnant mother. Effects include chemically induced maternal hypoxia or secondary nutritional deficiencies. An example of the former is the case of diflunisal (5-(2,4-difluorophenyl) salicylic acid, a nonsteroidal anti-inflammatory drug), which causes hemolytic anemia in pregnant rabbits. The anemia leads to adverse developmental effects (Clark et al. 1984). An example of secondary nutritional deficiencies is the functional zinc deficiency brought about by substantial induction of metallothionein in maternal liver as part of a systemic acute-phase response to a wide array of chemicals that have little in common other than their capacity to induce an acute-phase response, including de novo expression of metallothionein in the liver (Daston and Lehman-McKeeman 1996). The events that take place within the embryo after toxicant-induced zinc deficiency are equivalent to those occurring during dietary deficiency, but the salient point for developmental toxicology and risk assessment is the recognition that maternal factors might contribute substantially to embryonic response.
Other Mechanistic Considerations
There are other mechanisms that might be found to affect development. These might include such events as DNA intercalation, interaction with as yet unidentified targets, or complicated interactions that involve multiple changes, each of which is necessary—but not by itself sufficient—to initiate a pathogenetic cascade.
General Kinds of Pathogenesis
Once the toxicant has interacted with an endogenous molecule, the function of the endogenous molecule will be altered to an extent depending on the dose and duration. In the developing embryo, the function of the endogenous molecule can be seen as having a cellular and a developmental role, simply because the embryo is composed of cells whose activities are directed to a developmental outcome. From the current knowledge of cell biology, various general classes of function can be cited as susceptible to alteration. Any of these might be affected as part of a toxicant-induced pathogenic process. The classes of function include altered
-
gene expression,
-
patterns of apoptosis (programmed cell death),
-
replication, cell cycle, cell proliferation,
-
secretion, endocytosis, uptake, migration, adhesion, and
-
signal transduction.
Certain chemicals might affect more than one of these processes. There is mechanistic value in knowing which of these cell biological processes is affected.
All cells of all stages of development engage in the cell activities listed above. For an understanding of developmental consequences, however, more specific information is needed about which particular molecular components of which particular processes of development are affected. Some cell biological effects, such as failed cell proliferation or failed cell migration, might be several steps removed from the initial effect of the toxicant and several steps from the final effect of the altered cell behavior on development (e.g., a craniofacial defect). The challenge in recent years has been to identify particular molecular components of cellular and developmental processes, discern their activities, and understand the toxicant-caused alteration of activity. The recent information from cell and developmental biology has been essential for the progress in the understanding of mechanisms of toxicity.
Known Mechanistic Information on Selected Chemicals
The remainder of this chapter reviews the current hypotheses of mechanisms by which chemicals are thought to cause developmental toxicity. Eleven chemicals, listed alphabetically, are used to illustrate different mechanisms. For some chemicals, a great deal of evidence has been gathered supporting certain aspects of the mechanisms. For others, data are sparse and the understanding of the mechanism is incomplete. Experimental approaches used to study mechanisms of developmental toxicity are highlighted. In the near future, these approaches can be used in conjunction with new approaches from developmental biology and
genetics to elucidate further the mechanisms by which chemicals cause developmental toxicity.
Class III Antiarrhythmic Drugs
An example of a chemical interaction with a membrane receptor that leads to adverse developmental outcome is the case of class III antiarrhythmic drugs, such as dofetilide and almokalant. In rat embryos, these drugs block specific potassium channels in myocardial cells, presumably by high-affinity interactions with adrenergic or muscarinic receptors. This action on potassium flux leads to a profound bradycardia, which in turn progresses to malformations (probably through hypoxia in the affected structures) or death in utero (Webster et al. 1996). This example also illustrates the process of pathogenesis, as the molecular interaction produces an effect on embryonal organ function, which in turn affects the further development and viability of the embryo.
Cyclopamine
Binns et al. (1963) reported that an epidemic of cyclopia with associated holoprosencephaly in sheep was caused by teratogenic compounds present in the subalpine lily, Veratrum californicum. Subsequent work by Keeler and Binns (1968) showed that the active teratogenic agents in this plant were cyclopamine, its glycoside alkaloid X, jervine, and veratosine. Of these teratogenic agents, cyclopamine and jervine are the most active. In addition, it was noted that these two compounds closely resemble cholesterol in structure. In an early study, Roux and Aubry (1966) showed that alterations in cholesterol metabolism induced by AY-9944, an inhibitor of the final step in cholesterol synthesis, induced holoprosencephaly in rats. Almost 30 years then passed before additional insights were gained into the teratogenic mechanism of cyclopamine-induced holoprosencephaly.
In the 1990s, new data from several unrelated fields converged to suggest that cyclopamine-induced holoprosencephaly was caused by interference with cholesterol metabolism and SHH signaling. First, SHH is synthesized as a precursor that must be cleaved and covalently linked with cholesterol to be active (Roelink et al. 1995; Porter et al. 1996). Second, SHH is necessary and sufficient for patterning the ventral neural tube (Tanabe and Jessell 1996). Third, mutations in SHH were shown to cause holoprosencephaly in mice (Chiang et al. 1996). Fourth, holoprosencephaly associated with Smith-Lemli-Opitz syndrome results from a genetic decrease in Δ7-DHC (Δ7-dehydrocholesterol) reductase activity (Kelley et al. 1996; Tint et al. 1994). These discoveries, coupled with earlier studies linking cylopamine to holoprosencephaly, led to the hypothesis that cyclopamine causes holoprosencephaly by interfering with SHH signaling. Direct confirmation of this hypothesis came in 1998 from two independent labora-
tories (Cooper et al. 1998; Incardona et al. 1998). Although many of the mechanistic details are lacking, both groups showed that cyclopamine inhibits SHH signaling.
Diethylstilbesterol (DES)
DES was prescribed from the 1940s to the 1970s to prevent pregnancy loss. DES was found to be a transplacental carcinogen and teratogen (affecting the hypothalamo-hypophysial axis and reproductive organs) in humans approximately 25 years after its introduction into women’s health care (Herbst et al. 1971; Herbst 1981; Kaufman et al. 1980; Goldberg and Falcone 1999). Even more time passed before it was demonstrated to be a transplacental carcinogen in the mouse (McLachlan et al. 1980; C. Miller et al. 1998; Walker and Haven 1997), rat (Baggs et al. 1991; Henry and Miller 1986), and hamster (Khan et al. 1998).
Mechanistically, DES has produced a rich field for investigating mechanisms of teratogenic and carcinogenic action. Such animal and human observations place DES among the agents that can modify not only the estrogen receptor activity but also expression of uterine lactoferrin through signal transduction mechanisms (Newbold et al. 1997). More recent evidence has implicated chromosomes 3 and 6 as sites for gene control resulting in not only carcinogenesis but also teratogenesis (Hanselaar et al. 1997).
Recent investigations have coupled the effects of DES in the developing mouse female reproductive tract with downregulation of WNT7A, resulting in abnormal smooth-muscle proliferation (C. Miller et al. 1998). WNT7A is normally expressed in the luminal epithelium of the uterus. Following DES exposure in utero, low levels of WNT7A transcripts were detected at birth. Such alterations in the reproductive tract following DES exposure are consistent with knockout mice lacking Wnt7a having malformed female reproductive tracts (Miller and Sassoon 1998).
All of these investigations implicate the role of gene control and modification by estrogenic agents that might be more effective not only because of their estrogenic properties but also because of their pharmacokinetics and metabolism (Miller et al. 1982; Henry et al. 1984; Henry and Miller 1986). Thus, in the human, further questions are being raised about the gene-environment interactions based on the collection of experience with the use of DES during pregnancy (Hanselaar et al. 1997).
Diphenylhydantoin
Diphenylhydantoin (DPH; common name, phenytoin) is an anticonvulsant used to treat epilepsy. It produces abnormal development in fetuses whose mothers take the drug during pregnancy. Abnormalities include facial dysmorphism
(epicanthal folds, hypertelorism, broad, flat bridge of the nose, upturned tip of nose, and prominent lips), distal digital hypoplasia, intrauterine growth retardation, and mental retardation. This cluster of defects has been termed the fetal hydantoin syndrome and occurs in about 11-17% of pregnancies in which the mother has taken the drug (Hanson et al. 1976; van Dyke et al. 1988).
It appears necessary for DPH to be metabolized by cytochrome P450 (CYP) enzymes to reactive intermediates that form adducts with DNA or protein within the embryo (for a review, see Wells et al. 1997). The most likely intermediate is an arene oxide. An alternative hypothesis suggests that DPH is metabolized by prostaglandin synthetase to a teratogenic intermediate. This hypothesis is supported by the observation that DPH teratogenicity in mice can be mitigated by cotreatment with aspirin, an inhibitor of prostaglandin synthetase (Wells et al. 1989). It has been observed that DPH treatment in rodents decreases the expression of the mRNAs for a number of important growth factors, including TGFβ, NT3 and WNT1 (Musselman et al. 1994). Whether the decrease is due to an effect on gene expression or a degradation of RNA by reactive intermediates of DPH is not known.
Methotrexate
Methotrexate is a cancer chemotherapeutic drug. It is a competitive inhibitor of dihydrofolate reductase, which converts folate to tetrahydrofolate. Tetrahydrofolate is then metabolized to various coenzymes that play a role in the synthesis of purines and amino acids and conversion of deoxyuridylate to thymidylate. Exposure of rabbits to methotrexate during gestation causes craniofacial defects, limb deformities, and decreased fetal weight in the offspring (DeSesso and Goeringer 1991, 1992). Similar defects have been observed clinically in babies of mothers who had been given methotrexate between 35 and 50 days of gestation (Milunsky et al. 1968; Warkany 1978). Using a metabolic derivative of folinic acid, the authors (DeSesso and Goeringer 1991, 1992) demonstrated that methotrexate causes developmental toxicity by inhibition of dihydrofolate reductase. The metabolic derivative replaced the normal product of the inhibited enzyme and eradicated the developmental toxicity.
Methylmercury
Methylmercury (MeHg) is an environmental toxicant that primarily affects the central nervous system (CNS) and, to a lesser extent, the liver and kidneys. To reach the brain, it must cross the blood-brain barrier by traversing the brain capillary endothelial cells. MeHg possesses a high affinity for thiol groups and will bind to endogenous sulfhydryl-containing ligands, such as proteins, and low-molecular-weight compounds, such as glutathione, found in blood and tissue (for a review, see Clarkson 1993). Hirayama (1980, 1985) reported that intravenous injection of MeHg chloride and cysteine in rats increased the rate of
MeHg uptake into brain tissue, an effect that was reversed by administration of a neutral amino acid. The author concluded that MeHg was transported across the blood-brain barrier by an amino acid carrier. Subsequent studies by Kerper et al. (1992, 1996) and Mokrzan et al. (1995) showed that the amino acid carrier is an L (leucine-preferring) amino acid transporter and that MeHg is released from the brain capillary endothelial cells into the brain interstitial space as a glutathione complex.
The brains of both humans and experimental animals (rodents and primates) exposed in utero to MeHg show changes in neuronal migration and distribution patterns, cell loss, low neuronal abundance, and microcephaly, changes consistent with effects on the microtubular cytoskeleton and inhibition of cell-cycle progression (Burbacher et al. 1990). The effects of MeHg on mitotic activity and mitotic spindle function both in vivo and in cell culture have been characterized (Imura et al. 1980; Rodier et al. 1984; Brown et al. 1988; Wasteneys et al. 1988). It has been shown that MeHg directly binds to tubulin and inhibits microtubule formation (Vogel et al. 1986). Ponce et al. (1994) conducted in vitro studies using primary embryo neuronal cells to characterize MeHg’s effect on cell cycling and its role in developmental toxicity. Exposure at concentrations of 2 µM MeHg causes G2/M phase cell-cycle inhibition, and at 4 µM, all cell-cycle phases are inhibited, suggesting that the cytoskeleton and mitotic spindles might be particularly sensitive to MeHg.
Two recent studies have further characterized steps in the mechanism by which MeHg affects the cell cycle in embryos. Ou et al. (1997) used primary rodent embryonic neuronal cells to determine mRNA expression levels of two genes involved in a checkpoint pathway of cell-cycle arrest, Gadd45 and Gadd153, in response to MeHg. Exposure at 2 µM caused both GADD45 and GADD153 mRNA levels to increase. The authors concluded that activation of these Gadd genes could be a mechanism by which MeHg causes cell-cycle arrest in embryos. The same laboratory investigated the involvement of p21 (a cell-cycle regulatory gene of a checkpoint pathway of arrest of G1 and G2 phases of the cell cycle) in primary embryonic cells exposed to MeHg (Ou et al. 1999). The embryonic cells responded to MeHg exposure with a concentration-dependent increase in p21 mRNA, indicating that activation of cell-cycle regulatory genes could be one mechanism by which MeHg disrupts the cell cycle in embryos.
Retinoic Acid
Vitamin A (retinol) and the structurally related retinoids have a special place in developmental toxicology, both currently and historically. Nutritional deficiency of vitamin A was the first chemical manipulation to produce congenital malformations in a mammal (Hale 1933) and thus began the whole field of experimental mammalian teratology. Now, the biologically active metabolites of vitamin A, the retinoic acids (RAs), and their synthetic derivatives have become
the most thoroughly studied of all teratogens. Because natural retinoids are signaling molecules, critical to many developmental processes, exogenous retinoids or vitamin A deficiencies are teratogenic in all animals studied, including humans (for review, see Collins and Mao 1999).
In higher animals, vitamin A is an essential vitamin, requiring absorption from the diet or synthesis from dietary retinyl esters, β-carotene, or other carotenoids. The all-trans form of retinol is most abundant, but there are a number of isomers, which generate the corresponding active retinoid isomers, including 9-cis and 11-cis, following metabolism. The absorption and distribution of retinol involves serum (RBP) and cellular (CRBP-I and CRBP-II) binding proteins. A number of enzymes are capable of converting retinol to retinoic acid, including CYP monooxygenases, alcohol dehydrogenases, and aldehyde dehydrogenases. Mutation of the mouse NAD-dependent retinaldehyde dehydrogenase-2 (ALDH2) (Niederreither et al. 1999) causes severe developmental defects, a result that shows this enzyme to be essential for embryonic RA synthesis. Further metabolism of RA is complex, involving multiple oxidation and conjugation pathways, some also CYP dependent (e.g., CYP26) (Kraft and Juchua 1993; Kraft et al. 1993; Nau et al. 1994; Trofimova-Griffin et al. 2000). Cellular binding proteins (CRABP-I and -II) are thought to influence intracellular levels of RA, but their exact role is unclear. The mouse knockout of CRABP-I is without phenotype, and CRABP-II null mice have polydactyly (Lampron et al. 1995). This phenotype is also the phenotype of the double-knockout mice, which do not, however, differ from wild-type animals in sensitivity to RA teratogenicity (Lampron et al. 1995).
Although there are some strain and species variations in developmental sensitivity and responses to exogenous retinoids, they are usually not profound. The effective oral dose of all-trans-RA in all mammals tested is broadly similar. In contrast, the potency of 13-cis-RA varies by two orders of magnitude. The explanation for this difference lies in species differences in metabolism, coupled with metabolite-specific placental transfer (see Collins and Mao 1999), and is a good illustration of the importance of toxicokinetics.
Some of the dysmorphogenic effects of retinoids are very well conserved across species. For example, RA-induced truncation of the forebrain, with posteriorization in the hindbrain, has been observed in mammals, birds, amphibia, and fish. In addition to CNS and craniofacial malformations, RA also affects the limbs, cardiovascular system, gut, and thymus; the predominant defects depend upon the phase of organogenesis exposed (Collins and Mao 1999). In mice, preorganogenesis RA treatment around the time of implantation induces body axis duplication and supernumerary limbs (Rutledge et al. 1994), whereas fetal exposures can cause functional and behavioral abnormalities (Nolen 1986). Human exposure to the pharmaceutical retinoids 13-cis-RA (isotretinoin) or etretinate predominantly affect CNS and cranial neural-crest development (Coberly et al. 1996).
A large number of whole-animal and in vitro studies have characterized the relationships between structure and developmental toxicity of retinoids. As for any chemical, the in vivo potency results from a combination of pharmacokinetic and pharmacodynamic properties. These studies suggest that the teratogenicity of retinoids is receptor-mediated, but there are other possibilities (see below). The structural requirements for teratogenicity have been reviewed (Willhite et al. 1989; Collins and Mao 1999) and show a wide diversity in the polyene side-chain and β-cyclogeranylidene ring modifications that still retain activity, although an acidic polar terminus appears indispensable. Some aromatic retinoids (arotinoids), such as TTNPB, are 1,000-fold more potent in vivo teratogens than RA. This potency appears to be due predominantly to slower elimination and reduced affinity for CRABPs (Pignatello et al. 1999). Another aromatic retinoid, etretinate, has a very long half-life in humans after multiple exposures, with measurable concentrations in serum 2 years after cessation of intake, probably because of storage and slow release from adipose tissue (Eisenhardt and Bickel 1994). Experimental studies show that the critical pharmacokinetic characteristic for retinoid teratogenicity is the area under the curve (AUC) (Tzimas et al. 1997), rather than a transient high dose.
The receptors for retinoids are of the nuclear hormone ligand-dependent transcription-factor superfamily (Nuclear Receptors Committee 1999). They are of two types: RARs (subclass NR1B) and RXRs (subclass NR2B) (see Chambon 1996). For each type, there are three receptors, α, β, and γ (NR1B1, -2, and -3 and NR2B1, -2, and -3), each encoded by a separate gene. For all these genes, with the exception of RXR, multiple isoforms have been detected (e.g., NR1B2a, -b, -c, -d), generated by differential promoter usage and alternative splicing. Most of the embryonic effects of retinoids seem to be mediated by RAR-RXR heterodimers, but RXRs can form homodimers and can also form heterodimers with a number of other nuclear receptors, the most important being those for thyroid hormones and for peroxisome proliferators (Mangelsdorf and Evans 1995). Several isomers of RA are agonists for RARs, including all-trans-RA, 9-cis-RA, 4-oxo-RA, and 3,4-didehydro-RA, and 9-cis-RA seems to be the predominant RXR agonist (Collins and Mao 1999).
Each receptor, and in some cases each isoform, has been knocked out in mice to test for its function in development. Many combinations of knockouts have also been generated. Loss of RARβ (all isoforms), RAR 1, or RAR 2 has no phenotypic effect (Li et al. 1993; Lohnes et al. 1993; Luo et al. 1995). In contrast, disruption of all isoforms of RAR or RAR causes many of the effects of vitamin A deficiency, including growth deficiencies and male sterility (Lohnes et al. 1993; Lufkin et al. 1993). Compound RAR null mice display all the malformations induced by vitamin A deficiency, including defects of the eyes, limbs, and heart and the craniofacial, urogenital, and reproductive systems (Lohnes et al. 1994; Mendelsohn et al. 1994). An interesting recent example is the compound RAR-RARβ null mouse, which causes syndactyly and demonstrates a role of RA
in interdigit cell death (Dupe et al. 1999). Mice lacking RXR have a hypoplastic ventricular myocardium and placental defects (Sucov et al. 1994; Kastner et al. 1994). Combining RAR and RXR mutants causes considerably more severe defects, suggesting that normal embryonic retinoid signaling is mediated by RAR-RXR heterodimers (Kastner et al. 1997a,b).
Null mutant mice have been used to correlate individual receptors with specific teratogenic effects of retinoids. RAR appears to be essential for the RA-induced defects of truncation of the posterior axial skeleton and is partially required for neural-tube and craniofacial defects (Lohnes et al. 1993; Iulianella and Lohnes 1997). In contrast, RXR is required for RA-induced limb defects (Sucov et al. 1995). It is intriguing that in both cases the receptor is not required for normal development of the affected tissues but does mediate the teratogenic action, a result indicating that the receptor, when activated by exogenously added RA, is affecting gene expression at abnormal times and places, as compared with that done by endogenous retinoids. RARβ does not appear to directly mediate any teratogenic action of retinoids (Luo et al. 1995). Expression of a constitutively active RAR mimics the action of excess RA. For example, expression of active RAR 1 in limb causes the same limb defects as exogenous RA (Cash et al. 1997).
In general, the receptor specificity of retinoids correlates with their teratogenic actions; RAR agonists are potent teratogens and RXR agonists are ineffective and mixed agonists having intermediate activity (Kochhar et al. 1996). Although the lack of action of RXR agonists shows that RXR homodimers are not involved in RA teratogenicity, such ligands can potentiate some of the teratogenic effects of RAR agonists. For example, an RXR agonist increased the effects of an RAR agonist on some organs but not others (Elmazar et al. 1997). Comparative studies of selective RAR, β, and agonists showed a correlation of potency with receptor affinity and transactiviation as well as receptor-organ specificity (Elmazar et al. 1996). As expected, retinoid-receptor antagonists can reduce the developmental effects of agonists (Elmazar et al. 1997). In addition, pan-receptor antagonists, capable of blocking all types of retinoid receptor, reproduce the actions of vitamin A deficiency (Kochhar et al. 1998; Chazaud et al. 1999).
Of direct relevance to this report is that the relationship between structure and teratogenic activity of retinoids is accurately reflected in several in vitro systems. Indeed, rodent limb-bud micromass cultures have been used extensively to screen for the activities of retinoids (e.g., Kistler and Howard 1990). In addition, the dysmorphogenic action of the retinoids in vivo is faithfully reproduced in mammalian whole-embryo culture, and this system has contributed much to our current understanding of mechanism (e.g., Bechter et al. 1992; Chazaud et al. 1999).
The DNA target sequences for RAR-RXR heterodimers are termed RAREs (retinoic acid response elements) and are usually two direct sequence repeats, spaced by 1 to 5 bases (DR1-5), although there are other arrangements (Harmon
et al. 1995; Chambon 1996). For example, a functional inverted repeat with zero nucleotides in the spacer (IR0) was recently described (Lee and Wei 1999). A large number of genes containing RAREs have now been identified, and the transcription of many has been shown to be modulated by RA treatment (Chambon 1996; Collins and Mao 1999). Similarly, the expression of numerous genes has been observed to change following embryonic RA exposure. How many of these changes are directly controlled by RAREs and which are critical for teratogenicity is largely unknown. The HOX genes, however, represent one class of functionally important downstream RA targets in teratogenesis.
The homeobox-containing HOX transcription factors are involved in patterning of the CNS, limbs, axial skeleton, and other organ systems, where their expression encodes positional identity. As discussed in Chapter 6, HOX genes are arranged on chromosomes in clusters in which the genes are colinear with their expression domains (Duboule 1998). For example, in the early CNS and somites, 3′ HOX genes are expressed rostrally and 5′ caudally. This colinearity is also manifest in responsiveness to RA, the induction of 3′ HOX genes being more rapid and abundant (Marshall et al. 1996). Endogenous retinoid signaling might be responsible for the progressive expression of HOX genes in vivo. The primitive (Hensen’s) node, for example, is a site of RA synthesis and might pattern the paraxial mesoderm as it egresses through the primitive streak (Hogan et al. 1992). Exogenous retinoids can induce ectopic, expanded, or reduced HOX expression domains, which then establish abnormally arranged compartments of positional identity. These abnormal compartments then result in abnormal cell fate and morphogeneisis (Marshall et al. 1996).
There is good evidence for HOX-mediated retinoid teratogenicity in the axial skeleton, craniofacies, and limb, but perhaps the best understood example is the developmental effect in the hindbrain. The different HOX genes encode transcription factors that control the different identities of the rhombomeres (r) of the hindbrain. Hoxb2 has a rostral expression boundary at r2-3, Hoxb3 at r4-5, Hoxb4 at r6-7, and Hoxb1 in a band at r4 (Marshall et al. 1996; Studer et al. 1996). At early neural-plate stages in the mouse, exogenous retinoid treatment results in rostral expansion of these domains. In some cases, the treatment results in a transformation of r1-3 to an r4 identity with expansion of r5 (Conlon and Rossant 1992). In other cases, r2-3 is transformed into r4-5, and both the trigeminal motor nucleus and adjacent trigeminal ganglion are transformed into structures having a facial nucleus or ganglion appearance (Marshall et al. 1992). Analyses of the 3′ Hox genes reveal multiple RAREs that cooperate with other positive and negative regulatory elements to regulate spatial and temporal expression of the HOX genes (Marshall et al. 1996).
Retinoid-induced changes in HOX gene expression probably result in abnormally arranged compartments of HOX expression. The misexpressed HOX gene then activates and represses many other genes in abnormal places and thereby initiates abnormal development. Pathogenetic changes observed in retinoid-
treated embryos include abnormal differentiation, migration, proliferation and apoptosis (Collins and Mao 1999). Some of these effects might not be receptor-mediated. For example, disruption of membranes, changes in phosphorylation, and increases in reactive oxygen species might play roles in retinoid teratogenicity.
The significance of the retinoids in developmental toxicology might extend further, because a range of chemicals likely mediate their developmental toxicities by interfering with endogenous retinoid signaling. Ethanol can act as a competitive inhibitor of retinol dehydrogenase activity, thus lowering retinoid synthesis, and this effect might be a component of fetal alcohol effects (Duester 1991). Similar mechanisms have been proposed for the anticonvulsants phenytoin, phenobarbital, carbamazepine, ethosuximide, and valproic acid (Nau et al. 1995; Fex et al. 1995). Inhibition of retinoid catabolism has also been implicated. Metabolism of retinoic acid via NADPH-dependent cytochrome P450 is inhibited by azole antifungal drugs (Vanden Bossche et al. 1988; Schwartz et al. 1995), which can induce retinoid-like craniofacial defects (Wang and Brown 1994). None of these examples is wholly convincing, but the overall concept is plausible, particularly because the retinoids are unusual as secreted signals in being small lipophilic molecules. Because many synthetic chemicals are also small and lipophilic, the potential for interaction might be higher than that for other peptide-based signaling pathways.
TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin)
The mechanism by which TCDD induces developmental toxicity has been studied extensively (for a review, see Wilson and Safe 1998) and is one of the best understood. It is summarized here to provide an example of how a chemical interacts with an endogenous cytoplasmic receptor (in this case, a basic helix-loop-helix receptor (bHLH)) and alters the expression of several dozen genes, one or more of which might result in an adverse developmental outcome.
TCDD, an environmental pollutant, is a byproduct of the production of chlorinated products such as herbicides and wood preservatives, and is developmentally toxic in many species. Exposure in utero to TCDD causes increased mortality and growth retardation, and structural and behavioral abnormalities, including the induction of cleft palate and hydronephrosis in mice. Evidence supports the hypothesis that TCDD binds the aryl hydrocarbon receptor (AHR), allowing the receptor to bind with AH-responsive elements on DNA and leading to changes in gene expression. For example, mice with wild-type high-affinity AHR exposed to TCDD have a higher incidence of developmental abnormalities than do mice with low-affinity receptors. (The mouse Ahr gene contains a mutation that lowers the affinity of the encoded protein for DNA (Chang et al. 1993; Poland et al. 1994)). Large amounts of AHR have been localized to the palatal shelves in normal mice susceptible to the effects of TCDD (i.e., mice having high-affinity
receptors) and much lower amounts in resistant mice (i.e., mice having low-affinity receptors). Additional evidence of the relationship between TCDD and the AHR comes from a study in which mRNA and protein levels for the AHR were found to be decreased in TCDD-exposed mouse embryos compared with that in control embryos (Abbott et al. 1994). Changes in gene expression of epidermal growth factor (EGF), transforming growth factor α (TGFα), EGF receptor, transforming growth factor β1 (TGFβ1), and TGFβ2 have been reported in conjunction with TCDD-induced cleft palate (Abbott et al. 1989; Abbott and Birnbaum 1990). Genetically modified AHR-deficient mice are reported to be relatively unaffected by doses of TCDD that are 10-fold higher than the dose found to induce toxic and pathological effects in mice expressing functional AHR (Fernandez-Salguero et al. 1996). The results suggest that the TCDD-induced toxic effects are mediated by AHR.
Recently, comparative experiments by Abbott et al. (1998; 1999a,b) showed that formation of the palate, which divides the oral cavity from the nasal cavity in mouse and human embryos, involves homologous processes at the morphological, cellular, and molecular level. In organ culture, developing mouse and human embryo palates respond similarly to TCDD. Exposure to the chemical causes excessive epithelial cell proliferation, via several steps, which interferes with fusion of the opposing palatal shelves. Several factors that regulate proliferation in this fusion process have been identified, among them EGF, EGF receptor, TGFα, TGFβ1, TGFβ2, and TGFβ3. Expression patterns of protein and mRNA of each of the growth factor and receptor genes were examined during palatogenesis for mouse and human palates maintained in organ culture. The effects of TCDD exposure on expression of those genes as well as the AHR- and the AH-receptor nuclear translocator (ARNT) and the glucocorticoid receptor (GR), were also defined in both species. To compare the human and mouse palatal responses to TCDD, the induction of a dioxin-responsive gene, CYP1A1, was quantified and compared across species. This comparison required the generation of tissue-level concentration-response profiles across dose and time for both mouse and human profiles. Quantification of AHR and ARNT levels revealed differences between species in the expression levels of the effector molecules. The mouse and human responses at the same target-tissue concentration could thus be compared, and a sensitive molecular marker gene (CYP1A1 induction) could be correlated with gross morphological outcomes and changes in growth-factor expression. Overall, the data indicate that human palates expressed all of these regulatory genes, that responses to TCDD were detected, and that comparison between mouse and human palates revealed interspecies variation that might be a factor in each species’ response to TCDD.
Comparison of in vitro exposure levels between human and mouse palate tissues have revealed profound species differences. At comparable stages of development, human embryo palates are much less sensitive to TCDD than mouse embryos. These studies allow the conclusion that human embryonic palatal tis-
sue expresses much lower levels of AHR and ARNT, or has the poor affinity AHR, and the expression and induction of CYP1A1 were lower. A situation emerges in which approximately 350 times fewer receptors are expressed in the human embryo than in the mouse, approximately 200 times more TCDD is required in human embryo tissue than in mouse tissue to produce the critical effects, and the response of a transcriptionally regulated gene (CYP1A1) is approximately 1,500 times lower in human embryo palates than in mouse palates under identical exposure conditions.
Valproic Acid
Valproic acid (VPA) is an anticonvulsant drug used to treat epilepsy with the major side-effect of hepatotoxicity. VPA is unusual in that its human teratogenicity was predicted from laboratory animal studies, without any knowledge of mechanism (Brown et al. 1980; Kao et al. 1981). In all species, including humans, neural-tube-closure defects are a consistent component of the teratogenic effects, but many other organ systems are affected and their sensitivity varies among species (Kao et al. 1981; Robert 1992). The pharmacological effect of VPA appears to involve several mechanisms, including actions on γ-aminobutyric acid (GABA) synthesis and release; the release of γ-hydroxybutyric acid; attenuation of N-methyl-D-aspartate- (NMDA) type glutamate receptors, and direct effects on excitable membranes (Löscher 1999). The mechanisms leading to hepatotoxicity and teratogenicity are distinct and also differ from the pharmacological mechanisms. The exact mechanism of teratogenicity is unclear, but it too might be multifaceted. Suggested actions include effects on the cytoskeleton and cell motility (Walmod et al. 1998, 1999); several aspects of zinc, folate, methionine, homocysteine, and glutathione metabolism (Alonso-Aperte et al. 1999; Hishida and Nau 1998; Bui et al. 1998; Finnell et al. 1997b); peroxisome proliferation-activated receptor δ interaction (Lampen et al. 1999); and gene expression (Wlodarczyk et al. 1996). Despite the initial site of action (“receptor”) being unknown, there is a wealth of information on structure-teratogenicity relationships.
VPA is a simple short-chain carboxylic acid, 2-propylpentanoic acid. The following features affect teratogenicity (Nau 1994): (1) A free carboxylic acid is required. Amides, such as valpromide, are inactive (Spiegelstein et al. 1999; Radatz et al. 1998), as are stable esters. (2) The C2 carbon must be bonded to one hydrogen and two alkyl chains, as well as the carboxyl group. Substituting the hydrogen with any group abolishes activity, and a single chain or unsaturated derivatives (e.g., 2-en-VPA) are also inactive. (3) Activity is greatest when the two alkyl chains are unbranched (Bojic et al. 1996) and contain three carbons (Bojic et al. 1998). (4) Introducing a side-chain double or triple bond terminally (between C3 and C4) enhances teratogenicity but, in any other position, abolishes activity. (5) When one side-chain has a terminal unsaturation, C2 is asymmetric
and the enantiomers have markedly different potencies. In the cases of both 4-en-VPA and 4-yn-VPA, the S-enantiomer is more potent than the racemate and the R-enantiomer is virtually inactive (Andrews et al. 1997, 1995; Hauck and Nau 1992).
These structure-activity relationships are not due to pharmacokinetic differences, as shown by direct measurements of tissue levels and by the activities of VPA and analogs in embryo culture (Brown et al. 1987; Nau 1994; Andrews et al. 1997, 1995). They are also consistent across species (Andrews et al. 1997, 1995). The overall impression is that the teratogenic effect of valproids requires an interaction with a specific site, at which one alkyl chain becomes located in a hydrophobic pocket, thus enabling ionic bonding of the carboxyl group and the interaction of the second chain with a region that favors the high electron density of terminal unsaturation (Bojic et al. 1998).
Chemicals That Might Induce Apoptosis
More than 1,000 agents have been identified as teratogens in animal studies (Shepard 1998); moreover, a variety of studies has now shown that cell death is an early, common event in the teratogenic process initiated by many, if not all, teratogens (Scott 1977; Knudsen 1997). Often, teratogen-induced cell death occurs preferentially in areas of normal programmed cell death, suggesting that there might be a mechanistic link between programmed and teratogen-induced cell death (Alles and Sulik 1989). The importance of an appropriate amount of programmed cell death to normal development is highlighted by mouse mutants, such as Hammertoe, in which insufficient programmed cell death underlies abnormal limb development (Zakeri et al. 1994). Likewise, excessive teratogen-induced cell death is directly linked to abnormal development by the finding that 2-chloro-2′-deoxyadenosine-induced eye defects are associated with excessive teratogen-induced cell death (Wubah et al. 1996).
Recent research has shown that cell death induced by a variety of stimuli occurs by a process termed apoptosis. Although many of the details are still lacking, it is known that apoptosis is a tightly controlled process, triggered either internally or externally, by which a cell self-destructs in a manner that does not lead to destruction of neighboring cells. Key components in the execution phase of the apoptotic pathway are the intracellular cysteinyl-aspartate proteases known as caspases, particularly caspase-3 (Colussi and Kumar 1999). These enzymes are normally present in all cells as inactive precursors that become activated by cleavage at specific internal motifs. Once activated, these caspases function to degrade specific target substrates, such as poly(ADP-ribose)polymerase (PARP), DNA-PKs, and lamins. A recent report shows that developmental toxicants, such as hyperthermia, cyclophosphamide (an alkylating agent), and sodium arsenite (a thiol oxidant), induce increased cell death characterized by activation of caspase-3, cleavage of PARP, and fragmentation of DNA (Mirkes and Little 1998). Although some of the downstream events in the apoptotic pathway activated by
developmental toxicants are now known, upstream events that initiate and regulate this pathway in mammalian postimplantation embryos are unknown but oxidative stress clearly plays a role (Nebert et al. 2000). In particular, it is not known how cells in the embryo perceive exposure to a developmental toxicant and then respond to the insult. The ability of cells to perceive a stimulus or perturbation and then transduce these events into appropriate intracellular responses is commonly referred to as signal transduction. Although little is known about the interaction between developmental toxicants and signaling pathways in mammalian embryos, a recent report showed that heat shock can rapidly activate the stress-activated protein kinase pathways mediated by c-Jun terminal kinase (JNK) and p38 (Wilson et al. 1999), as well as the unfolded protein stress pathway involving a variety of chaperon proteins (Welch 1991; Sidrauski et al. 1998).
Chemicals That Might Induce Autism and Their Role in Understanding the Disorder
Recent discoveries regarding mutations responsible for the genetic susceptibility to autism spectrum disorders (ASDs) are an example of how studies of developmental toxicity can lead to breakthroughs in understanding the etiology of human birth defects, even when the defects are ones with strong genetic components (for a review, see Rodier 2000).
ASDs are among the most common congenital anomalies, occurring at a rate of 2-5 per 1,000 births (Bryson et al. 1988; Bryson and Smith 1998). Until recently, little was known about the causes and even less about the nature of the CNS injury underlying the symptoms. Family studies had indicated that unknown genetic factors account for about 90% of the variance (Bailey et al. 1995), but linkage studies provided few regions, and no genes, unambiguously linked to autism (Myers et al. 1998; Philippe et al. 1999). The family studies suggested that environmental factors are also involved (Le Couteur et al. 1996) but could not identify any of those factors. In 1994, it was discovered that exposure of the closing neural tube of the human embryo to thalidomide, a well-known teratogen, could produce autism at a high rate (Strömland et al. 1994). Valproic acid (Christianson et al. 1994) and ethanol (Nanson 1992) have also been implicated as teratogens that increase the risk of autism. The critical period for induction of autism by thalidomide was determined from the somatic defects of the patients with autism, each of whom also had malformed ears and hearing deficits. This stage of development—days 20-24 of gestation, which is the period of neural tube closure—is much earlier than the periods usually considered in studies of neuroteratology, because only a few neurons of the brain stem form so early. Most of these are motor neurons for cranial-nerve nuclei (Bayer et al. 1993), and cranial-nerve dysfunctions are indeed present in the thalidomide cases. Figure 4-1 shows a comparison of brain-stem neuroanatomy of a control and a patient with autism.
Using the information about the critical period, Rodier and colleagues were
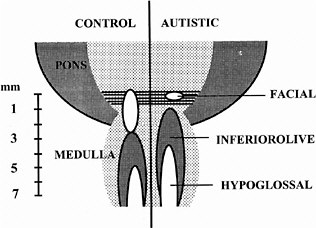
FIGURE 4-1 Comparison of brain-stem neuroanatomy of a control and a patient with autism. In the brain on the right, the number of neurons in the facial nucleus is greatly reduced, and a region caudal to it appears to be missing. The more caudal nuclei are shifted rostrally. The missing area is similar to the embryonic fifth rhombomere, from which most of the facial nucleus arises. Source: Adapted from Rodier et al. (1996).
able to describe alterations of the human brain stem related to autism and to create an animal model of the initial insult using exposure to valproic acid (Rodier et al. 1996). In addition, in cases of autism of unknown cause, they documented the existence of minor physical anomalies that were similar to those reported in the thalidomide-exposed cases (Rodier et al. 1997). The craniofacial symptoms had been reported before in the autism literature, but ignored because they seemed trivial in comparison to the disabling behavioral symptoms. However, the craniofacial defects speak directly to the embryological origin of the disorder. The anatomical studies described effects almost identical to those seen in mice with null mutations of the gene HoxA1 (Chisaka et al. 1992; Carpenter et al. 1993), which is essential to brain-stem and ear development and expressed only during the period of neural tube closure. No variants of HoxA1 had ever been detected in any mammalian species, but the teratological findings suggested the hypothesis that defective versions of the gene must exist and must contribute to the genetic etiology of autism.
Remarkably, an alternate allele of HoxA1 was discovered in a substantial number of people diagnosed with ASD (Rodier 1998). The variant allele not only appeared significantly more frequently in familial cases than in historical controls or parent controls, but the number of homozygotes was significantly reduced from the expected value in all groups, suggesting that homozygosity for the variant reduces viability. (The mouse knockout of the same gene is lethal soon after birth.) A second variant of HoxA1 has since been detected in cases of autism and is under investigation (Stodgell et al. 1999). Further, it has now been
determined that valproic acid selectively alters the expression of Hoxa1 in the embryo, providing an explanation for the similarity between the phenotypes of teratological cases and genetic cases of autism (Ingram and Rodier 1998). The increased understanding of ASDs is an example of how several research paths have converged to provide evidence supporting an interesting genotype-environment hypothesis.
SUMMARY
Early researchers of the causes of abnormal development used many of the same methods and animal models as developmental biologists studying normal development. Knowledge about normal developmental processes was essential to understand the developmental pathogenesis induced by chemical and physical agents, and, reciprocally, such agents at that time were used to disrupt normal development in order to understand the processes.
Our ability to understand the mechanisms by which chemicals act at the cellular and molecular levels to affect development has improved greatly during the past 2 decades. The improvement has occurred in concert with the advances in cell and developmental biology. As discussed in Chapter 6, progress in developmental biology came from the systematic analysis of developmental mutants of model animals, and progress in cell biology came from biochemical and molecular biological techniques, particularly gene cloning and sequence analysis.
Mechanism is an inclusive term for developmental toxicologists. To be complete, it should include information about (1) the toxicant’s kinetics of uptake, distribution, storage, metabolism, and excretion as it gains access to the conceptus; (2) its interaction with a molecular component of a cellular or developmental process; (3) the consequence of that interaction for the component’s function; and (4) the consequence of that altered function for the operation of cellular and developmental processes (pathogenesis), leading to a structural or functional developmental defect. Thus, a full description of a mechanism of toxicity would draw on molecular, cellular, and developmental knowledge.
The current hypotheses of the mechanisms of toxicity and the evidence supporting the hypotheses for 11 toxicants, or groups of toxicants, were reviewed. Of these, TCDD and retinoids are the exemplars at present. The hypotheses for the toxicity are quite complete and substantiated. The toxicants interact with known proteins, specific members of the bHLH and nuclear hormone receptor families, respectively, which are signal transduction components as well as genetic regulatory components. The liganded receptors activate (or perhaps repress) the expression of certain genes at abnormal times and places. The full range of genes is not yet known, but some genes are known. The products of the misexpressed genes affect developmental processes, such as cell migration, cell responses, and the expression of yet other genes. The connection of altered gene expression to defects of organogenesis is still somewhat weak, but the outlines
have been established. Some of the altered expressions include genes encoding signaling components involved in cell-cell interactions during organogenesis.
For the case of cyclopamine, a transmembrane signaling pathway is known to be disrupted by the toxicant, leading to failure of an induction needed to pattern the eye field of the diencephalon. This is an example of a transmembrane signaling pathway that is a direct target for a toxicant.
For several other toxicants (e.g., methylmercury and methotrexate), the molecular target is not known but is probably some component of a process of cellular proliferation (e.g., DNA damage, block to DNA synthesis, disrupted spindle formation, and energy depletion). In many cases, high-dose disruption of the process results in cell death by apoptosis, preceded by the cell’s attempts to restore viability by way of molecular stress and checkpoint pathways, some of which have been partially defined. Excessive cell death results in disrupted development. The developmental effects of these toxicants has been broadly defined, but most details are missing.
For a few toxicants, fetal organ function is probably compromised by processes similar to the agent’s pharmaceutical-physiological effect on the mother (e.g., ACE inhibitors). Functional defects are in the early stages of elucidation (e.g., the connection of autism to altered HOX gene expression and altered rhombomere development in the hindbrain) and the possible role of some toxicants (including thalidomide) in altering the expression of those genes). Finally, several others act by mechanisms still largely unknown, despite knowledge of the final developmental defect (e.g., diphenylhydantoin and valproic acid).
In conclusion, there are only a few examples where the molecular, cellular, and developmental information is complete enough to say the hypothesis of the mechanism of toxicity is well substantiated. In no case is the mechanism of cellular and developmental toxicity fully known both toxicokinetically and toxicodynamically. However, it should be appreciated how broad and deep the scientific understanding has to be in order to have all the facets of a hypothesized mechanism distinguished and substantiated. The variety of mechanisms by which environmental toxicants probably work should be noted: mechanisms for toxicity are cellular, developmental, or physiological. Some involve two or more of these three. Some mechanisms occur at embryonic stages, fetal stages, or both, and some affect the conceptus, the mother, or both. Recent advances in the understanding of normal development (e.g., signaling pathways and transcriptional regulatory circuits) and cell biology (e.g., the cell cycle and checkpoint pathways) have identified critical processes, which, if investigated for their alteration by developmental toxicants, can provide exciting new advances in mechanistic investigations.






























