
This paper was presented at a colloquium entitled “Vision: From Photon to Perception,” organized by John Dowling, Lubert Stryer (chair), and Torsten Wiesel, held May 20–22, 1995, at the National Academy of Sciences in Irvine, CA.
The biology of vision in Drosophila
(G protein/phospholipase C/ion channels/signal transduction)
CHARLES S.ZUKER
Howard Hughes Medical Institute and Departments of Biology and Neurosciences, University of California at San Diego, La Jolla, CA 92093-0649
ABSTRACT Phototransduction systems in vertebrates and invertebrates share a great deal of similarity in overall strategy but differ significantly in the underlying molecular machinery. Both are rhodopsin-based G protein-coupled signaling cascades displaying exquisite sensitivity and broad dynamic range. However, light activation of vertebrate photoreceptors leads to activation of a cGMP-phosphodiesterase effector and the generation of a hyperpolarizing response. In contrast, activation of invertebrate photoreceptors, like Drosophila, leads to stimulation of phospholipase C and the generation of a depolarizing receptor potential. The comparative study of these two systems of phototransduction offers the opportunity to understand how similar biological problems may be solved by different molecular mechanisms of signal transduction. The study of this process in Drosophila, a system ideally suited to genetic and molecular manipulation, allows us to dissect the function and regulation of such a complex signaling cascade in its normal cellular environment. In this manuscript I review some of our recent findings and the strategies used to dissect this process.
The Drosophila compound eye is made up of 800 ommatidia or unit eyes. Each ommatidium is composed of 20 cells including 8 photoreceptor neurons (for review, see ref.1). The eight photoreceptor neurons can be divided into three main classes depending on their spectral sensitivity: the six outer photoreceptors are blue sensitive and contain the major rhodopsin in the retina, Rh1 (2,3). Of the two central neurons, the distal R7 cells express one of two different UV-sensitive opsins (4), and the proximal R8 cell expresses a blue-green rhodopsin (5). Each photoreceptor cell contains a specialization of the plasma membrane, known as a rhabdomere, composed of ≈60,000 microvilla; these are the functional equivalent of the discs in the rod outer segments and contain rhodopsin and the machinery involved in phototransduction (the large increase in surface area provided by the rhabdomeres allows the photoreceptor neurons to pack > 100 million molecules of rhodopsin per cell).
Fig. 1 shows a highly schematized, summarized view of the phototransduction cascade in Drosophila. The light receptor molecule rhodopsin (R) is composed of a protein, opsin, covalently linked to a chromophore, 3-hydroxy-11-cis-retinal. Upon absorption of a light photon the chromophore is isomerized from the 11-cis to the all-trans configuration. This change in the conformation of the chromophore leads to a change in the conformation of the protein and to the activation of its catalytic properties. Activated rhodopsin, or metarhodopsin (M), activates a heterotrimeric G protein of the Gq-family (6,7), which in turn activates a phospholipase C (PLC) encoded by the norpA gene (8). PLC catalyzes the breakdown of the minor membrane phospholipid phosphatidyl 4,5-bisphosphate (PIP2) into the two intracellular messengers inositol trisphosphate (I P3) and diacylglycerol (DAG). This reaction leads to the opening of cation-selective channels and the generation of a depolarizing receptor potential (Drosophila photoreceptors, like most invertebrates, depolarize as opposed to hyperpolarize in response to light). In addition to excitation, photoreceptor neurons have evolved sophisticated mechanisms to control termination of the light response (deactivation) and light and dark adaptation (for review, see D. Baylor in this issue). Molecular, genetic, and physiological studies suggest that as many as 50 different gene products are dedicated to the functioning and regulation of this one signaling cascade in the fruit fly Drosophila melanogaster (9–11).
Genetic and Molecular Dissection of Phototransduction
We and others have used three general strategies to identify molecules involved in phototransduction. The first relies on the expectation that many of the proteins involved in this process will be encoded by genes preferentially expressed in the visual system. This is not an unreasonable assumption because the high degree of specialization seen in photoreceptors was most likely accompanied by the evolution of dedicated components. Thus, by taking advantage of mutants lacking compound eyes and using highly sensitive subtraction hybridization protocols, it has been possible to isolate a large number of genes encoding eye-specific proteins. Examples include ninaA, eye-PKC and G-protein subunits. The second strategy, and perhaps the most powerful, relies on classical genetics and functional screens. Nearly 30 yr ago Seymour Benzer isolated the first Drosophila visual mutants by screening for defects in visual behavior (this was also the birth of the field of neurogenetics) (12). Several years later, Bill Pak and coworkers at Purdue University pioneered the use of electrophysiological screens to search for mutant flies with defects in visual physiology (13). Since then, several groups, including our own, have extended these screens to include a wide range of genetic, physiological, and behavioral assays of photoreceptor function. The molecular and physiological analysis of many of these mutants is providing fundamental insight into the biology of this process (see below). The third strategy relies on enhancer trap screens, a technique developed in Walter Gehring's laboratory several years ago. In essence, by using a bacterial β-galactosidase reporter element and assaying for flies with blue eyes, it has been possible to tag and isolate genes that are preferentially expressed in the visual system. Using combinations of these three strategies we have been able to identify a number of genes involved in phototransduction (9–11). Of particular interest are those whose role could have not been predicted on
The publication costs of this article were defrayed in part by page charge payment. This article must therfore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: IP3, inositol, trisphosphate; DAG, diacylglycerol; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; PKC, protein kinase C; [Ca2+]i, intracellular Ca2+ concentration.
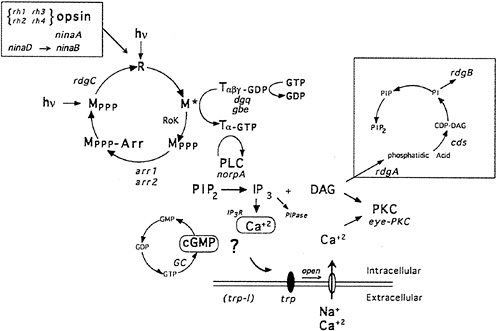
FIG. 1. Phototransduction in Drosophila photoreceptors. Absorption of a photon of light causes a conformational change in the rhodopsin molecule (R) and activates its catalytic properties. Active metarhodopsin (M*) catalyzes G protein activation. The G protein exchanges GDP for GTP and releases the inhibitory βγ subunits. Active G protein catalyzes the activation of the norpA-encoded PLC. PLC hydrolyzes PIP2 into the intracellular messengers IP3 and DAG. cGMP has also been implicated as a possible intracellular messenger mediating excitation. Extracellular sodium and calcium enter the cell through the light-activated conductance and cause the depolarization of the photoreceptor cells. The light-activated conductance appears to be composed of at least two types of channels. The trp gene is required for a class of channels with high calcium permeability. DAG is thought to modulate a photoreceptor cell-specific PKC (encoded by inaC) that regulates deactivation and desensitization of the light response. Metarhodopsin is inactivated via phosphorylation by rhodopsin kinase (RoK) and arrestin binding (encoded by the arr1 and arr2 genes). Inactive metarhodopsin is photoconverted back to rhodopsin and then presumably dephosphorylated by the rdgC-encoded phosphatase. The box in the upper right indicates a pathway likely to be required for synthesis of PIP2. rdgA encodes DAG-kinase and rdgB encodes a protein with significant sequence homology to phosphatidylinositol-transfer protein, cds refers to CDP-DAG synthase. dgq and gbe are the genes encoding the photoreceptor cell-specific isoforms of Gα and Gβ subunits, respectively. ninaB and ninaD are genes required for retinal biogenesis, and ninaA is a cyclophilin homolog required for rhodopsin biogenesis, rh1, rh2, rh3, and rh4 are the structural genes for the four known rhodopsins. IP3R and PIPase refer to the IP3 receptor and inositol polyphosphate phosphatase (an enzyme required to break down IP3). Mutations in all gene products highlighted in red are now available (refs.11 and 22, and unpublished work from C.S.Z. laboratory).
biochemical grounds but in which a genetic approach provided fundamental insight as to their functional requirement. Examples are the cyclophilin homologue ninaA and its role in rhodopsin biogenesis (14–17), an eye-specific protein kinase C (PKC) required for deactivation and calcium feedback regulation (18,19), the role of Gβ in the termination of the light response (20), and a number of enzymes involved in inositol phospholipid metabolism and shown to be required for photoreceptor cell excitation (10,21).
Inositol Phospholipids and Phototransduction
Drosophila phototransduction is one of the best model systems for the study of G protein-coupled PLC signaling (10,22,23). Not only is the system amenable to molecular genetic analysis but also it can report activity with exquisite sensitivity and specificity: photoreceptor cells are sensitive to single photons, and the signaling pathway can be turned on and off with millisecond kinetics (phototransduction in Drosophila is the fastest known G protein cascade, taking just a few tens of milliseconds to go from light activation of rhodopsin to the generation of a receptor potential). As described above, active PLC catalyzes the hydrolysis of the minor membrane phospholipid PIP2 into the second messengers inositol IP3 and DAG. IP3 mobilizes calcium from internal stores, which affects and modulates many cellular processes, and DAG activates members of the PKC family of proteins. Given the central role of PIP2 in signaling, its levels may be expected to be tightly regulated in the cell.
Fig. 2 shows an expanded view of the PIP2 cycle. CDP-DAG synthase (CDS) is an enzyme required to convert phosphatidic acid into CDP-DAG, the acceptor for the inositol head group. Using enhancer trap technology, we identified an eye-specific form of CDP-DAG and isolated mutations in this gene (eyecds) (21). To determine whether eye-cds mutants have a defect in their signaling properties, wild-type and mutant animals were assayed for their ability to maintain a continuously activated state of the photoreceptor cells because such a state would require the continuous availability of the second messenger PIP2. Our results demonstrated that light activation depletes a pool of PIP2 necessary for excitation that cannot be replenished in eye-cds mutants (Fig. 3a–d). This phenotype is due exclusively to a defect in eye-cds, because introduction of the wild-type eye-CDS cDNA into mutant hosts fully restores wild-type physiology (Fig. 3e–f). Furthermore, inclusion of PIP2 in the patch pipette is sufficient to restore signaling in the depleted eye-cds mutants (Fig. 3g–h). These results suggest, contrary to expectations, that the pool of PIP2 required for signaling is quite small and likely synthesized “on demand”. On the basis of these findings, we reasoned that it should be possible to modulate the output of this cascade by experimentally manipulating the levels of eye-CDS. Indeed, we made
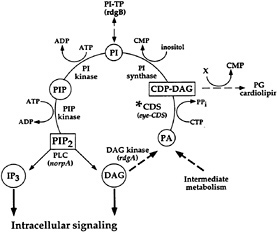
FIG. 2. PIP2 regeneration cycle. Upon activation, PLC hydrolyzes PIP2 to yield IP3 and DAG. To regenerate the PIP2, DAG is phosphorylated by DAG kinase (dgk) to produce phosphatidic acid (PA). CDP-DAG synthase (CDS*) adds on CMP to phosphatidic acid. This product, CDP-DAG, is the activated donor of the phosphatidyl group to inositol. The phosphatidylinositol (PI) is phosphorylated by PI kinase and PIP kinase to yield PIP2. Listed in parentheses are the sites of action of known photoreceptor cell-specific proteins in Drosophila (adapted from ref.21).
transgenic animals that overexpressed the enzyme and generated photoreceptor neurons that now display responses that are 300–400% larger than wild-type cells (21). These results open up the possibility of genetically and pharmacologically manipulating PIP2 signaling in vivo and highlight three unexpected aspects of PLC signaling. (i) PIP2 pools required for signaling are distinct from the general pool. This is demonstrated by the observation that eye-cds mutants are only defective in signaling and only in response to light activation. (ii) PIP2 levels involved in signaling are low and likely generated “on-line”. (iii) PIP2 availability is rate limiting and regulates the output of this signaling pathway. This is further demonstrated by the observation that eye-cds mutants display a reduction in the amplitude of their response as a function of the number of light flashes (and thus their state of depletion).
A search for second-site mutations that enhance or suppress the eye-CDS phenotype should produce mutations in other components of this cycle and make it possible to carry out a comprehensive genetic dissection of the various players required for the functioning and regulation of PIP2 and its metabolites.
Calcium in Drosophila Photoreceptors
An important and unresolved issue in the study of invertebrate phototransduction has been the identification of the intracellular messenger(s) that mediate the opening of the lightactivated ion channels. IP3, calcium, and cGMP have been implicated in this process (23,24). Although the messenger(s) that actually gates the plasma membrane ion channels remains elusive, patch clamp studies have provided strong evidence implicating calcium in the regulation of the light response (18,25,26). For example, extracellular calcium influx is both sufficient and necessary to regulate activation and deactivation kinetics of the light-activated conductance. In the absence of external calcium, photoreceptors display slow activation and deactivation kinetics. Conversely, high extracellular calcium solutions, or release of caged-intracellular Ca2+ ([Ca2+]i) during a light response (27), cause a transient acceleration in activation kinetics followed by rapid deactivation.
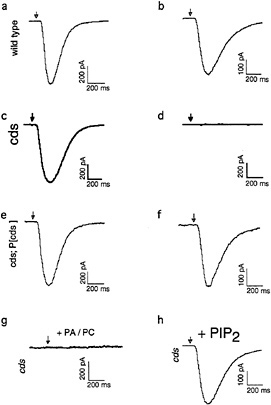
FIG. 3. Defects shown by eye-cds mutants in photoreceptor cell function. To determine whether eye-cds mutants have a defect in their signaling properties, we assayed wild-type and mutant animals for their ability to maintain a continuous supply of the second-messenger PIP2. Control and mutant cells were dissected and transferred to a bath solution with nominally zero calcium. The excitation mechanisms were then depleted before patching by subjecting the cells to 40 min of a light pulse protocol, consisting of 3 sec of intense light pulses followed by 3 min in the dark. If wild-type cells are patched after the depletion protocol with 700 nM [Ca2+]i in the patch pipette, the light response reliably recovers (a, b). If the same depletion protocol is applied to cds mutant cells, the light response does not recover (c, d). This phenotype is due exclusively to a defect in eye-cds because introduction of the wild-type eye-CDS cDNA into mutant hosts fully restores wild-type physiology (e, f). Depleted cds mutants can be rescued by supplying PIP2 through the patch pipette (g). (a, c, and e) Responses of wild-type, eye-cds and P[cds+] photoreceptors before depletion, respectively. Arrows indicate the position of the stimulating light flash. See ref.21 for further details.
Despite the role of calcium in regulation, all available data indicate that calcium release from internal stores is neither the signal nor is it required for the opening of the light-activated channels ( 28,29). However, internal calcium stores are required both for the developmental maturation of the lightactivated currents (30) and for maintaining a responsive state (31). For instance, mature currents (e.g., adult-like) can be induced in immature pupae by artificially raising [Ca2+]i, and photoreceptors depleted for their internal stores show a dramatic loss of sensitivity that can be rescued by raising [Ca2+]i.
In efforts to directly image calcium changes in intact photoreceptors, Rama Ranganathan, Brian Backsai, Roger Tsien, and myself (29) developed a preparation suitable for simultaneous recording of light-activated currents and the dynamics of [Ca2+]i using fluorescent calcium indicators. Past attempts to measure simultaneously light-induced current and fluores
cence signals in vertebrate (32,33) and invertebrate (28,34) photoreceptors have been compromised by the inability to functionally separate the stimulating light from that required for fluorescence excitation. Two possible solutions to this problem are as follows: (i) to use a fluorescent calcium indicator whose excitation spectrum is well separated from the action spectra of the cell (not currently available) (35,36), or (ii) to retune the cell's response to become spectrally separated from available fluorescent calcium indicators. Indeed, we genetically engineered flies that express a UV-specific rhodopsin in place of the normal rhodopsin (4), so that long wavelength light can be used to image [Ca2+]i while minimally exciting the photoreceptor cell. To achieve high temporal and spatial resolution in recording [Ca2+]i, the cells were imaged on a high-speed laser scanning confocal microscope acquiring images at 30 frames per sec (37). Using this preparation, we showed that influx of external calcium is responsible for all measured light-dependent changes in [Ca2+]i (29).
The first response to the UV stimulus is seen as a highly localized elevation in [Ca2+]i at the junction of the rhabdomere and cell body (Fig. 4); this is consistent with localization of light-sensitive channels at the base of the rhabdomeres. This initial response is rapidly followed by a general increase in [Ca2+]i at the rhabdomere, which generates a dramatic gradient of calcium levels within the cell. The existence of functionally relevant, ultramicro domains of high [Ca2+]i has been postulated in a number of signaling processes (for review, see ref.38). Theoretical calculations indicate that [Ca2+]i can reach tens to hundreds of micromolarity within 10 to 100 nm from each conducting channel. Such ultramicro domains would, of course, be too small to be resolved by present optical techniques. Using a combination of calcium chelators, electrophysiology, and calcium imaging, we showed that the Ca2+ influx initially generates large-amplitude submicroscopically localized [Ca2+]i transients that are >6 µM and are crucial for negative regulation. These studies provided experimental support for a functional requirement of spatially localized [Ca2+]i and suggest that the transducing machinery may be organized and compartmentalized as a “transducisome” responding to highly localized signals. This provides an elegant avenue to prevent signal cross-talk in intracellular signaling pathways.
What are the targets of calcium in mediating negative regulation? An electrophysiological screen for Drosophila phototransduction mutants with defects in deactivation kinetics demonstrated that photoreceptors from inaC mutants (39) are specifically defective in the calcium-dependent negative regulatory mechanisms (40). When compared to wild-type controls, inaC cells have deactivation kinetics that are > 20 times slower. Molecular cloning of the inaC locus showed that it encodes an eye-specific isoform of PKC (eye-PKC) (19). eye-PKC is found only in photoreceptors, and within photoreceptors, the protein localizes to the light-sensitive microvillar membranes (19). These results suggest a model in which the light-dependent generation of DAG (from the breakdown of PIP2) together with the influx of external calcium activates eye-PKC. Interestingly, eye-PKC is also required for normal kinetics of the return of [Ca2+]i to resting levels (29). Thus, this is a wonderful regulatory loop in which activation of regulatory mechanisms is intimately tied to the productive activation of the signaling cascade. Active PKC could then phosphorylate specific target(s) and mediate termination of the light response by catalyzing the inactivation of active intermediates. The target(s) of eye-PKC have not yet been identified.
Light-Activated Ion Channels
The Drosophila light-activated conductance is nonselective for cations and is primarily permeable to calcium ions (18,25). Unexpectedly, the light-activated conductance is composed of at least two biophysically distinct channels. One of these is encoded by the trp gene product and is responsible for the majority of the calcium permeability (31). Although the molecular identity of the non-trp-dependent light-activated channels is not known, Phillips et al. (41) identified a protein with

FIG. 4. Spatial localization of light-induced calcium transients in transgenic animals expressing a UV rhodopsin in the blue photoreceptor cells. Sequential images of a cell in response to a 33-msec UV stimulus at t = 0 are shown. Calcium changes relative to preflash levels are displayed using a pseudocolor scale. Images were acquired at a video frame rate of 30 Hz. The rhabdomere of this cell (rhabdo) is distinctly resolved from the cell body (nucl. = nucleus). By the third image (t = 67 msec), the first response in [Ca2+]i is noted as a highly localized increase at the rhabdomere base. This time window (between 33 and 67 msec) is consistent with the time latency of the light response (11). The subrhabdomeric calcium burst is rapidly followed by a wave of elevated [Ca2+]i largely localized to the rhabdomere. The [Ca2+]i in the cell body also rises, but to a lesser degree. To enhance signals above noise, we time averaged images from five identical flash trials, and each resulting image was subjected to spatial averaging. The bottom, right-hand panel shows a black and white photograph of the same cell at low contrast levels to emphasize the difference between cell body and rhabdomere. [Reproduced with permission from ref.29 (copyright Cell Press).]
significant sequence similarity to the Trp protein that is also expressed in photoreceptors. This gene, called Trp-like (trpl), encodes a protein that displays 39% amino acid identity with Trp.
trp mutants were isolated over 25 yr ago (42) and shown to be defective in maintenance of the light response (thus the name transient-receptor-potential). We now know that internal calcium is required to maintain a receptor potential and that trp mutants have lost the major light-activated calcium entry pathway (28,31,34). On the basis of these findings, Hardie and Minke (23) have suggested that the Trp phenotype results from a failure in the refilling of the internal stores, trp mutant photoreceptors are also inactivated after a strong light stimulus. This is likely due to the emptying of the stores and a subsequent decrease in the efficiency of the excitation process ( 43,44).
Analysis of the Trp sequence showed regions of weak similarity with neuronal voltage-gated Ca2+ channels (45,46), consistent with the notion that trp encodes a plasma membrane channel with high calcium permeability. Interestingly, a number of studies suggest that Trp may be related to the elusive vertebrate Icrac ion channel (calcium-release-activated-channel) (11,23), and thus Trp homologs may be critically important in the regulation of intracellular calcium. In efforts to determine whether Trpl also encodes a component of the light-activated conductance, we set up to isolate mutations in this gene. A difficulty in setting up a screen for mutations in trpl is the lack of a reliable, predictable phenotype that defines its loss of function and the possibility that trp and trpl may serve redundant functions. Because of these concerns, we used a screening strategy that was based on the loss of Trpl antigen on immunoblots (20,47). The advantage of this screen is that it does not rely on a hypothetical physiological or behavioral defect but only on the presence or absence of Trpl protein. After screening several thousand chromosomes, we isolated a knock-out mutation in trpl. These mutants are now being subjected to detailed genetic, biochemical, and physiological characterization. Interestingly, Trp and Trpl are not subunits of the same channel (B. Niemeyer and C.S.Z., in preparation). Recently, vertebrate homologs of Trp and Trpl have been cloned. The analysis of these channels and the trp and trpl mutants is likely to provide important insight into the biology of this novel class of ion channels and their role in calcium homeostasis.
Future Challenges
Phototransduction has proven to be an ideal model system for the study of G protein-coupled signaling cascades. Basic cellular phenomena like signal amplification and integration, response deactivation, and adaptation have been first addressed in this signaling pathway. The study of this process also resulted in the molecular cloning of the first seven transmembrane domain receptor (48), the first cyclic nucleotide-gated ion channel (49), and the crystal structure of the first Gα protein subunit (50). Furthermore, the genetic dissection of this pathway in humans and flies has provided fundamental insight into the molecular and cellular basis of inherited retinal disorders (51,52). However, despite these great advances, many important questions still remain. For example, what are the determinants of the kinetics of activation? What are the detailed molecular mechanisms of light and dark adaptation? How do the different signaling molecules interact with each other and regulate their output? How is response deactivation controlled? What are the intracellular messengers in invertebrate phototransduction? How is signal cross-talk prevented?
A complete understanding of the phototransduction process will have to wait until all the gene products that have a role in this process are identified and studied by the physiological effect of their loss or dysfunction. It is here where the study of phototransduction in Drosophila offers unprecedented versatility. The study of this signaling cascade in the fruit fly Drosophila melanogaster makes it possible to use powerful molecular genetic techniques to identify novel transduction molecules and then to examine the function of these molecules in vivo, in their normal cellular and organismal environment. Recent advances in mouse knockout technology also offer an exciting opportunity for a genetic dissection of this process in vertebrates. The combination of these two systems of study may provide the answer to the biggest challenge for the future: how is the entire response of a photoreceptor cell orchestrated in vivo?
I deeply thank past and present members of my laboratory for their contributions. This research was funded by grants from the National Eye Institute, the Pew Foundation, the McKnight foundation, and the March of Dimes. C.S.Z. is an investigator of the Howard Hughes Medical Institute.
1. Wolff, T. & Ready, D. ( 1993) in The Development of Drosophila melanogaster, eds. Bate, M. & Arias, A. M. (Cold Spring Harbor Lab. Press, Plainview, NY), p. 1277.
2. O'Tousa, J. E., Baehr, W., Martin, R. L., Hirsh, J., Pak, W. L. & Applebury, M. L. ( 1985) Cell 40, 839–850.
3. Zuker, C. S., Cowman, A. F. & Rubin, G. M. ( 1985) Cell 40, 851–858.
4. Feiler, R., Bjornson, R., Kirshfeld, K., Mismer, D., Rubin, G. M., Smith, D. P., Socolich, M. & Zuker, C. S. ( 1992) J. Neurosci. 12, 3862–3868.
5. Harris, W. A., Stark, W. S. & Walker, J. A. ( 1976) J. Physiol. (London) 256, 415–439.
6. Lee, Y.-J., Dobbs, M. B., Verardi, M. L. & Hyde, D. R. ( 1990) Neuron 5, 889–898.
7. Scott, K., Leslie, A., Sun, Y., Hardy, R. & Zuker, C. ( 1995) Neuron 15, 919–927.
8. Bloomquist, B., Shortridge, R., Schneuwly, S., Perdew, M., Montell, C., Steller, H., Rubin, G. & Pak, W. ( 1988) Cell 54, 723–733.
9. Smith, D. P., Stamnes, M. A. & Zuker, C. S. ( 1991) Annu. Rev. Cell Biol. 7, 161–190.
10. Zuker, C. S. ( 1992) Curr. Opin. Neurobiol. 2, 622–627.
11. Ranganathan, R., Malicki, D. M. & Zuker, C. S. ( 1995) Annu. Rev. Neurosci. 18, 283–317.
12. Benzer, S. ( 1967) Proc. Natl. Acad. Sci. USA 58, 1112–1119.
13. Pak, W. L., Grossfield, J. & Arnold, K. ( 1970) Nature (London) 227, 518–520.
14. Stamnes, M. A., Shieh, B.-H., Chuman, L., Harris, G. L. & Zuker, C. S. ( 1991) Cell 65, 219–227.
15. Stamnes, M., Rutherford, S. & Zuker, C. ( 1992) Trends Cell Biol. 2, 272–276.
16. Colley, N., Baker, E., Stamnes, M. & Zuker, C. ( 1991) Cell 67, 255–263.
17. Baker, E., Colley, N. & Zuker, C. ( 1994) EMBO J. 13, 4886–4895.
18. Ranganathan, R., Harris, G. L., Stevens, C. F. & Zuker, C. S. ( 1991) Nature (London) 354, 230–232.
19. Smith, D. P., Ranganathan, R., Hardy, R. W., Marx, J., Tsuchida, T. & Zuker, C. S. ( 1991) Science 254, 1478–1484.
20. Dolph, P. J., Man, Son, Hing, H., Yarfitz, S., Colley, N. J., Deer, J. R., Spencer, M., Hurley, J. B. & Zuker, C. S. ( 1994) Nature (London) 370, 59–61.
21. Wu, L., Niemeyer, B., Colley, N., Socolich, M. & Zuker, C. ( 1994) Nature (London) 373, 216–222.
22. Minke, B. & Selinger, Z., eds. ( 1991) in Progress in Retinal Research, eds. Osborne, N. & Chader, G. (Pergamon, New York), Vol. 11, pp. 99–123.
23. Hardie, R. C. & Minke, B. ( 1993) Trends Neurosci. 16, 371–376.
24. Minke, B. & Selinger, Z. ( 1992) in Progress in Retinal Research, eds. Osborne, N. & Chader, G. (Pergamon, Oxford), pp. 99–124.
25. Hardie, R. ( 1991) Proc. R. Soc. London B 245, 203–210.
26. Hardie, R. C. & Minke, B. ( 1994b) J. Gen. Physiol. 103, 389–407.
27. Hardie, R. ( 1995) J. Neurosci. 15, 889–902.
28. Peretz, A., Suss-Toby, E., Rom-Glas, A., Arnon, A., Payne, R. & Minke, B. ( 1994) Neuron 12, 1257–1267.
29. Ranganathan, R., Bacskai, B., Tsien, R. & Zuker, C. ( 1994) Neuron 13, 837-848.
30. Hardie, R. C., Peretz, A., Pollock, J. A. & Minke, B. ( 1993) Proc. R. Soc. London B 252, 223–229.
31. Hardie, R. & Minke, B. ( 1992) Neuron 8, 643–651.
32. Ratto, G. M., Payne, R., Owen, W. G. & Tsien, R. Y. ( 1988) J. Neurosci. 8, 3240–3246.
33. Gray-Keller, M. & Detwiler, P. ( 1994) Neuron 13, 849–861.
34. Peretz, A., Sandler, C., Kirschfeld, K., Hardie, R. & Minke, B. ( 1994) J. Gen. Physiol. 104, 1057–1077.
35. Tsien, R. Y. ( 1980) Biochemistry 19, 2396–2404.
36. Tsien, R. Y. ( 1989) Annu. Rev. Neurosci. 12, 227–253.
37. Tsien, R. & Bacskai, B. ( 1994) in Handbook of Biological Confocal Microscopy, ed. Pawley, J. (Plenum, New York), pp. 459–478.
38. Augustine, G. J. & Neher, E. ( 1992) Curr. Opin. Neurobiol. 2, 302–307.
39. Pak, W. L. ( 1979) in Neurogenetics, Genetic Approaches to the Nervous System, ed. Breakfield, X. O. (Elsevier, New York), pp. 67–99.
40. Ranganathan, R., Harris, G. L., Stevens, C. F. & Zuker, C. S. ( 1991) Nature (London) 354, 230–235.
41. Phillips, A., Bull, A. & Kelly, L. ( 1992) Neuron 8, 631–642.
42. Cossens, D. & Manning, A. ( 1969) Nature (London) 224, 285– 287.
43. Minke, B., Wu, C.-F. & Pak, W. L. ( 1975) Nature (London) 258, 84–87.
44. Minke, B. ( 1982) J. Gen. Physiol. 79, 361–385.
45. Montell, C. & Rubin, G. M. ( 1989) Neuron 2, 1313–1323.
46. Jan, L. & Jan, Y. ( 1992) Cell 69, 715–718.
47. Dolph, P. J., Ranganathan, R., Colley, N. J., Hardy, R. W., Socolich, M. & Zuker, C. S. ( 1993) Science 260, 1910–1916.
48. Nathans, J. & Hogness, D. ( 1983) Cell 343, 807–814.
49. Kaupp, U., Niidome, T., Tanabe, T., Terada, S., Bonigk, W., Stühmer, W., Cook, N., Kangawa, K., Matsuo, H., Hirose, T., Miyata, T. & Numa, S. ( 1989) Nature (London) 342, 762–766.
50. Noel, J. P., Hamm, H. E. & Sigler, P. B. ( 1993) Nature (London) 366, 654–663.
51. Dryja, T. ( 1992) Eye 6, 1–10.
52. Colley, N., Cassill, A., Baker, E. & Zuker, C. ( 1994) Proc. Natl. Acad. Sci. USA 92, 3070–3074.






