
11
Pulmonary Hypertension
In this chapter, the committee recommends that a new listing in the cardiovascular system listings be created to address pulmonary hypertension. Pulmonary hypertension, which is incurable despite treatment, is associated with functional limitations and significantly increases patient mortality. Most patients will meet criteria for disability based on the condition generally being fairly advanced at the time of diagnosis.
DESCRIPTION
The right side of the heart collects venous blood from the body and pumps it into the lungs, where oxygen is taken up from inspired air and carbon dioxide is released in the expired air. This critical circulation of blood through the vessels of the lungs—the pulmonary arteries, capillaries, and veins—normally occurs at much lower pressures than blood flow through the systemic circulation to the rest of the body. The normal pulmonary artery systolic pressure is 20 mm Hg or less, and the normal mean (average) pulmonary artery pressure is 12 mm Hg. A number of disease processes affect the pulmonary circulation and increase the pressure levels in the pulmonary arteries and right ventricle. If these pressure elevations are sufficiently severe or sustained, right-sided heart failure may develop.
Pulmonary hypertension is present when the pulmonary artery pressures are elevated above normal. The term applies particularly to diseases that affect the small pulmonary arteries and markedly increase their resistance to blood flow. Pulmonary hypertension of this type may be of unknown cause
(so-called primary pulmonary hypertension) or secondary to conditions, such as connective tissue disease, sarcoidosis, and certain inherited conditions. Pulmonary hypertension may develop as the result of chronic pulmonary thromboembolism. The condition may also develop secondary to lung disease or hypoxemia, such as chronic obstructive pulmonary disease, interstitial lung disease, and obstructive sleep apnea. Pulmonary hypertension secondary to lung disease is termed cor pulmonale (cor pulmonale is not discussed further because disability criteria are established in the pulmonary disease listings). Pulmonary hypertension is a relatively common complication of left-sided heart disease, such as mitral valvular disease or cardiomyopathy, or due to congenital heart disease. Pulmonary hypertension that develops as a secondary manifestation of another primary form of heart disease should be evaluated using the disability criteria established for the primary disease, and so is not discussed further in this chapter.
Pulmonary hypertension is associated with high mortality and with marked functional limitations. The condition is often quite advanced at the time of diagnosis, largely because its symptoms are nonspecific and the diagnosis is difficult to make.
EPIDEMIOLOGY
Pulmonary hypertension is an uncommon disease with an estimated prevalence of 2 per million in the United States. Women are affected much more often than men, and the typical age at the time of diagnosis is between 30 and 50. The disease is associated with genetic markers (mutations and polymorphisms in the Type II bone morphogenic protein receptor), and with the use of appetite suppressants.
DIAGNOSTIC CRITERIA AND METHODS
The definitive method for the diagnosis of pulmonary hypertension is a right heart catheterization with hemodynamic measurements.
Right Heart Catheterization
Pressures in the pulmonary artery, right ventricle, and right atrium can be measured directly during a right heart catheterization. Cardiac output and pulmonary capillary wedge pressure should also be measured to characterize fully the state of the pulmonary vessels.
Pulmonary hypertension is considered present when the mean pulmonary artery pressure is greater than 25 mm Hg at rest. Pulmonary arterial hypertension is a more specific diagnostic term that requires a pulmonary capillary wedge pressure of 15 mm Hg or less with a pulmonary vascular
resistance of greater than 3 Wood units (mm Hg/liters/minute) in the setting of an elevated mean pulmonary artery pressure (McLaughlin et al., 2009).
Doppler Echocardiography
Most patients with pulmonary hypertension have some degree of regurgitation of the tricuspid valve. The velocity of tricuspid regurgitation can be measured using Doppler echocardiography, and this can then be used to estimate the systolic pulmonary artery pressure. This estimated pressure correlates with the measured pressure, but the mean pulmonary artery pressure, the pulmonary capillary wedge pressure, and the pulmonary vascular resistance cannot be estimated with echocardiography.
Other Diagnostic Methods
The chest radiograph and the 12-lead electrocardiogram are commonly abnormal among patients with pulmonary hypertension, but are neither sensitive nor specific diagnostic markers. No biomarker has yet been established to be useful for the diagnosis of pulmonary hypertension.
Functional Assessment
The distance a patient can walk in 6 minutes (the “6-minute walk test”) has been used in many clinical trials to assess efficacy of treatment, but it is not commonly used in clinical practice. Although age and gender-based community norms have not been established for the 6-minute walk test, walking 500 meters or more in 6 minutes is considered normal.
Cardiopulmonary exercise testing can establish objectively the functional work capacity of the patient by establishing the patient’s anaerobic threshold and maximum oxygen uptake with exercise. However, formal exercise testing is used infrequently among patients with pulmonary hypertension.
TREATMENT
In 2009, the American College of Cardiology Foundation and American Heart Association issued an expert consensus document on pulmonary hypertension that reviewed in detail the recent advances in treatment (McLaughlin et al., 2009).
Patients with pulmonary artery hypertension are advised to avoid heavy physical exertion or isometric exercise, which may lead to loss of consciousness. Continuous intravenous infusion of prostacyclin analogs improves functional status and survival in patients with primary pulmonary hypertension. Other prostanoid drugs that are easier to administer have subse-
quently been developed (treprostinil, iloprost) and have promise for the treatment of primary pulmonary hypertension. Endothelin receptor antagonists (bosentan, sitaxsentan, ambrisentan) and phosphodiesterase inhibitors (sildenafil, tadalafil) also improve the symptoms and functional status of patients with pulmonary arterial hypertension. Severe cases of pulmonary hypertension can be treated with heart-lung transplantation.
Secondary forms of pulmonary hypertension may be treated by addressing the root cause of the disorder. Chronic pulmonary embolism may be treated surgically with pulmonary thromboendarterectomy in selected patients.
Recent Advances
Several new drug therapies have been developed over the past decade, with particular interest in the endothelin receptor antagonists and phosphodiesterase inhibitors.
Side Effects of Treatment
Continuous intravenous therapy with epoprostenol carries the risk of infection and difficulties of maintaining access for the drug. All drugs used to lower pulmonary artery pressures may also lower systemic blood pressures. Endothelin antagonists may cause liver damage.
DISABILITY
Functional Limitations
Patients with pulmonary hypertension have been shown to have marked limitations on standard functional status measures, such as the RAND Short-Form 36 scales (Chen et al., 2008; Rubenfire et al., 2009). Patients with pulmonary hypertension also have had evidence of reduced functional capacity on standard heart failure measures, such as the Minnesota Living with Heart Failure Scale (Zlupko et al., 2008).
Work Disability
Patients with pulmonary hypertension may be unable to work because of either functional limitations or their use of continuous intravenous treatment.
CURRENT LISTING
Pulmonary hypertension has no current listing. The current listing for cor pulmonale from the respiratory system listings (3.00) is:
3.09 Cor pulmonale secondary to chronic pulmonary vascular hypertension. Clinical evidence of cor pulmonale (documented according to 3.00G) with:
-
Mean pulmonary artery pressure greater than 40 mm Hg, or
-
Arterial hypoxemia. Evaluate under the criteria in 3.02C2 (SSA, 2008).
CONCLUSIONS AND RECOMMENDATION
Pulmonary hypertension is associated with functional limitations to exercise, and it significantly increases patient mortality. Pulmonary hypertension can be treated with drugs, but such treatment does not cure the disease. Because the disease is generally at a fairly advanced stage by the time of diagnosis, most patients with pulmonary hypertension will meet criteria for disability (Figure 11-1).
RECOMMENDATION 11-1. The Social Security Administration should establish a new listing in the cardiovascular system for pul-
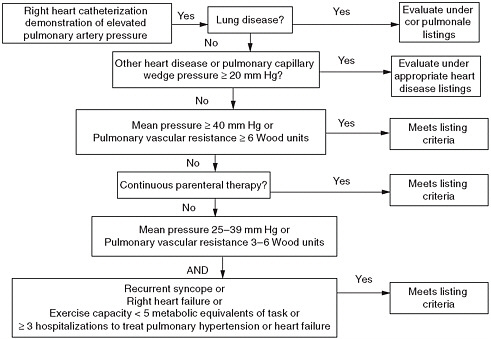
FIGURE 11-1 Meeting criteria for disability due to pulmonary hypertension.
monary hypertension. The new listing should allow claimants with pulmonary hypertension documented by right heart catheterization to meet the listing if (A) there is evidence of severe pulmonary hypertension OR (B) there is evidence of moderate pulmonary hypertension AND of marked functional limitations.
-
Evidence of severe pulmonary hypertension, which is associated with severe functional limitation, includes any of the following:
-
Mean pulmonary artery pressure of 40 mm Hg or greater; or
-
Pulmonary vascular resistance of 6 Wood units (mm Hg per liter per minute) or greater; or
-
Continuous parenteral therapy with prostacyclin analogs.
-
OR
-
Evidence of moderate pulmonary hypertension, which imposes severe functional limitation on many but not all individuals, includes any of the following:
-
Recurrent syncope secondary to pulmonary hypertension; or
-
Right heart failure (same criteria as for heart failure listing); or
-
Mean pulmonary artery pressure between 25 and 39 mm Hg; or
-
Pulmonary vascular resistance above 3 and below 6 Wood units.
-
AND
Evidence of marked functional disability provided by either of the following:
-
An exercise capacity of less than 5 metabolic equivalents of task; or
-
Three or more hospital admissions within a consecutive 12-month period to treat right heart failure or pulmonary hypertension.
REFERENCES
Chen, H., D. B. Taichman, and R. L. Doyle. 2008. Health-related quality of life and patient-reported outcomes in pulmonary arterial hypertension. Proceedings of the American Thoracic Society 5(5):623–630.
McLaughlin, V. V., S. L. Archer, D. B. Badesch, R. J. Barst, H. W. Farber, J. R. Lindner, M. A. Mathier, M. D. McGoon, M. H. Park, R. S. Rosenson, L. J. Rubin, V. F. Tapson, and J. Varga. 2009. ACC/AHA 2009 expert consensus document on pulmonary hypertension: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians, the American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Journal of the American College of Cardiology 53(17):1573–1619.
Rubenfire, M., G. Lippo, B. D. Bodini, F. Blasi, L. Allegra, and E. Bossone. 2009. Evaluating health-related quality of life, work ability, and disability in pulmonary arterial hypertension: An unmet need. Chest 136(2):597–603.
SSA (Social Security Administration). 2008. Listing of impairments—Adult listings (Part A). Disability evaluation under Social Security (Blue Book). http://www.socialsecurity.gov/disability/professionals/bluebook/AdultListings.htm (accessed July 22, 2010).
Zlupko, M., M. O. Harhay, R. Gallop, J. Shin, C. Archer-Chicko, R. Patel, H. I. Palevsky, and D. B. Taichman. 2008. Evaluation of disease-specific health-related quality of life in patients with pulmonary arterial hypertension. Respiratory Medicine 102(10):1431–1438.








