
CONCLUSIONS
If a revised neutron dosimetry confirms the trend that is indicated by the available activation measurements for Hiroshima, the solid-cancer data from Hiroshima could cease to be proof of a finite risk coefficient for photons. That would be a major change in the evidence supporting or refuting the linear non-threshold hypothesis, and it would add importance to the data either from Nagasaki, where the neutron doses are smaller and are unlikely to be affected by dosimetric reassessment, or from other studies where photons alone were involved.
The exploratory computations referred to here are tentative, not only because any changes in the neutron dosimetry that may be required are still uncertain, but also because they use the assumed correction factors only in a summary fashion (for details, see Kellerer and Nekolla 1997). In spite of the limitations of the computations, they demonstrate clearly the potentially important implication of new findings on the neutron doses in Hiroshima and they show the need for more-detailed numerical analyses.
6
MATHEMATICAL MODELS OF RADIATION CARCINOGENESIS
One recent development in radiation-protection research that has implications for risk assessment is the use of mathematical models of the cancer process based on a multistage mechanism. The concept of the multistage process of cancer dates back to the early part of the century, but it was in the 1950s that approaches to modeling the process gained momentum with the models developed by Armitage and Doll (1954, 1957) and the multiple-mutation approaches of Fisher (1958) and Burch (1960). Knudson (1971) derived a two-stage explanation of retinoblastoma in children from a study of the occurrence of sporadic unilateral tumors and familial bilateral tumors, which led to the concept of antioncogenes or tumor-suppressor genes (Knudson 1985, 1991). Support for the concept of tumor-suppressor genes came also from cell-fusion experiments; fusion of a tumor cell with a normal cell was found to suppress the malignant phenotype (Harris 1971; Stanbridge 1976). In the meantime, the retinoblastoma (Rb) gene has been identified, and the molecular biologic implications of the analysis of retinoblastoma have been verified (Knudson and others 1976; Cavenee and others 1983, 1985; Dunn and others 1988; Benedict and others 1988, 1990). Tumors arise from a biallelic mutation of the Rb gene in accordance with the two-stage model. Children with bilateral tumors carry an inherited mutation in the Rb gene in all their cells, so a spontaneous mutation in the normal Rb gene in a retinoblast cell leads to the tumors, but the rare sporadic unilateral tumors are associated with two spontaneous mutations in a retinoblast cell. Many other tumor-suppressor genes have been identified in recent years, and several reviews document these developments (Marshall 1991; Weinberg 1991; Skuse and Ludlow 1995).
As a consequence of the analysis of the occurrence of retinoblastoma, Moolgavkar and Knudson (1981) proposed a two-stage model with clonal expansion of ''intermediate" cells for human carcinogenesis. The model has been shown to provide a qualitative description of the age-dependent incidence of all human cancers for both children and adults (Moolgavkar and Venzon 1979; Moolgavkar 1983) and has been applied to the epidemiology of carcinomas of the breast and lung (Stevens and Moolgavkar 1979; Moolgavkar and others 1979, 1980, 1989, 1993) and radon-induced lung tumors in rats (Moolgavkar and others 1990). The mathematical nature of the model has been investigated (Moolgavkar and others 1988; Moolgavkar and Luebeck 1990; Moolgavkar 1992; Heidenreich 1996; Heidenreich and others 1997, in press) The model, or slight modifications of it, is gaining increasing use for the analysis of radiation-induced cancer in both epidemiologic and animal studies (Kai and others 1993; Leenhouts and Chadwick 1994a,b, 1997; Little 1995; Venema and others 1995; Holt 1997; Moolgavkar 1997), although some workers continue to take the Armitage-Doll multistage model into account (Little and others 1992, 1994; Chen 1993; Little and Charles 1994; Little 1995, 1996).
THE TWO-MUTATION MODEL
Although the modifications of the two-mutation model lead to some quantitative differences in analyses, a global description that covers the essence of the model can be used to gain an insight into the cancer process and derive some generally applicable implications. A schematic representation of the two-mutation model based on the developments of Moolgavkar and Knudson (1981) is given in figure 5.
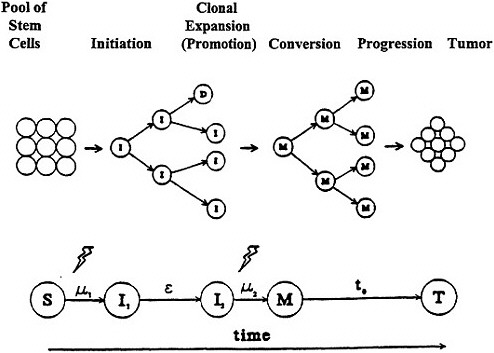
Figure 5. Moolgavkar and Knudsen two-stage model with clonal expansion of "intermediate" cells for human carcinogenesis. Adapted with permission from Chadwick and Leenhouts (1995).
The model includes two mutational steps, μ1 (initiation) and μ2 (conversion), which convert a normal stem cell via an intermediate stage to a malignant cell, which can then grow out (progression) into a detectable tumor. An important aspect of the model is that it incorporates cellular turnover of the stem cells, the intermediate cells (I), and the malignant cells (M), as well as taking cell death (D) and differentiation info account. Thus, an intermediate cell can divide in a non-linear, exponential-like, clonal expansion (promotion) to create, with the passage of time (ε), an increasing number of potential target cells for the second mutation. To increase the biologic plausibility of the model, a lag time(t0), often held constant, is invoked for the time from the generation of the first malignant cell to the detection of the tumor, although this should not be confused with latency. The model thus incorporates the various steps associated with the development of cancer, such as initiation, promotion, conversion, and progression; but the terminology and characterization of these steps are not unambiguously identified in the cancer literature.
The mathematical equations that can be derived from the model to describe the age-dependent incidence of a specific cancer do not always have an explicit solution, but by calculating the number of cells in each compartment—that is, the stem cells, the intermediate cells, and the malignant cells—for small intervals in an iterative process
starting from zero, or birth, we can derive the age-specific incidence. The calculations rely on assumptions about the starting conditions at time zero, such as the number and time-dependent expansion in the number of stem cells, the levels of the mutation rates, and the expansion rate of the intermediate cells. Spontaneous cancer is inherently assumed to develop with time as a consequence of two "spontaneous" mutations occurring in the initiation and conversion steps, and analysis of age-specific incidence curves for a given unexposed population can be used to define the background against which a radiation effect must be determined. Radiation is assumed to be able to induce mutations in both steps, although the relative importance of radiation in each step is not defined a priori and radiation-induced killing of stem cells, intermediate cells, and malignant cells is also taken into account. In this way, dose-effect relationships determined in cellular radiation biology are introduced into the model, and lead to the simultaneous calculation of the age-specific incidence and the dose-dependent incidence of a cancer. The model can be used for both acute and very protracted (such as lifetime) exposures; radiation-induced mutations increase the mutation rates, μ1 and μ2, instantaneously in the case of an acute dose or for the duration of exposure for a chronic dose. Thus, in accordance with cellular radiation biology a linear-quadratic dose-effect relationship might be used for the cellular effects of an acute exposure in general, with a linear dose-effect relationship for chronic and very protracted exposures; in addition, the cellular RBEs of different kinds of radiation can be taken into account. Radiation is normally not assumed to act as a promoter except at very high acute doses, when cell depletion might stimulate increased division of stem cells and intermediate cells.
A consequence of radiation increasing the mutation rates in both the initiation and conversion steps of the sample model previously described and the interdependence of one mutational step on the other for the development of cancer, is that in most cases, a radiation-induced mutation in one step will interact with a "spontaneous" mutation in the other step on the path to cancer. That actually implies that the level of the radiation effect is related to the "spontaneous" cancer incidence. It also means that at low doses radiation will usually be a cocarcinogen, inasmuch as a second mutation induced "spontaneously" will be needed to complete the process, and only rarely will it be a complete carcinogen.
Thus, models might be used in the analysis of epidemiologic cohort studies that have good age-specific incidence data for both the cohort and the control populations. It is less suited to the analysis of epidemiologic case-control studies. Its use for the analysis of results of animal experiments is often hindered by the fact that many animal tumors are not directly lethal. Unless animals are sacrificed throughout the experiment, the tumors are not detected until the animals are moribund and age-specific incidence data are not normally available.
IMPLICATIONS OF THE MODEL FOR RISK ESTIMATION
The two-mutation model—which is, as Knudson (1991) points out, "a minimal model for carcinogenesis," is also remarkably effective and has several implications for radiation risk. The model, which calculates simultaneously the age-specific incidence and the dose-dependent incidence of cancers over the whole lifetime, can be used for acute exposures (Little and Charles 1994; Moolgavkar 1997), very protracted exposures such as those of uranium miners (Moolgavkar and others 1993), and lifetime exposures such as to indoor radon. The model has therefore been used by some investigators to provide a basis for lifetime extrapolations of radiation risk. A BEIR VII phase-2 committee should examine all relevant models and consider how appropriate models might contribute to risk assessment.
If an acute exposure affects the first mutation step (initiation), the model predicts that the risk will resemble a relative-risk projection; if the exposure affects the second mutation (conversion) the model predicts that the risk will resemble an absolute-risk projection (figure 6).
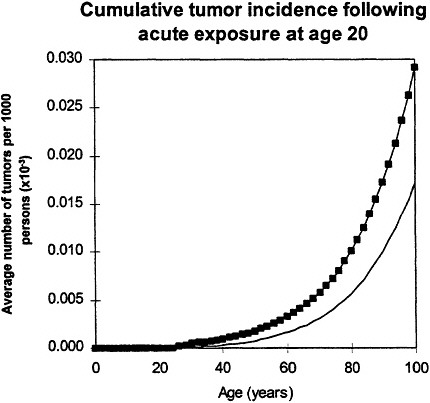
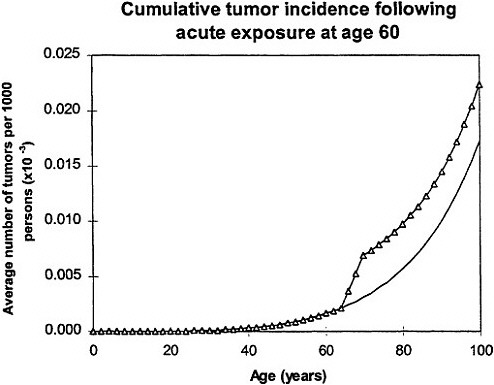
Figure 6. Upper panel: calculated age dependence of tumor incidence after acute exposure at age 20 ![]() compared with spontaneous incidence (continuous line) resembles relative risk.
compared with spontaneous incidence (continuous line) resembles relative risk.
Lower panel: calculated age dependence of tumor incidence after acute exposure at age 60 (Δ) compared with spontaneous incidence (continuous line) resembles absolute risk. Adapted with permission from Chadwick and Leenhouts (1995).
In all calculations, it was assumed that cellular radiation sensitivity was co tam throughout lifetime and that the radiation sensitivity was equal for both mutational steps.
If the exposure is over a lifetime, the model predicts that the risk will resemble a relative-risk projection (figure 7), which implies that, in this case, exposure at an early age is the defining factor (Leenhouts and Chadwick 1994a; Chadwick and Leenhouts 1995).
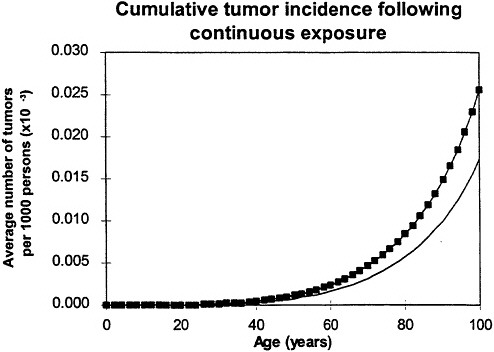
Figure 7. Calculated age dependence of tumor incidence after protracted lifetime exposure ![]() compared with spontaneous incidence (continuous line) resembles relative risk. In all calculations, it was assumed that cellular radiation sensitivity was constant throughout lifetime and that the radiation sensitivity was equal for both mutational steps. Adapted with permission from Chadwick and Leenhouts (1995).
compared with spontaneous incidence (continuous line) resembles relative risk. In all calculations, it was assumed that cellular radiation sensitivity was constant throughout lifetime and that the radiation sensitivity was equal for both mutational steps. Adapted with permission from Chadwick and Leenhouts (1995).
Inherent in the model is the interplay between the radiation-induced mutations and the spontaneous mutations and the fact that, at low doses, the radiation will always be a cocarcinogen. Because the background cancers must arise from "spontaneous" mutations, and there is interplay between the radiation-induced mutations and the "spontaneous" mutations, the model predicts that the risk following exposure to radiation depends on the background incidence of cancer associated with both acute and protracted exposures (figure 8).
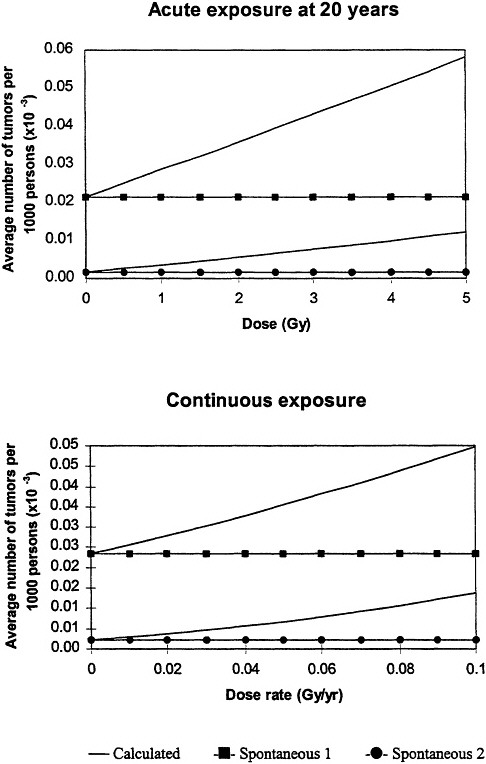
Figure 8. Upper panel: calculated low-dose incidence at end of life after acute, low-dose-rate exposure at age 20 for two levels of spontaneous cancer incidence as a function of lifetime dose. Initial slope increases as spontaneous-cancer incidence increases. All calculations assume constant cellular radiation ensitivity throughout lifetime and equal radiation sensitivity for both mutational steps.
Lower panel: calculated low-dose incidence at end of life after protracted lifetime exposure for two levels of spontaneous-cancer incidence as a function of average annual dose rate. Initial slope increases as spontaneous-cancer incidence increases; both curves exhibit slight upward curvature. In all calculations, it was assumed that cellular radiation sensitivity was constant throughout lifetime and that the radiation sensitivity was equal for both mutational steps. Adapted with permission from Chadwick and Leenhouts (1995).
For example, the probability that a particular radon exposure leads to a lung cancer in a nonsmoker is lower than the probability that it leads to a lung cancer in a smoker, even though the relative risk in nonsmokers is higher. With respect to cancers that are extremely rare, the model predicts that radiation will have little chance of inducing a cancer except after protracted exposure to very high doses (for example, bone cancers in radium-dial painters). An important consequence of this dependence of risk following exposure to radiation on spontaneous-cancer incidence is that different cancer types with different spontaneous incidences cannot be grouped for analysis. This implication of the model provides a theoretical basis, which can be tested, for the extrapolation of radiation risk for a specific cancer across populations (such as, breast cancer from Japan to America) on the basis of the background level of the cancer.
In the comparison of exposure conditions (such as age at first exposure, length of exposure, and radiation effect on second mutations), the model implicitly predicts an "inverse dose-rate effect" that is more related to an exposure-time effect but, in any case is independent of any considerations of inverse dose-rate effect at the cellular level. It seems likely that the inverse dose-rate effect revealed by the model is the one responsible for the indications of such an effect found in some epidemiologic studies (such as studies of uranium miners).
The model provides a basis for the study of the combined effect of two carcinogenic agents by adding the mutagenic contributions of the two agents to the spontaneous mutation rate in both the initiation and the conversion steps. In addition, the model offers the possibility of including the effect of agents that are thought to be promoters rather than mutators.
The model implies that the shape of the dose-effect relationships for induction of the more commonly occurring cancers will reflect the shape of the cellular dose-effect relationships for the induction of chromosomal mutations and that the modifying effects of dose rate and radiation type at the cellular level will be reflected in the changes in the shape of the cancer-induction curves (Leenhouts and Chadwick 1994b). In the case of very rare cancers, the model implies that induction by radiation will be rare and have a curvilinear dose-effect relationship.
CONCLUSIONS
Recent developments in the application of the two-mutation model of carcinogenesis to the analysis of radiation epidemiologic and animal studies have suggested differing approaches and different insights into the action of radiation. A BEIR VII committee should critically examine the status of models that might be relevant to risk assessment. Multistage models might provide a tool that can be used to relate the molecular investigations on radiation mechanisms at the cellular level to the epidemiologic studies of exposed populations. The models also might provide a basis for extrapolating radiation effects to low doses and low dose rates and across populations which could be useful and meaningful for risk assessment. It must always be borne in mind that any model is a simple representation of the facts and, while the use of the two-mutation model of carcinogenesis might suggest different insights into the action of radiation, considerable care must be taken in applying such models to experimental data and in interpreting the results of such analyses.










